

Abstract
Background
Pituitary development and GH secretion are orchestrated by multiple genes including GH1, GHRHR, GLI2, HESX1, LHX3, LHX4, PROP1, POU1F1, and SOX3. We aimed to assess their mutation frequency and clinical relevance in children with severe GH deficiency (GHD).
Methods
The Genetics and Neuroendocrinology of Short Stature International Study (GeNeSIS; Clinical Trial Registry Number: NCT01088412) was a prospective, open-label, observational research program for pediatric patients receiving GH treatment, conducted in 30 countries between 1999 and 2015. The study included a sub-study to investigate mutations in the genes listed above. PCR products from genomic blood cell DNA were analyzed by Sanger sequencing. DNA variants were classified as pathogenic according to the recommendations of the American College of Medical Genetics and Genomics. Demographic, auxologic, and endocrine data at baseline and during GH treatment were documented and related to the genotyping results.
Findings
The analysis comprised 917 patients. In 92 patients (10%) 33 mutations were found, 16 previously described and 17 novel (52%). Mutation carriers were significantly younger, shorter, and more slowly growing than non-carriers. In general, their peak values in GH stimulation tests were very low; however, in 15/77 (20%) patients with GH1, PROP1, and SOX3 mutations they were only moderately diminished (3-6 μg/L). Two patients with a GH1 mutation developed TSH deficiency and one ADH deficiency. Using logistic multi-regression analysis, significant indicators of a mutation were combined pituitary hormone deficiency, greater patient-parent height difference (SDS), low GH peak, and young age. Final height SDS gain in mutation carriers (mean ± SD 3.4 ± 1.4) was greater than in non-carriers (2.0 ± 1.4; P < .001) and in patients with non-GHD short stature.
Interpretation
DNA testing for mutations in children with severe GHD shows a positive finding in approximately 10%. Phenotypes of mutation carriers can be variable. The benefit for clinical practice justifies DNA testing as an important component in the diagnostic work-up of patients with severe GHD.
Fund
Eli Lilly and Company, Indianapolis, IN, USA.
ClinicalTrials.com registration: NCT01088412.
Research in context.
Evidence before this study
We searched PubMed for articles published before the start of GeNeSIS in November 1999, and continuously thereafter until October 2017, for the terms: growth failure, dwarfism, short stature, pituitary development, hypopituitarism, growth hormone, GH, transcription factor genetics, GH deficiency, GH treatment, multiple pituitary hormone deficiency, combined pituitary hormone deficiency, mutations, GH1, GHRHR, HESX1, GLI2, LHX3, LHX4, POU1F1, PROP1, and SOX3. Short stature is the most frequent reason why children are presented to the pediatric endocrinologist and growth hormone (GH) deficiency (GHD) is the most frequent diagnosis that leads to GH treatment. However, diagnosing GHD is not trivial, because severity of GHD can range from no GH secretion to almost normal. In severe GHD, the metabolic GH effects, e.g. preventing hypoglycemia, may acutely be even more important, possibly life-saving, than the long-term growth-promoting effects. However, available data on genotype-phenotype correlations in patients with genetic causes of GHD are mainly based on cohorts of limited size. Although it is accepted that pathogenic mutations in genes controlling pituitary development or GH synthesis and secretion confirm the diagnosis of GHD better than any other diagnostic measures, DNA testing of such genes is not yet part of medical care in many countries. The reasons may be manifold: lack of available laboratories, cost, belief in rarity of mutations, and limited awareness of the clinically important information that can be gained from genetic findings.
Added value of this study
This study was a prospective, open-label, observational research program conducted in 30 countries and >800 study sites. To our knowledge this was the largest study (N = 917) to screen, under conditions of clinical practice, for DNA variants in nine “classical” candidate genes involved in pituitary development or GH secretion. Ninety-two (10%) of the screened patients (8.1% of kindreds) had a pathogenic or likely pathogenic mutation as defined by the American College of Medical Genetics and Genomics (ACMG). Novel DNA variants were found frequently, 17 (52%) in a total of 33 mutations. As expected, mutations in genes involved in pituitary development (GLI2, HESX1, LHX3, POU1F1, and PROP1) caused deficiency of other pituitary hormones besides GH before study entry or during follow-up. Unexpected at the time, however, was development of TSH and ADH deficiency in patients with GH1 mutation. A number of patients with GH1, PROP1, and SOX3 mutations had stimulated GH peak values between 3 and 6 μg/L. Indicators of a mutation were combined pituitary hormone deficiency, diminished height standard deviation score (SDS) minus target height SDS, very low GH concentration in GH stimulation tests, and manifestation of GHD at a young age. When results from pituitary imaging were also considered, the frequency of pathologic findings of DNA testing increased for certain genes up to 17%. Regional clustering of mutations was seen only with PROP1 mutations, which were frequent in East Central or Eastern European countries (up to 62% of kindreds in Lithuania). The short-term growth response to GH treatment over four years was greater in mutation carriers compared to non-carriers. The long-term response till final height (near-adult height) was significantly greater in mutation carriers with a height SDS gain of 3.4 ± 1.4 versus 2.0 ± 1.4 in non-carriers tested for DNA variants or in non-tested patients with organic (1.5 ± 1.6) or idiopathic (1.4 ± 1.0) GHD.
Implications of all the available evidence
The novel mutations identified in this study provide new insights into the biology of the affected genes and proteins and the highly variable clinical phenotypes. The findings may assist in clinical practice in selecting the patients appropriate for DNA testing, which should not necessarily be precluded by low normal height or “measurable” stimulated GH values. Testing for mutations in GH1 should be part of the test panel even if other pituitary hormones in addition to GH are also deficient. The patient and parent benefits of a genetic diagnosis are manifold and include information for genetic counselling and state-of-the-art clinical management. A mutation in any of the genes tested in this study should alert the physician to monitor indefinitely for development of pituitary hormone deficiencies other than GHD. A genetic diagnosis for GHD also predicts an excellent growth outcome of GH therapy. Balancing the cost of genetic testing against the frequency of pathogenic findings and the value of the information, the authors recommend making DNA testing an important component in the diagnostic work-up of GHD.
Alt-text: Unlabelled Box
1. Introduction
Since the early 1980s, numerous single gene defects associated with impaired growth have been identified, including defects in genes related to pituitary development, growth hormone (GH) secretion and GH action [[1], [2], [3], [4], [5]]. Specific cells in the anterior pituitary gland produce GH, thyroid-stimulating hormone (TSH), gonadotropins (luteinizing hormone [LH]/follicle stimulating hormone [FSH]), adrenocorticotropin (ACTH), and prolactin (PRL). The development of the pituitary gland is complex with highly organized temporal and spatial expression of transcription factors including GLI2, HESX1, LHX3, LHX4, PROP1, POU1F1, and SOX3 that shape the developing pituitary and specify hormone producing cells.
Defects in the GH1 and GHRHR genes, involved in the control of GH secretion, typically cause isolated GH deficiency (IGHD) [1,2]. However, the phenotypes can be variable and IGHD may evolve to combined pituitary hormone deficiencies (CPHD) with time in patients with a GH1 mutation [3,6]. Genetic IGHD has been classified into four categories: type IA (severe, autosomal recessive), IB (less severe, autosomal recessive), II (autosomal dominant) and III (X-linked) [3,7]. Pathogenic mutations in pituitary transcription factors typically cause a wide range of phenotypes, from IGHD to severe, life-threatening CPHD. Mutations in genes expressed early in pituitary development - as in HESX1, SOX3 and GLI2 - often entail additional midline and facial anomalies [1,2,5].
The diagnosis of pediatric GHD is based commonly on clinical history, physical examination, auxology, GH stimulation tests, serum insulin-like growth factor-I (IGF-I), and pituitary imaging. Although genetic evaluation is becoming increasingly important, auxological and GH treatment outcome data in genetic GHD result mostly from cohorts of limited size [8]. A large cohort of pediatric patients with severe GHD was tested for disease-related mutations within the prospective, observational Genetics and Neuroendocrinology of Short Stature International Study (GeNeSIS). The analyses intended to identify novel DNA variants, to determine the frequency of mutations, their geographic distribution, clinical phenotypes, and likely GH treatment outcomes.
2. Patients and methods
2.1. Study design and patients
GeNeSIS was a prospective, open-label, observational research program conducted in 30 countries at >800 study sites (Clinical Trial Registry Number: NCT01088412) between 1999 and 2015. It was set up as a Core Study on safety and effectiveness of GH treatment for growth, with topic-focused sub-studies that included a DNA Analysis Sub-study for investigating mutations in genes involved in pituitary development or GH secretion (Supplementary Table S1). Patients with severe GHD according to the discretion of the investigator were invited to provide a blood sample for DNA analysis. Patients were classified as having IGHD or CPHD based on clinical and biochemical information reported by the site. The algorithms for diagnosing deficiencies of TSH, gonadotropins, ACTH, PRL or ADH have been previously described in detail [9]. The study was conducted in accordance with the Declaration of Helsinki and approved by ethics review boards of the participating institutions. Written informed consent for data collection, data processing, DNA testing and publication was obtained from the parents or legal guardians, in accordance with national regulatory requirements.
2.2. Data collection
Data were collected by investigators according to their common practices and entered on case report forms. They included aetiology of GHD, demographic, auxologic and biochemical data, status of other pituitary hormones, and information on abnormal brain imaging [10]. Baseline data were collected before initiation of GH treatment and follow-up data were typically collected at 6-month intervals. The diagnosis of GHD in an individual patient was accepted as provided by the investigator irrespective of results of GH testing. If several GH stimulation tests were reported, the highest GH peak value was used in statistical analyses. Serum IGF-I and IGFBP-3 concentrations were determined either centrally (University Children's Hospital, Giessen, Germany) or at local laboratories and converted to central laboratory equivalent values using previously published algorithms [10].
2.3. Genetic analysis
DNA analyses were performed at University Hospital for Children and Adolescents, Leipzig, Germany, or Institut National de la Santé et de la Recherche Médicale (INSERM), Créteil then Paris, France. EDTA blood samples were sent to the central laboratories together with information on diagnosis, dysmorphic signs, MRI findings, and pituitary secretory status. Patients with IGHD as stated in the transmittal form were typically tested for mutations in GH1 and GHRHR, while patients with CPHD were typically tested for mutations in GLI2, HESX1, LHX3, LHX4, POU1F1, PROP1, and SOX3. DNA testing was also performed in a number of parents and siblings (27% of kindreds), if a mutation was found in the index case and DNA was provided. All genes were analyzed in a targeted re-sequencing approach using genomic DNA (QIAamp DNA Blood Mini Kit; Qiagen, Hilden, Germany). Coding exons and neighbouring intron sequences were amplified by PCR as described previously with some modifications (Supplementary Table S1). For GH1 a long-range pre-PCR was performed to identify large deletions [11]. SOX3 was analyzed only partially for the sequence encoding the polyalanine tract (c.700 to c.744) [12]. Primer sequences can be obtained upon request. PCR products were prescreened for any sequence difference to a reference DNA sample from a healthy subject by single-stranded conformation polymorphism technique (SSCP; n = 64) until 2001 and thereafter by denaturing HPLC (n = 801; WAVE, Transgenomic, Glasgow, UK). PCR products with suspicious electrophoretograms or chromatograms were further analyzed by dideoxy (Sanger) sequencing (BigDye Terminator; 310 or 3730 XL Genetic Analyzer; Thermo Fisher Scientific, Waltham, MA). The sensitivity of the methodology was approximately 85% for SSCP and 96% for dHPLC [13]. PCR products of 52 patients were directly sequenced. Sequences were compared to the human genomic reference (GRCh37/hg19) obtained from the UCSC Genome Browser and variant positions were annotated according to gene-specific transcript and protein references given in Supplementary Tables S1 and S2.
Variants with a minor allele frequency (MAF) of <1% in at least two of the three large-scale variant databases accessible at the time of evaluation (Supplementary Table S2) were pre-selected followed by classification as pathogenic, likely pathogenic, uncertain significance, likely benign, or benign according to the recommendations of the American College of Medical Genetics and Genomics (ACMG) [14]. All in silico analyses were performed in April 2017. Patients with variants classified as pathogenic or likely pathogenic constitute the cohort of “patients with mutations” or “carriers”, whereas patients without variants or with variants classified as likely benign, benign or uncertain significance constitute the cohort of “patients without mutations” or “non-carriers”. A mutation was annotated as “novel”, if all of the following criteria were met: no record in the disease-related variant databases ClinVar and HGMD, no relevant result in a text search using the three- and one-letter codes for amino acids in PubMed, Google and Google Scholar (Supplementary Table S2), no relevant findings on inspection of recent reviews [2,3,5,15]. All surveys were performed in July 2017.
2.4. Statistics
Three patient populations were defined for statistical analysis: (i) the baseline population (N = 917) comprised all patients with GHD in the DNA Analysis Sub-study; (ii) the four-year population (N = 240) included all GH-naïve patients with available height SD score (HtSDS) at baseline and at one, two, three and four years of GH treatment; (iii) the final height (FH [near adult height, last measured height]) population (N = 215) included all patients who fulfilled at least one of the following criteria: closed epiphyses, height velocity < 2 cm/y, or bone age ≥ 14 y in girls and ≥ 16 y in boys. Patients from the GeNeSIS Core Study with a diagnosis of organic or idiopathic GHD or with other non-GHD forms of short stature approved for GH treatment were analyzed the same way for comparison of FH outcomes. Age and sex-based SDS values for height (US National Center for Health Statistics), height velocity, body mass index, target height (TH) according to Tanner, bone age, serum IGF-I, and IGFBP-3 were calculated as described previously [9,10]. Results are presented as mean ± SD unless otherwise stated. Comparisons among groups were performed by analysis of variance (ANOVA) for continuous variables and by Fisher's exact test for categorical variables. For identifying indicators of mutations, logistic multi-regression analysis was performed with varied sets of explanatory variables, applying full models and stepwise selection models. Model quality was tested using the Hosmer & Lemeshow goodness-of-fit test. Results were considered different at a two-sided significance level of 0.05. Statistical analyses were performed using SAS software (version 8.2, SAS Institute Inc., Cary NC).
2.5. Data sharing
Lilly provides access to all individual participant data collected during the study, after anonymization, with the exception of pharmacokinetic or participant-specific genetic data. Data are available to request in a timely fashion after the indication studied has been approved in the US and EU and after primary publication acceptance. No expiration date of data requests is currently set once they are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment for up to 2 years per proposal. For details on submitting a request, see the instructions provided at www.clinicalstudydatarequest.com.
3. Results
3.1. Patients
The selected cohort that underwent DNA testing represented 6.5% (917/14,138) of GeNeSIS participants who had a diagnosis of GHD. These 917 patients comprised 440 with idiopathic GHD (48%) and 475 with organic GHD (52%), most of the latter with congenital GHD (457 [96%]); 475 (52%) had IGHD, 415 (45%) CPHD and 27 (3%) could not be classified. Samples for genetic testing were received from 25 countries (Supplementary Table S3) with mutations identified in up to 70% of the patients and 64% of kindreds (Lithuania). There was no conspicuous regional accumulation of gene mutations except for PROP1 in East Central or Eastern European countries.
3.2. DNA variants
Mutations were found in 92 patients (10.0% [Table 1]; 70 unrelated, 8.1% of kindreds [Supplementary Table S4]). Thirty-three distinct mutations that met the ACMG criteria for pathogenicity were identified in 31 unique genotypes (Table 2, Table 3). Sixteen mutations had been described previously, including all 6 PROP1 mutations, while 17 mutations (52%) were newly identified in GeNeSIS. Seven of these 17 novel mutations, including all five LHX3 mutations, were published separately before (Table 2).
Table 1.
Number and percentage of patients with mutations.
| Gene | All in study |
IGHD |
CPHD |
Unknowna |
Patients tested |
Mutations in tested patients |
|---|---|---|---|---|---|---|
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | |
| All studied | 917 (100.0%) | 475 (100.0%) | 415 (100.0%) | 27 (100.0%) | ||
| All without mutation | 825 (90.0%) | 444 (93.5%) | 354 (85.3%) | 27 (100.0%) | ||
| All with mutation | 92 (10.0%) | 31 (6.5%) | 61 (14.7%) | 0 (0.0%) | ||
| GH1 | 26 (2.8%) | 23 (4.8%) | 3 (0.7%) | 0 (0.0%) | 531 (57.9%) | 26 (4.9%) |
| GHRHR | 5 (0.5%) | 5 (1.1%) | 0 (0.0%) | 0 (0.0%) | 526 (57.4%) | 5 (1.0%) |
| GLI2 | 1 (0.1%) | 0 (0.0%) | 1 (0.2%) | 0 (0.0%) | 123 (13.4%) | 1 (0.8%) |
| HESX1 | 2 (0.2%) | 0 (0.0%) | 2 (0.5%) | 0 (0.0%) | 417 (45.5%) | 2 (0.5%) |
| LHX3 | 5 (0.5%) | 0 (0.0%) | 5 (1.2%) | 0 (0.0%) | 396 (43.2%) | 5 (1.3%) |
| LHX4 | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 224 (24.4%) | 0 (0.0%) |
| POU1F1 | 2 (0.2%) | 0 (0.0%) | 2 (0.5%) | 0 (0.0%) | 393 (42.9%) | 2 (0.5%) |
| PROP1 | 49 (5.3%) | 1 (0.2%) | 48 (11.6%) | 0 (0.0%) | 418 (45.6%) | 49 (11.7%) |
| SOX3 | 2 (0.2%) | 2 (0.4%) | 0 (0.0%) | 0 (0.0%) | 445 (48.5%) | 2 (0.4%) |
Patients are split by pituitary secretory status (isolated GHD [IGHD] or combined pituitary hormone deficiencies [CPHD]).
Pituitary secretory status could not be unequivocally assigned.
Table 2.
Mutations identified within the study.
| no. | cDNA change | Protein change (shown or predicted) | Type of variation⁎ | Zygosity† | Ref.‡ | Genotype frequency [% (n)] | Minor allele frequency (%) this study (n); [1kG/ESP/ExAC/gnomAD]§ | Predicted impact PolyPhen/SIFT/MutationTaster§ |
|---|---|---|---|---|---|---|---|---|
| GH1 | ||||||||
| 1 | 6.7 kB gene deletion | p.0 | Del | Ho | [45] | 0.407(2) | 0.407 (4); [n.a.] | n.a. |
| 2 | 7.0 kB gene deletion | p.0 | Del | Ho | [46] | 0.203 (1) | 0.203 (2); [n.a.] | n.a. |
| 3 | c.178G>A | p.Ala60Thr (see text) | missense | He | Novel | 0.203 (1) | 0.102 (1); [ – /– /– /– ] | Benign /benign /benign |
| 4 | c.275A>G | p.Glu92Gly (see text) | missense | He | Novel | 0.203 (1) | 0.102 (1); [ – /– /– /– ] | Damaging /damaging /benign |
| 5 | c.291+1G>A | p.Glu58_Ser97del¶ | SD | He | [47] | 2.033 (10) | 1.016 (10); [ – /– /– /– ] | n.a. /n.a. /damaging |
| 6 | c.291+2T>C | p.Glu58_Ser97del¶ | SD | He | [48] | 0.203 (1) | 0.102 (1); [ – /– /– /– ] | n.a. /n.a. /damaging |
| 7 | c.291+3G>C | p.Glu58_Ser97del¶ | SD | He | Novel | 0.407 (2) | 0.203 (2); [ – /– /– /– ] | n.a. /n.a. /damaging |
| 8 | c.291+5G>A | p.Glu58_Ser97del¶ | SD | He | [49] | 0.203 (1) | 0.102 (1); [ – /– /– /– ] | n.a. /n.a. /damaging |
| 9 | c.292-37_292-16del | p.Glu58_Ser97del¶ | BS | He | Novel [50] | 0.203 (1) | 0.102 (1); [ – /– /– /– ] | n.a. /n.a. /benign |
| 10 | c.499C>T | p.Gln167Ter | nonsense | He | Novel | 0.203 (1) | 0.102 (1); [ – /– /– /– ] | n.a. /n.a. /damaging |
| GHRHR | ||||||||
| 11 | c.57+1G>T | p.? | SD | cHe[S5:5] | Novel | 0.204 (1) | 0.102 (1); [ – /– /– /– ] | n.a. /n.a. /damaging |
| 12 | c.214G>T | p.Glu72Ter | nonsense | cHe17 | [51] | 0.204 (1) | 0.102 (1); [ 0.02 /– /0.048 /0.019] | n.a. /n.a. /damaging |
| 13 | c.335G>A | p.Cys112Tyr | missense | cHe16 | Novel | 0.204 (1) | 0.102 (1); [ – /– /– /– ] | Damaging /benign /damaging |
| 14 | c.758C>T | p.Pro253Leu | missense | cHe15 | Novel | 0.204 (1) | 0.102 (1); [ – /– /– /– ] | Damaging /damaging /damaging |
| 15 | c.812+1G>A | p.? (see citation) | SD | cHe14 | [27] | 0.204 (1) | 0.102 (1); [ – /– /8.2E–4 /4.1E–4] | n.a. /n.a. /damaging |
| 16 | c.1089_1093del | p.Leu364Phefs⁎21 | Del-FS | cHe13 | [52] | 0.204 (1) | 0.102 (1); [ – /0.024 /7.0E–3 /5.1E–3] | n.a. /n.a. /damaging |
| 17 | c.1102C>T | p.Gln368Ter | nonsense | cHe12 | Novel | 0.204 (1) | 0.102 (1); [ – /– /– /– ] | n.a. /n.a. /damaging |
| GLI2 | ||||||||
| 18 | c.1807_1810dup | p.His604Argfs⁎15 | Ins-FS | He | Novel | 0.826 (1) | 0.413 (1); [ – /– /– /– ] | n.a. /n.a. /damaging |
| HESX1 | ||||||||
| 19 | c.385_386ins315 | p.? (see citation) | Ins-FS | Ho | Novel [30] | 0.254 (1) | 0.254 (2); [ – /– /– /– ] | n.a. |
| 20 | c.479G>A | p.Arg160His | missense | dHe[S5:12] | [34] | 0.270 (1) | 0.127 (1); [ – /– /– /1.1E–3] | Damaging /damaging /damaging |
| LHX3 | ||||||||
| 21 | c.252-3C>G | p.? (see citation) | SA | cHe23 | Novel [53] | 0.269 (1) | 0.134 (1); [ – /– /– /– ] | n.a. /n.a. /damaging |
| 22 | c.287_288delinsTCCT | p.Gly96Valfs⁎78 | Ins-Del-FS | Ho | Novel [29] | 0.269 (1) | 0.269 (2); [ – /– /– /– ] | n.a. /n.a. /damaging |
| 23 | c.353G>A | p.Cys118Tyr | Missense | cHe21 | Novel [53] | 0.269 (1) | 0.134 (1); [ – /– /– /– ] | Damaging /damaging /damaging |
| 24 | c.629C>T | p.Ala210Val | Missense | Ho | Novel [29] | 0.269 (1) | 0.269 (2); [ – /– /– /– ] | Damaging /damaging /damaging |
| 25 | c.672G>A | p.Trp224Ter | Nonsense | Ho | Novel [29] | 0.269 (1) | 0.269 (2); [ – /– /– /– ] | n.a. /n.a. /damaging |
| POU1F1 | ||||||||
| 26 | c.427C>T | p.Arg143Ter | nonsense | Ho | Novel | 0.271 (1) | 0.271 (2); [ – /– /– /– ] | n.a. /n.a. /damaging |
| PROP1 | ||||||||
| 27 | gene deletion | p.0 | Del | Ho | [19] | 0.255 (1) | 0.255 (2); [n.a.] | n.a. |
| 28 | c.150del | p.Arg53Aspfs⁎112 | Del-FS | Ho, cHe32 | [54] | 2.551 (10) | 1.403 (11); [ – /– /– /9.7E–3] | n.a. /n.a. /damaging |
| Ho: 0.255 (1) | ||||||||
| cHe: 2.296 (9) | ||||||||
| 29 | c.211C>T | p.Arg71Cys | missense | cHe32 | [55] | 0.255 (1) | 0.128 (1); [ – /– /3.3E–3 /2.0E–3] | Damaging /damaging /damaging |
| 30 | c.217C>T | p.Arg73Cys | missense | Ho | [56] | 0.510 (2) | 0.510 (4); [ – /– /8.3E–4 /7.2E–4] | Damaging /damaging /damaging |
| 31 | c.295C>T | p.Arg99Ter | nonsense | Ho, cHe32 | [57] | 0.510 (2) | 0.383 (3); [ – /– /– /7.2E–4] | n.a. /n.a. /damaging |
| Ho: 0.255 (1) | ||||||||
| cHe: 0.255 (1) | ||||||||
| 32 | c.301_302del | p.Leu102Cysfs⁎8 | Del-FS | Ho, cHe28,29,31 | [58] | 7.908 (31) | 6.505 (51); [ – /0.072 /0.014 /0.018] | n.a. /n.a. /damaging |
| Ho: 5.102 (20) | ||||||||
| cHe: 2.806 (11) | ||||||||
| SOX3 | ||||||||
| 33 | c.712_744dup | p.Ala238_Ala248dup | Ins (in frame) | X-Hem | [59] | 0.233 (1) | 0.172 (1); [ – /– /– /– ] | n.a. /n.a. /benign |
Numbers of mutations and genotypes and corresponding percentages refer to kindreds. Mutations in bold = transitions at CpG sites. n.a. = not applicable.
BS = branch site affected (splicing); Del = deletion; ESE = exonic splicing enhancer affected; FS = frameshift; Ins = insertion; SA = splice acceptor site affected; SD = splice donor site affected.
Ho = homozygous; He = heterozygous; X-Hem = X-linked hemizygous; cHe = compound heterozygous; dHe = double heterozygous, the no. (1st column) of the co-occurring variant/s is shown as superscript; numbers in brackets refer to variants listed in Supplementary Table S5.
Citations for variants identified in this study and previously published elsewhere are shown as superscript.
For details see Supplementary Table S2.
GH1 p.Glu58_Ser97del results from exon 3 skipping (17.5 k isoform).
Table 3.
Demographic data and phenotypic features of unpublished patients with previously undescribed (novel) mutations.⁎
| Pat. | Genotype (allele 1 /allele 2) | Inheritance (allele 1/allele 2) | Nationality | Sex | Mother's height (SDS) | Father’s height (SDS) | BL age (y) | Pre-HV (SDS) | BL Ht (SDS) | Max. GH peak (μg/L) | Additional pit. hormone deficiencies | Reported hypothalamic-pituitary morphology and other anomalies |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GH1 | ||||||||||||
| A | c.[499C>T];[?] | ND /ND | Russia | F | ND | ND | 13.1 | −1.4 | −3.4 | 1.5 | ||
| p.[Q167⁎];[?] | ||||||||||||
| B | c.[178G>A];[=] | Ma /ND | Spain | M | −3.7 | −0.8 | 9.2 | ND | −0.4 | 4.7 | ADH | Histiocytosis |
| p.[A60T];[A60=] | ||||||||||||
| C | c.[291+3G>C];[=] | ND /ND | Germany | M | −0.1 | −0.1 | 1.7 | −2.7 | −4.2 | 0.2 | Acromicria | |
| p.[E58_S97del];[E58_S97=] | ||||||||||||
| D | c.[291+3G>C];[=] | ND /ND | Belgium | M | 2.3 | 1.1 | 1.3 | ND | −4.6 | 0.7 | Prominent forehead | |
| p.[E58_S97del];[E58_S97=] | ||||||||||||
| E1 (index) | c.[275A>G];[=] | Ma /Pa | France | M | −1.4 | 1.0 | 1.7 | −1.7 | −3.4 | 5.9 | Prominent forehead | |
| p.[E92G];[E92=] | ||||||||||||
| E2 (brother) | c.[275A>G];[=] | Ma /Pa | France | M | −1.4 | 1.0 | 3.2 | −2.7 | −3.3 | 3.8 | Prominent forehead | |
| p.[E92G];[E92=] | ||||||||||||
| E3 (cousin) | c.[275A>G];[=] | Ma /Pa | France | M | −1.9 | −0.7 | 2.1 | −1.7 | −3.1 | 2.7 | Prominent forehead | |
| p.[E92G];[E92=] | ||||||||||||
| GHRHR | ||||||||||||
| F | c.[1102C>T];[214G>T] | Ma /ND | Thailand | M | −0.4 | −1.2 | 7.4 | −4.0 | −4.0 | <0.5 | Trunk obesity | |
| p.[Q368⁎];[E72⁎] | ||||||||||||
| G | c.[758C>T];[812+1G>A] | Ma /Pa | Thailand | M | −0.2 | −1.0 | 6.9 | ND | −4.8 | 0.7 | PH | |
| p.[P253L];[(see ref. 27)] | ||||||||||||
| H | c.[335G>A];[1089_1093del] | Ma /Pa | Australia | F | −1.7 | −1.0 | 5.5 | ND | −3.9 | 0.3 | Mild jaundice | |
| p.[C112Y];[L364Ffs⁎21] | ||||||||||||
| I1 (index) | c.57+1G>T(;)177G>C | ND /ND | USA | M | −0.1 | 0.5 | 7.8 | −6.2 | −4.1 | 1.1 | ||
| p.(unknown)(;)W59C | ||||||||||||
| I2 (sister) | c.57+1G>T(;)177G>C | ND /ND | USA | F | −0.1 | 0.5 | 5.4 | −3.2 | ND | 1.3 | ||
| p.(unknown)(;)W59C | ||||||||||||
| GLI2 | ||||||||||||
| J | c.[1807_1810dupGTGC];[=] | Ma /ND | India | M | −0,9 | −1,6 | 6.2 | ND | −3.8 | 0.1 | TSH, ACTH | EPP, PH Hypoglycemia, micropenis, acromicria, late dentition, late closure of fontanelles, dry skin, trunk obesity |
| p.[H604Rfs⁎15];[H604=] | ||||||||||||
| POU1F1 | ||||||||||||
| K1 (index) | c.[427C>T];[427C>T] | Ma /Pa | India | F | −0,5 | −2,3 | 3.2 | ND | −10.8 | 0.1 | TSH | Hypoglycemia, prominent forehead, late dentition |
| p.[R143⁎];[R143⁎] | ||||||||||||
| K2 (sister) | c.[427C>T];[427C>T] | Ma /Pa | India | F | −0,5 | −2,3 | 2.0 | ND | −8.3 | 0.1 | TSH | |
| p.[R143⁎];[R143⁎] | ||||||||||||
For details of published patients with novel mutations identified in GeNeSIS see references in Table 2. BL = baseline; SDS = standard deviation score; Pre-HV = height velocity before start of GH treatment; Ht = height. Ma = maternal. Pa = paternal. F = female. M = male; ND = not determined; PH = pituitary hypoplasia; EPP = ectopic posterior pituitary.
Most sequence aberrations were single nucleotide substitutions (22/33; 67%) with a large proportion being transitions affecting highly mutable CpG dinucleotides (7/22, 32% of substitutions, 6/7 reported elsewhere, [Table 2]) [16,17]. Only two distinct whole GH1 deletions in 3 kindreds and 1 PROP1 deletion in an Indian patient were found. The latter differed in its breakpoints from the PROP1 deletion found frequently in Kurdish populations [18,19]. The most prevalent mutations in our cohort were GH1:c.291 + 1G > A (n = 10), PROP1:c.150del (n = 11), and PROP1:c.301_302del (n = 51). Although the majority of the remaining mutations occurred once (n = 19), only 11 were presumably private; 8 were published elsewhere and/or are listed in the large variant databases. Twelve DNA variants classified as “uncertain significance or benign” are listed in Supplementary Table S5 and further details are described in the text which follows that table. These patients were considered as non-carriers in statistical analyses.
3.3. Demographics and phenotypes
The total number of girls (348/917 [38%]) was smaller than that of boys (569/917 [62%]), whereas the percentage of girls with a mutation was somewhat greater (43 [12.4%] versus 49 [8.6%]). Although all patients were expected to have severe GHD, most clinical and biochemical parameters before the start of GH treatment were significantly different between mutation carriers and non-carriers (Table 4): they were younger with a more retarded bone age, they were shorter compared to their peers (HtSDS) or parents (HtSDS – THSDS) and their growth rate was more compromised (height velocity SDS). Moreover, stimulated GH, serum IGF-I SDS, serum IGFBP-3 SDS, and fasting blood glucose were significantly smaller in mutation carriers. Patients with GH1 mutations (3.2y) were younger at baseline than patients with GHRHR (6.6y) or PROP1 (6.6y) mutations (Supplementary Table S6). Moreover, the average THSDS (mid-parental height SDS) of patients with GH1 or LHX3 mutations were decreased compared to that of patients with PROP1 mutations. Heights below the fifth percentile of either the mother or father were present in 7 of 22 (32%) kindreds with a GH1 mutation and in 3 of 5 (60%) kindreds with a LHX3 mutation.
Table 4.
Baseline characteristics of patients with GH deficiency with or without mutations.
| Parameter | With mutation |
Without mutation |
P⁎ | ||
|---|---|---|---|---|---|
| n | Mean ± SD | n | Mean ± SD | ||
| Chronological age (y) | 90 | 5.7 ± 4.2 | 811 | 7.3 ± 4.7 | 0.004 |
| Bone age SDS | 54 | −3.7 ± 2.0 | 487 | −2.7 ± 1.8 | <0.001 |
| Height SDS | 74 | −4.1 ± 2.2 | 734 | −2.9 ± 1.5 | <0.001 |
| Target height SDS | 87 | −0.2 ± 1.0 | 748 | −0.5 ± 1.0 | 0.013 |
| Height SDS – target height SDS | 70 | −4.0 ± 2.0 | 674 | −2.4 ± 1.6 | <0.001 |
| Height velocity SDS | 35 | −2.3 ± 2.0 | 365 | −1.5 ± 1.9 | 0.020 |
| BMI SDS | 75 | −0.6 ± 2.2 | 686 | −0.7 ± 2.0 | 0.898 |
| Max. stimulated GH peak (μg/L)† | 85 | 1.1 (0.5; 2.1) | 699 | 3.8 (1.3; 6.8) | <0.001 |
| IGF-I SDS | 27 | −5.9 ± 3.5 | 285 | −3.4 ± 3.3 | <0.001 |
| IGFBP-3 SDS | 26 | −4.7 ± 3.0 | 271 | −1.5 ± 2.6 | <0.001 |
| Fasting glucose (mg/dL) | 23 | 65.4 ± 14.7 | 172 | 75.3 ± 15.9 | 0.005 |
P-value by ANOVA.
Because of skewed distribution values are provided as median (1st quartile; 3rd quartile); P-value for rank-transformed data. y = year; SDS = standard deviation score; BMI = body mass index.
While most mutation carriers had very low reported GH peak levels, there were exceptions (Fig. 1): 10 of 47 patients with PROP1, 4/23 with GH1, and 1 of 2 with SOX3 mutation had values between 3 and 6 μg/L. When mutation carriers were grouped by type of IGHD, those with type I were significantly shorter than those with type II or III (Fig. 2A). HtSDS correlated significantly with log(GH peak) with clustering of IGHD type III and type II patients bearing mutations in GH1 exonic splicing enhancer sites in the upper range (Fig. 2B).
Fig. 1.

Maximum GH peak in stimulation tests by chronological age in patients with mutations. If more than one test was performed in a patient, the overall highest peak is presented.
Fig. 2.

Baseline height and stimulated GH peak serum concentrations of patients with IGHD depend on the type of mutation.
(A) Mean ± SD of height SDS in patients (n = 21) and affected relatives (n = 12, framed symbols) with IGHD type I, type II and type III. IGHD type IA with a very severe phenotype was due to GH1 deletions (#1, and #2 in Table 2) while IGHD type IB with a less severe phenotype was due to GHRHR mutations (#11–17 in Table 2). IGHD type II resulted from GH1 mutations #3–8 in Table 2, and IGHD type III from a SOX3 mutation (#33). The difference between IGHD type I vs. types II and III was significant by ANOVA (P < .001). (B) Height SDS versus log2(maximum GH peak) in patients (n = 19) and relatives (n = 6) with IGHD types I, II, and III. Pearson correlation (r = 0.51, P = .0099) and non-linear regression fitting the log2 presentation of the X-axis (hatched line) were calculated using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA). ESE = carriers of mutations affecting putative exonic splicing enhancers. Intronic = invariant and other intronic splice site mutations.
Sixty-six percent of mutation carriers had additional pituitary hormone deficiencies vs. 43% of non-carriers (P < .001). The most frequent was TSH deficiency (64% vs. 34%, P < .001) followed by gonadotropin deficiency (27% vs. 13%, P < .001), ACTH deficiency (16% vs. 25%, P = .072), PRL deficiency (13% vs. 5%, P = .008) and ADH deficiency (1% vs. 3%, P = .349). One patient with PROP1 mutation had IGHD at baseline and six had ACTH deficiency (12%). Two patients with a GH1c.291 + 1G > A mutation, known to cause exon 3-skipping, developed TSH deficiency. One patient with a family history of GHD had ADH deficiency and received a diagnosis of histiocytosis during the study, which is unlikely to be related to his GH1 mutation.
Morphological anomalies of the pituitary gland were reported in 324 (39%) patients without a mutation and in 22 (24%) with a mutation (Supplementary Table S7). Patients had single anomalies or combinations of the following: septo-optic dysplasia (SOD), ectopic posterior pituitary (EPP), stalk defect (SD; hypoplasia or interruption), pituitary hypoplasia (PH), pituitary aplasia (PA), or pituitary enlargement (PE). Four patients with PROP1 mutations (4/49; 8%) had PE. None of the mutation carriers showed the classical triad (EPP, SD, PA/PH).
Significant indicators of mutation, by logistic multi-regression analysis in all patients, were CPHD, low baseline HtSDS minus THSDS, and low GH peak. In the subset of patients with IGHD, indicators of mutations in GH1 or GHRHR were young age and low GH peak. In patients with CPHD, the only significant indicator of mutations in GLI2, HESX1, LHX3, POU1F1, PROP1 or SOX3 was low HtSDS minus THSDS. Sex, baseline bone age SDS, or IGF-I SDS were not significant indicators in any model. Excluding patients with EPP, SOD, or SD - which are not caused by GH1 or PROP1 mutations - increased positive GH1 mutation screening from 4.8% to 5.5% in patients with IGHD, and positive PROP1 mutation screening from 11.6% to 17.0% in patients with CPHD. Four of six patients with PE had a PROP1 mutation.
3.4. Growth response to GH treatment
The growth response in the longitudinal four-year population is shown in Fig. 3 and in Supplementary Table S8. Patients with a mutation were more than two years younger at start of GH treatment. Their annualized mean increase in height velocity, height velocity SDS, and change in HtSDS during the first two years was significantly greater than in non-carriers despite significantly smaller GH doses. In the FH population, mean GH doses at the start and the end of treatment were smaller in mutation carriers (Table 5). Though starting from lower HtSDS at baseline, patients with a mutation ended up at a similar FHSDS as non-carriers because of a significantly greater HtSDS gain (3.4 ± 1.4 vs. 2.0 ± 1.4, P < .001), and the percentage of patients who achieved a FH within the normal range (>−2 SDS) did not differ (83% vs. 84%). Comparison between all patients selected for DNA testing (with and without mutations) and other GeNeSIS diagnostic groups revealed a greater HtSDS gain in the tested patients despite smaller average GH doses (Fig. 4).
Fig. 3.

Growth response to GH treatment versus year of therapy. (A) Height SDS, (B) height SDS – target height SDS, (C) height velocity SDS, (D) annualized change in height SDS per year. Patients with mutation (circles, red) versus without mutation (squares, blue). The graphs show the data of the longitudinal four-year population with available height SDS at each time-point (N = 27 with a mutation, N = 213 without a mutation). Data are presented as mean ± SEM, P-values by year, ns = not significant.
Table 5.
Baseline characteristics and final height outcome of GH treated patients with or without mutations.
| Parameter | With mutation |
Without mutation |
P⁎ | ||
|---|---|---|---|---|---|
| n | Mean ± SD | n | Mean ± SD | ||
| Females/males† | 24 | 8/16 (33%/67%) | 191 | 71/120 (37%/63%) | |
| Chronological age (y) at baseline | 24 | 7.4 ± 4.3 | 191 | 9.4 ± 4.2 | 0.029 |
| Height SDS at baseline | 24 | −4.1 ± 2.3 | 191 | −2.9 ± 1.2 | <0.001 |
| Target height SDS | 23 | −0.1 ± 1.2 | 179 | −0.7 ± 1.0 | 0.014 |
| Height SDS – target height SDS | 23 | −4.1 ± 2.1 | 179 | −2.3 ± 1.5 | <0.001 |
| Max. stimulated GH peak (μg/L)‡ | 24 | 1.0 (0.5; 2.3) | 175 | 4.7 (1.9; 7.7) | <0.001 |
| GH dose at start (mg/kg/week) | 23 | 0.19 ± 0.06 | 181 | 0.20 ± 0.07 | 0.239 |
| Last reported GH dose (mg/kg/week) | 24 | 0.17 ± 0.07 | 186 | 0.21 ± 0.09 | 0.037 |
| Age at final height (y) | 24 | 18.5 ± 1.7 | 191 | 17.7 ± 2.5 | 0.144 |
| Years on GH therapy (y) | 24 | 10.7 ± 4.4 | 190 | 7.7 ± 4.0 | <0.001 |
| Final height SDS | 24 | −0.7 ± 1.7 | 191 | −0.9 ± 1.3 | 0.386 |
| Final height SDS – target height SDS | 23 | −0.6 ± 1.3 | 179 | −0.3 ± 1.1 | 0.160 |
| Final height SDS gain§ | 24 | 3.4 ± 1.4 | 191 | 2.0 ± 1.4 | <0.001 |
| Patients with final height SDS > −2† | 24 | 20 (83%) | 191 | 161 (84%) | |
Values are provided as mean ± SD unless otherwise specified.
P-value by ANOVA; bold = significant differences.
Values are provided as n (%).
Because of skewed distribution, values are provided as median (1st quartile, 3rd quartile); P-value for rank-transformed data.
Final height SDS – height SDS at baseline. y = year; SDS = standard deviation score.
Fig. 4.
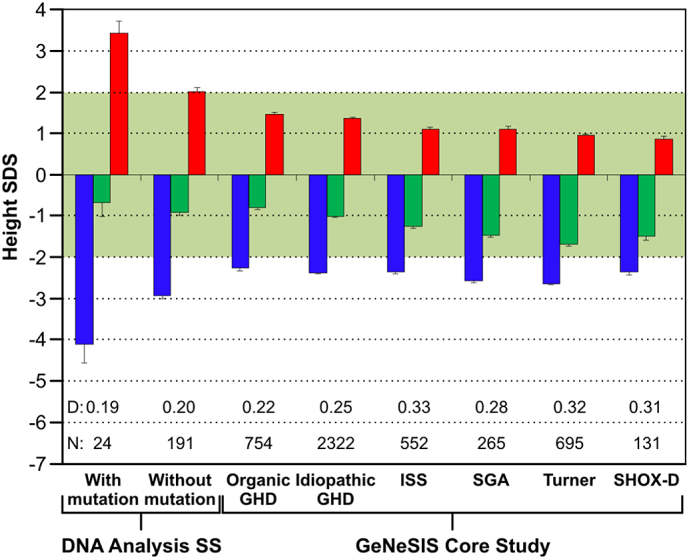
Final growth outcome. Height SDS at baseline (blue), final height SDS (green) and change in height SDS from baseline to final height (red) in various GeNeSIS cohorts: Patients with and without mutation tested in the DNA Analysis Sub-study; organic GH deficiency (GHD) including patients with genetic defects, intracranial tumors, and abnormal pituitary development; idiopathic GHD; idiopathic short stature (ISS); short for gestational age (SGA); Turner syndrome (Turner); short-stature homeobox-containing gene deficiency (SHOX-D). D indicates GH dose (mg/kg/week), N indicates cohort size. Data are shown as mean + or – SEM. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
To our knowledge, this was the largest study screening, under conditions of clinical practice, for DNA variants in genes regulating pituitary development or GH secretion. About 10% of the patients had a mutation with a preponderance of GH1 and PROP1, which is within the magnitude reported by others [2,3,8], although frequencies may vary substantially depending on patient origin and selection [8]. Novel DNA variants were found frequently.
For most mutations identified in the study the pathophysiological impact is well established, such as nonsense mutations or splice-site mutations that lead to the shortened, in-frame deleted GH1 17.5 k isoform due to exon 3 skipping. Others have been investigated in vitro (citations in Table 2). Some novel mutations, however, may provide new insights into the biology of the affected genes or proteins. GH1:c.178G > A leads to a benign predicted amino acid substitution p.Ala60Thr, whereas the GH1:c.275A > G (p.Glu92Gly) mutation is predicted to produce a protein with disturbed function. However, both mutations which confer autosomal dominant IGHD type II in at least one additional relative, reside within known exon 3 splicing regulatory elements (ESE1 [20] and pESE2 [21], respectively). Because T for G nucleotide substitution at position 178 was experimentally shown to alter splicing in favor of exon 3 skipping [20], and c.275A > G is predicted to introduce a new splicing suppressor and to break an existing splicing enhancer site [22], these mutations are likely to convey their pathogenic impact – at least in part – at the level of mRNA processing.
Another novel mutation conferring IGHD II, GH1:c.291 + 3G > C, confirms the intron 3 splice donor site as a mutational “hot spot” [23]. The only GH1 nonsense mutation c.499C > T (p.Gln167Ter) is located in exon 5 and was identified in a heterozygous state with no parental clinical information or DNA available. The few previously reported GH1 nonsense mutations affecting the mature peptide either map to exon 3 and are suggested to act on mRNA processing rather than acting as protein-truncated variants [24,25], or were described in the homozygous state associated with IGHD type IB. [26] Hence, it remains elusive whether the c.499C > T mutation provokes dominant IGHD II or whether a mutation on the second allele escaped genetic analysis.
GHRHR mutations were found exclusively in the compound heterozygous state with c.214G > T and c.812 + 1G > A, identified in two kindreds from Thailand, confirming previous IGHD reports from Asia (Exome Aggregation Consortium ExAC and [27,28]) and c.335G > A being the first reported missense mutation disrupting a GHRHR disulfide bond (p.Cys112Tyr).
Of three novel nonsense mutations that result from frameshift insertions and/or deletions, details of patients carrying HESX1:c.385_386ins315 and LHX3:c.287_288delinsTCCT have been published before [29,30]; the heterozygous GLI2:c.1807_1810dup mutation manifested in a family with incomplete penetrance [15], with the mother being normal whereas a mother's maternal first cousin (height 112 cm at 29 years) had a central incisor and panhypopituitarism.
The only identified pathogenic POU1F1 mutation, c.427C > T (p.Arg143Ter), causing a truncated protein lacking the POU domains essential for DNA binding [4], resides at a CpG site, which is probably a mutational hotspot because the same dinucleotide was previously found mutated in patients with a different ethnic background [31,32].
An affected child from Belgium carried a HESX1:c.479G > A (p.Arg160His) mutation that was transmitted from his unaffected father. This CpG transition has most likely a detrimental effect on pituitary function as shown previously in patients carrying the same or the similar p.Arg160Cys mutation [33,34]. However, the latter patients were found to carry the HESX1 mutation in the homozygous state, which is not the case with our patient who is heterozygous for the HESX1:c.479G > A (p.Arg160His) mutation. In this context, it is worth noting that (i) the majority of reported HESX1 mutations have a dominant inheritance with highly variable penetrance [35] and (ii) PROP1 and HESX1 are known to interact genetically during pituitary development and have also been suggested to interact physically as transcription factors [36]. Because disease manifestation of variably penetrant mutations may depend on the genetic background, it is therefore tempting to speculate that the concomitant heterozygous PROP1:c550G > C (p.Ala184Pro) variant with uncertain significance (Supplementary Table S5), inherited from the mother, may have promoted manifestation of panhypopituitarism in our patient.
PROP1 mutations showed a conspicuously high frequency in East Central and Eastern European countries with a Baltic or Slavic ethnic background due to two founder events about 101 and 44 generations ago [37], but were rare in other countries that participated in GeNeSIS.
Clinical and biochemical data indicated severe GHD in both mutation carriers and non-carriers with greater severity in carriers. GHD manifested at a very young age in children with GH1 mutation, in whom GH secretion is directly affected, and at an older age in children with PROP1 mutation, whose somatotrophs develop malfunction during childhood [38,39]. The smaller mean THSDS of non-carriers suggests an admixture of familial short stature. Mean THSDS was also decreased in patients with GH1 or LHX3 mutation, suggesting that some parents in these cohorts may also have been affected. At least for GH1 mutations, a dominant negative effect is known in patients with IGHD type II which may explain the relatively high frequency of such a genetic defect.
“Measurable” stimulated GH concentrations in patients with IGHD were consistent with the underlying defect: SOX3 and GH1 mutations that were presumed to interfere with exonic splicing enhancer (ESE) site functionality were associated with a milder phenotype, whereas patients with biallelic GH1 deletion mark the most severe end of the phenotypic spectrum. Additionally, heterozygous skipping of GH1 exon 3 and production of a 17.5 k GH isoform, which damages somatotrophs and other pituitary cell types through by-stander effects, may ultimately cause CPHD [3]. Since moderately diminished stimulated peak GH was also found in some patients with PROP1 mutations, the presence of a pathogenic mutation does not necessarily imply a complete lack of GH secretion; instead, the biochemical and growth phenotype can be variable [1,3]. These findings call for caution regarding the use of strict stimulated GH cut-off values for the diagnosis of GHD. Consensus guidelines advise to take a more flexible approach including history, growth rate, other pituitary hormones, and genetic testing [40].
Associations between pituitary morphological anomalies and certain mutated genes were consistent with previous reports [8,41]. It seems plausible that mutations in genes involved in pituitary development are more likely to cause morphological anomalies than mutations in genes regulating GH synthesis and secretion (GH,GHRHR). Pituitary enlargement in patients with PROP1 mutations may be singled out because of its clinical relevance; it regresses spontaneously with time and neurosurgical intervention should not be undertaken [42]. Additional pituitary hormone deficiencies may evolve in all patients with mutations in the tested genes including GH1 [[1], [2], [3], [4], [5]]. The consequence for clinical practice is important and potentially life-saving in the case of ACTH deficiency: life-long monitoring of all pituitary hormones is mandatory.
The cost-benefit balance is a factor when deciding to test or not to test for genetic causes of GHD. Clinical or biochemical indicators of a mutation may therefore be helpful in this decision. Logistic multi-regression analysis indicated that CPHD, low stimulated GH peak, HtSDS decrease compared to THSDS, and young age at growth retardation may increase the likelihood of detecting a genetic cause of GHD. Although very low GH peak values were an important indicator of a mutation, “measurable” serum GH should not preclude DNA testing. Lastly, the frequency of positive findings for certain genes can be increased further by taking the absence or presence of certain pituitary morphological anomalies into account. A very strong indicator for a PROP1 mutation was an enlarged pituitary gland.
The growth response to GH was greater in mutation carriers compared to non-carriers, possibly due to their greater height deficit from parental height and their younger age at treatment initiation. In GHD and other indications, GH therapy is known to be more effective at younger age [43]. The temporal pattern of GH response was similar to other forms of short stature, with greatest catch-up growth in the first year and a decline thereafter. GH treatment beyond four years aims primarily at maintaining achieved height gain and avoiding catch-down growth that may occur after discontinuation of GH resulting in loss of investment (cost, daily injections, and exposure to potential risks). While height gain in this study cohort, especially in mutation carriers, was significantly greater than in cohorts with less severe GHD or non-GH deficient forms of short stature, the necessity for GH treatment in patients with severe GHD reaches beyond growth. The majority of such patients, especially those with mutations, require life-long GH replacement for its metabolic effects [44].
The current study had some limitations: Clinical data were collected as provided by the investigators with limited data monitoring as is common in observational studies. Methods of DNA analysis changed during the 13-year study duration. Samples were not tested for all genes. Data of relatives other than siblings were too scarce to perform useful analyses.
In summary, DNA testing of children with severe GHD identified pathogenic DNA variants in about 10% of the patients. The most frequently affected genes were GH1 and PROP1. The majority of GH1 mutations were heterozygous splice site mutations resulting in skipping of exon 3 and a shortened protein which has a dominant negative effect causing IGHD type II. PROP1 mutations were mainly observed in patients from Eastern European countries. Indicators of a mutation were CPHD, diminished HtSDS – THSDS, very low stimulated GH concentration, and manifestation of GHD at a young age. These findings may assist in selecting the appropriate patients for DNA testing, especially if there is no evidence for a mutation in the family history which would definitely require appropriate DNA testing. The patient and parent benefits are multiple and include information for genetic counselling and preemptive clinical management with increased vigilance for development of additional pituitary hormone deficiencies on a long-term basis. Due to the very small required sample volume, GHD or CPHD can be confirmed in very young children and life-saving therapy, e.g. cortisol, can be commenced in a timely manner. Presence of a mutation predicts an excellent response to GH therapy. An enlarged pituitary gland in combination with a PROP1 mutation must preclude neurosurgical intervention. Patients with a mutation will need GH therapy life-long, which should be communicated to the family early. Retesting for GHD after cessation of GH treatment for growth is not necessary as recommended by learned societies [44]. The large proportion of tested patients without mutation suggests that other genes influencing GH secretion were not included in this study or are still unknown, but new DNA analysis techniques may expand the range of tested genes [1,2,5]. Balancing the cost of genetic testing versus the frequency of pathogenic findings and the value of the information, the authors recommend making DNA testing an important component in the diagnostic work-up of GHD.
Contributors
WFB and JK contributed to the scientific literature search. WFB, JK, SA, CJC, CJ, AGZ, CAQ, GBC, CLD, JL, RGR, JSP and RWP contributed to the study design. JK, SA, HMP, ML, M-LS, M-PL, RWP contributed to the genetic analyses. SA, CLD, JL and RWP contributed to clinical data collection. WFB, JK, CJC, AGZ, CAQ and RWP contributed to data analyses. WFB, JK, SA, CJC and RWP drafted the manuscript and all authors provided critical revision of the manuscript.
Declaration of interests
GeNeSIS was sponsored by Eli Lilly and Company (Lilly, Indianapolis, IN, USA). The sponsor funded all aspects of study design, data collection, genetic analyses and statistical analyses, but did not impose any impediment, directly or indirectly, on the publication of the study results. CJC and CJ are employees and stockholders of Lilly, while WFB, AGZ, and CAQ are former employees and stockholders of Lilly, GBC is a former employee of Lilly. WFB also reports he is a consultant for Ammonett Pharma, Lilly Germany and Merck KGaA Darmstadt. CLD, JL, RGR, JSP and RWP were members of the GeNeSIS International Scientific Advisory Board and received consulting and speaker fees from Lilly. The laboratories of SA and RWP received fees and grants from Lilly for setting up and conducting the genetic analyses. JK, HMP, ML. M-L S, and M-PL have no conflicting interests to report.
Acknowledgements
The authors thank the patients, their families and the contributing physicians and site study coordinators for their participation and engagement in this study. Special thanks are due to Johannes Weigel and Gunter Flemming for their assistance in targeted sequencing. The authors thank also the Genome Aggregation Database (gnomAD) and the groups that provided exome and genome variant data to this resource. A full list of contributing groups can be found at http://gnomad.broadinstitute.org/about. The authors thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2018.09.026.
Appendix A. Supplementary data
Supplementary material
References
- 1.Wit J.M., Oostdijk W., Losekoot M., van Duyvenvoorde H.A., Ruivenkamp C.A.L., Kant S.G. Mechanisms in endocrinology: novel genetic causes of short stature. Eur J Endocrinol. 2016;174:R145-R173. doi: 10.1530/EJE-15-0937. [DOI] [PubMed] [Google Scholar]
- 2.Giordano M. Genetic causes of isolated and combined pituitary hormone deficiency. Best Pract Res Clin Endocrinol Metab. 2016;30:679–691. doi: 10.1016/j.beem.2016.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Alatzoglou K.S., Dattani M.T. Genetic causes and treatment of isolated growth hormone deficiency-an update. Nat Rev Endocrinol. 2010;6:562–576. doi: 10.1038/nrendo.2010.147. [DOI] [PubMed] [Google Scholar]
- 4.Pfaffle R., Klammt J. Pituitary transcription factors in the aetiology of combined pituitary hormone deficiency. Best Pract Res Clin Endocrinol Metab. 2011;25:43–60. doi: 10.1016/j.beem.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 5.Fang Q., George A.S., Brinkmeier M.L. Genetics of combined pituitary hormone deficiency: roadmap into the genome era. Endocr Rev. 2016;37:636–675. doi: 10.1210/er.2016-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cerbone M., Dattani M.T. Progression from isolated growth hormone deficiency to combined pituitary hormone deficiency. Growth Horm IGF Res. 2017;37:19–25. doi: 10.1016/j.ghir.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Dauber A., Rosenfeld R.G., Hirschhorn J.N. Genetic evaluation of short stature. J Clin Endocrinol Metab. 2014;99:3080–3092. doi: 10.1210/jc.2014-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Rienzo F., Mellone S., Bellone S. Frequency of genetic defects in combined pituitary hormone deficiency: a systematic review and analysis of a multicentre Italian cohort. Clin Endocrinol (Oxf) 2015;83:849–860. doi: 10.1111/cen.12849. [DOI] [PubMed] [Google Scholar]
- 9.Blum W.F., Deal C., Zimmermann A.G. Development of additional pituitary hormone deficiencies in pediatric patients originally diagnosed with idiopathic isolated GH deficiency. Eur J Endocrinol. 2014;170:13–21. doi: 10.1530/EJE-13-0643. [DOI] [PubMed] [Google Scholar]
- 10.Deal C., Hasselmann C., Pfäffle R.W. Associations between pituitary imaging abnormalities and clinical and biochemical phenotypes in children with congenital growth hormone deficiency. Data from an international observational study. Horm Res Paediatr. 2013;79:283–292. doi: 10.1159/000350829. [DOI] [PubMed] [Google Scholar]
- 11.Kamijo T., Phillips J.A., Ogawa M., Yuan L.-F., Y-F Shi, X-L Bao. Screening for growth hormone gene deletions in patients with isolated growth hormone deficiency. J Pediatr. 1991;118:245–248. doi: 10.1016/s0022-3476(05)80492-5. [DOI] [PubMed] [Google Scholar]
- 12.Woods K.S., Cundall M., Turton J. Over- and underdosage of SOX3 is associated with infundibular hypoplasia and hypopituitarism. Am J Hum Genet. 2005;76:833–849. doi: 10.1086/430134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones A.C., Austin J., Hansen N. Optimal temperature selection for mutation detection by denaturing HPLC and comparison to single-stranded conformation polymorphism and heteroduplex analysis. Clin Chem. 1999;45:1133–1140. [PubMed] [Google Scholar]
- 14.Richards S., Aziz N., Bale S. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arnhold I.J.P., França M.M., Carvalho L.R., Mendonca B.B., Jorge A.A.L. Role of GLI2 in hypopituitarism phenotype. J Mol Endocrinol. 2015;54:R141–R150. doi: 10.1530/JME-15-0009. [DOI] [PubMed] [Google Scholar]
- 16.Cooper D.N., Youssoufian H. The CpG dinucleotide and human genetic disease. Hum Genet. 1988;78:151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- 17.Lek M., Karczewski K.J., Minikel E.V. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertko E., Klammt J., Dusatkova P. Combined pituitary hormone deficiency due to gross deletions in the POU1F1 (PIT-1) and PROP1 genes. J Hum Genet. 2017;62:755–762. doi: 10.1038/jhg.2017.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hemchand K., Anuradha K., Neeti S. Entire prophet of Pit-1 (PROP-1) gene deletion in an Indian girl with combined pituitary hormone deficiencies. J Pediatr Endocrinol Metab. 2011;24:579–580. doi: 10.1515/jpem.2011.184. [DOI] [PubMed] [Google Scholar]
- 20.Ryther R.C., Flynt A.S., Harris B.D., Phillips J.A.I., Patton J.G. GH1 splicing is regulated by multiple enhancers whose mutation produces a dominant-negative GH isoform that can be degraded by allele-specific small interfering RNA (siRNA) Endocrinology. 2004;145:2988–2996. doi: 10.1210/en.2003-1724. [DOI] [PubMed] [Google Scholar]
- 21.Babu D., Mellone S., Fusco I. Novel mutations in the GH gene (GH1) uncover putative splicing regulatory elements. Endocrinology. 2014;155:1786–1792. doi: 10.1210/en.2013-2146. [DOI] [PubMed] [Google Scholar]
- 22.Desmet F.-O., Hamroun D., Lalande M., Collod-Béroud G., Claustres M., Béroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fofanova O.V., Evgrafov O.V., Polyakov A.V., Poltaraus A.B., Peterkova V.A., Dedov I.I. A novel IVS2 −2A>T splicing mutation in the GH-1 gene in familial isolated growth hormone deficiency Type II in the spectrum of other splicing mutations in the Russian population. J Clin Endocrinol Metab. 2003;88:820–826. doi: 10.1210/jc.2002-020269. [DOI] [PubMed] [Google Scholar]
- 24.Gucev Z., Tasic V., Saranac L. A novel GH1 mutation in a family with isolated growth hormone deficiency type II. Horm Res Paediatr. 2012;77:200–204. doi: 10.1159/000334643. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi I., Takahashi T., Komatsu M., Sato T., Takada G. An exonic mutation of the GH-1 gene causing familial isolated growth hormone deficiency type II. Clin Genet. 2002;61:222–225. doi: 10.1034/j.1399-0004.2002.610310.x. [DOI] [PubMed] [Google Scholar]
- 26.Alatzoglou K.S., Turton J.P., Kelberman D. Expanding the spectrum of mutations in GH1 and GHRHR: genetic screening in a large cohort of patients with congenital isolated growth hormone deficiency. J Clin Endocrinol Metab. 2009;94:3191–3199. doi: 10.1210/jc.2008-2783. [DOI] [PubMed] [Google Scholar]
- 27.Wang Q., Diao Y., Xu Z. Identification of a novel splicing mutation in the growth hormone (GH)-releasing hormone receptor gene in a Chinese family with pituitary dwarfism. Mol Cell Endocrinol. 2009;313:50–56. doi: 10.1016/j.mce.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 28.Maheshwari H.G., Silverman B.L., Dupuis J., Baumann G. Phenotype and genetic analysis of a syndrome caused by an inactivating mutation in the growth hormone-releasing hormone receptor: dwarfism of sindh. J Clin Endocrinol Metab. 1998;83:4065–4074. doi: 10.1210/jcem.83.11.5226. [DOI] [PubMed] [Google Scholar]
- 29.Pfaeffle R.W., Savage J.J., Hunter C.S. Four novel mutations of the LHX3 gene cause combined pituitary hormone deficiencies with or without limited neck rotation. J Clin Endocrinol Metab. 2007;92:1909–1919. doi: 10.1210/jc.2006-2177. [DOI] [PubMed] [Google Scholar]
- 30.Sobrier M.-L., Netchine I., Heinrichs C. Alu-element insertion in the homeodomain of HESX1 and aplasia of the anterior pituitary. Hum Mutat. 2005;25:503. doi: 10.1002/humu.9332. [DOI] [PubMed] [Google Scholar]
- 31.Ohta K., Nobukuni Y., Mitsubuchi H. Mutations in the Pit-1 gene in children with combined pituitary hormone deficiency. Biochem Biophys Res Commun. 1992;189:851–855. doi: 10.1016/0006-291x(92)92281-2. [DOI] [PubMed] [Google Scholar]
- 32.McLennan K., Jeske Y., Cotterill A. Combined pituitary hormone deficiency in Australian children: clinical and genetic correlates. Clin Endocrinol (Oxf) 2003;58:785–794. doi: 10.1046/j.1365-2265.2003.01781.x. [DOI] [PubMed] [Google Scholar]
- 33.Dattani M.T., Martinez-Barbera J.-P., Thomas P.Q. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet. 1998;19:125–133. doi: 10.1038/477. [DOI] [PubMed] [Google Scholar]
- 34.Durmaz B., Cogulu O., Dizdarer C., Stobbe H., Pfaeffle R., Ozkinay F. A novel homozygous HESX1 mutation causes panhypopituitarism without midline defects and optic nerve anomalies. J Pediatr Endocrinol Metab. 2011;24:779–782. doi: 10.1515/jpem.2011.162. [DOI] [PubMed] [Google Scholar]
- 35.McCabe M.J., Alatzoglou K.S., Dattani M.T. Septo-optic dysplasia and other midline defects: the role of transcription factors: HESX1 and beyond. Best Pract Res Clin Endocrinol Metab. 2011;25:115–124. doi: 10.1016/j.beem.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 36.Kato Y., Kimoto F., Susa T., Nakayama M., Ishikawa A., Kato T. Pituitary homeodomain transcription factors HESX1 and PROP1 form a heterodimer on the inverted TAAT motif. Mol Cell Endocrinol. 2010;315:168–173. doi: 10.1016/j.mce.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 37.Dusatkova P., Pfaffle R., Brown M.R. Genesis of two most prevalent PROP1 gene variants causing combined pituitary hormone deficiency in 21 populations. Eur J Hum Genet. 2015;24:415–420. doi: 10.1038/ejhg.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Böttner A., Keller E., Kratzsch J. PROP1 mutations cause progressive deterioration of anterior pituitary function including adrenal insufficiency: a longitudinal analysis. J Clin Endocrinol Metab. 2004;89:5256–5265. doi: 10.1210/jc.2004-0661. [DOI] [PubMed] [Google Scholar]
- 39.Deladoey J., Fluck C., Buyukgebiz A. “Hot spot” in the PROP1 gene responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab. 1999;84:1645–1650. doi: 10.1210/jcem.84.5.5681. [DOI] [PubMed] [Google Scholar]
- 40.Grimberg A., Divall S.A., Polychronakos C. Guidelines for growth hormone and insulin-like growth factor-I treatment in children and adolescents. Growth hormone deficiency, idiopathic short stature, and primary insulin-like growth factor-I deficiency. Horm Res Paediatr. 2016;86:361–397. doi: 10.1159/000452150. [DOI] [PubMed] [Google Scholar]
- 41.Dattani M.T. Growth hormone deficiency and combined pituitary hormone deficiency. Does the genotype matter? Clin Endocrinol (Oxf) 2005;63:121–130. doi: 10.1111/j.1365-2265.2005.02289.x. [DOI] [PubMed] [Google Scholar]
- 42.Obermannova B., Pfaeffle R., Zygmunt-Gorska A. Mutations and pituitary morphology in a series of 82 patients with PROP1 gene defects. Horm Res Paediatr. 2011;76:348–354. doi: 10.1159/000332693. [DOI] [PubMed] [Google Scholar]
- 43.Ranke M.B., Lindberg A. Predicting growth in response to growth hormone treatment. Growth Horm IGF Res. 2009;19:1–11. doi: 10.1016/j.ghir.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 44.Clayton P.E., Cuneo R.C., Juul A., Monson J.P., Shalet S.M., Tauber M. Consensus statement on the management of the GH-treated adolescent in the transition to adult care. Eur J Endocrinol. 2005;152:165–170. doi: 10.1530/eje.1.01829. [DOI] [PubMed] [Google Scholar]
- 45.Wagner J.K., Eble A., Hindmarsh P.C., Mullis P.E. Prevalence of human GH-1 gene alterations in patients with isolated growth hormone deficiency. Pediatr Res. 1998;43:105–110. doi: 10.1203/00006450-199801000-00016. [DOI] [PubMed] [Google Scholar]
- 46.Kamijo T., Phillips J.A. Detection of molecular heterogeneity in GH-1 gene deletions by analysis of polymerase chain reaction amplification products. J Clin Endocrinol Metab. 1992;74:786–789. doi: 10.1210/jcem.74.4.1548341. [DOI] [PubMed] [Google Scholar]
- 47.Cogan J.D., Ramel B., Lehto M. A recurring dominant negative mutation causes autosomal dominant growth hormone deficiency—a clinical research center study. J Clin Endocrinol Metab. 1995;80:3591–3595. doi: 10.1210/jcem.80.12.8530604. [DOI] [PubMed] [Google Scholar]
- 48.Binder G., Keller E., Mix M. Isolated GH deficiency with dominant inheritance: new mutations, new insights. J Clin Endocrinol Metab. 2001;86:3877–3881. doi: 10.1210/jcem.86.8.7757. [DOI] [PubMed] [Google Scholar]
- 49.Hayashi Y., Yamamoto M., Ohmori S., Kamijo T., Ogawa M., Seo H. Inhibition of growth hormone (GH) secretion by a mutant GH-I gene product in neuroendocrine cells containing secretory granules. An implication for isolated GH deficiency inherited in an autosomal dominant manner. J Clin Endocrinol Metab. 1999;84:2134–2139. doi: 10.1210/jcem.84.6.5736. [DOI] [PubMed] [Google Scholar]
- 50.Vivenza D., Guazzarotti L., Godi M. A novel deletion in the GH1 gene including the IVS3 branch site responsible for autosomal dominant isolated growth hormone deficiency. J Clin Endocrinol Metab. 2006;91:980–986. doi: 10.1210/jc.2005-1703. [DOI] [PubMed] [Google Scholar]
- 51.Wajnrajch M.P., Gertner J.M., Harbison M.D., Chua S.C., Leibel R.L. Nonsense mutation in the human growth hormone-releasing hormone receptor causes growth failure analogous to the little (lit) mouse. Nat Genet. 1996;12:88–90. doi: 10.1038/ng0196-88. [DOI] [PubMed] [Google Scholar]
- 52.Salvatori R., Fan X., Phillips J.A., Prince M., Levine M.A. Isolated growth hormone (GH) deficiency due to compound heterozygosity for two new mutations in the GH-releasing hormone receptor gene. Clin Endocrinol (Oxf) 2001;54:681–687. doi: 10.1046/j.1365-2265.2001.01273.x. [DOI] [PubMed] [Google Scholar]
- 53.Sobrier M.-L., Brachet C., Vié-Luton M.-P. Symptomatic heterozygotes and prenatal diagnoses in a nonconsanguineous family with syndromic combined pituitary hormone deficiency resulting from two novel LHX3 mutations. J Clin Endocrinol Metab. 2012;97:E503–E509. doi: 10.1210/jc.2011-2095. [DOI] [PubMed] [Google Scholar]
- 54.Riepe F.G., Partsch C.J., Blankenstein O., Monig H., Pfaffle R.W., Sippell W.G. Longitudinal imaging reveals pituitary enlargement preceding hypoplasia in two brothers with combined pituitary hormone deficiency attributable to PROP1 mutation. J Clin Endocrinol Metab. 2001;86:4353–4357. doi: 10.1210/jcem.86.9.7828. [DOI] [PubMed] [Google Scholar]
- 55.Paracchini R., Giordano M., Corrias A. Two new PROP1 gene mutations responsible for compound pituitary hormone deficiency. Clin Genet. 2003;64:142–147. doi: 10.1034/j.1399-0004.2003.00106.x. [DOI] [PubMed] [Google Scholar]
- 56.Duquesnoy P., Roy A., Dastot F. Human Prop-1: cloning, mapping, genomic structure. Mutations in familial combined pituitary hormone deficiency. FEBS Lett. 1998;437:216–220. doi: 10.1016/s0014-5793(98)01234-4. [DOI] [PubMed] [Google Scholar]
- 57.Vallette-Kasic S., Barlier A., Teinturier C. PROP1 gene screening in patients with multiple pituitary hormone deficiency reveals two sites of hypermutability and a high incidence of corticotroph deficiency. J Clin Endocrinol Metab. 2001;86:4529–4535. doi: 10.1210/jcem.86.9.7811. [DOI] [PubMed] [Google Scholar]
- 58.Wu W., Cogan J.D., Pfaffle R.W. Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nat Genet. 1998;18:147–149. doi: 10.1038/ng0298-147. [DOI] [PubMed] [Google Scholar]
- 59.Laumonnier F., Ronce N., Hamel B.C.J. Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am J Hum Genet. 2002;71:1450–1455. doi: 10.1086/344661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material