

Abstract
Isocitrate dehydrogenases 1 and 2 (IDH1/2) are enzymes that play a major role in the Krebs cycle. Mutations in these enzymes are found in the majority of lower gliomas and secondary glioblastomas, but also in myeloid malignancies and other cancers. IDH1 and IDH2 mutations are restricted to specific arginine residues in the active site of the enzymes and are gain-of-function, i.e. they confer a neomorphic enzyme activity resulting in the accumulation of D-2-hydroxyglutarate (2-HG). 2-HG is an oncometabolite causing profound metabolic dysregulation which, among others, results in methylator phenotypes and in defects in homologous recombination repair. In this review, we summarize current knowledge regarding the function of normal and mutated IDH, explain the possible mechanisms through which these mutations might drive malignant transformation of progenitor cells in the central nervous system, and provide a comprehensive review of potential treatment strategies for IDH-mutated malignancies, focusing on gliomas.
Keywords: Gliomas, Isocitrate dehydrogenases 1 and 2, IDH1, IDH2, myeloid leukemia, synthetic lethality, IDH-inhibitor, BCL2- inhibitor, review
In the past decade, our knowledge regarding the molecular events that drive malignant transformation in diffuse gliomas has increased significantly. This knowledge led to a major revision of the 2007 edition of the World Health Organization Classification of Tumors of the Central Nervous System that was published 2 years ago, the 2016 Classification of Tumors of the Central Nervous System. In the 2016 revision, molecular parameters are integrated with histology in order to clearly define glioma subtypes, providing at the same time, in some cases, prognostic information. IDH mutations are undoubtedly one of the most important molecular parameters in the current glioma classification (1-3).
IDH mutations in gliomas were first reported in 2008. Parsons et al., using next-generation sequencing technology, found IDH1 mutations to be present in 12% of patients with glioblastoma, mainly in younger patients and in patients with secondary glioblastoma. In this initial publication, it was reported for the first time that the presence of IDH1 mutations is also associated with improved prognosis in terms of overall survival, relative to patients with glioblastoma with wild-type IDH1 (4). Following this pivotal study, Yan et al. reported that IDH1 and IDH2 mutations occur in the majority (>70%) of WHO grade II/III astrocytomas, oligodendrogliomas, oligoastrocytomas and secondary glioblastomas, and are associated with improved prognosis (5). Multiple subsequent studies have confirmed these findings (6-13). Interestingly, two studies have shown that the presence of an IDH mutation in patients with astrocytoma/glioblastoma is a stronger predictor for overall survival than the histological type and grade of the tumor (14,15).
IDH1 and IDH2 mutations have also been found in patients with hematological malignancies and in a number of solid tumor types, including acute myeloid leukemia (AML), myelodysplastic syndromes, myeloproliferative neoplasms, cholangiocarcinoma, melanoma and cartilaginous tumors (16-24). In AML, IDH1/IDH2 mutations have been reported in 16-17% of patients, while the frequencies in all other entities remain much lower (23,25). Furthermore, IDH mutations have also been observed in intrahepatic cholangiocarcinomas (26), chondrosarcomas (16) and sporadically in other tumors as well (Table I).
Table I. Frequency of isocitrate dehydrogenase (IDH) mutations in different tumor types.

AML, Acute myeloid leukemia; MDS, myelodysplastic syndrome; MPN, myeloproliferative neoplasms.
In this review, we summarize current knowledge regarding the function of normal and mutated IDH, explain the possible mechanisms through which these mutations might drive malignant transformation of progenitor cells in the central nervous system, and provide a comprehensive review of potential treatment strategies for IDH-mutated malignancies, focusing on gliomas.
Biochemistry and Function of Wild-type IDH
In biochemistry, IDH enzymes are known for their role in the Krebs cycle (tricarboxylic or citric acid cycle). The IDH family in humans consists of three IDH isoenzymes, IDH1, IDH2 and IDH3, which all catalyze the oxidative decarboxylation of isocitrate into alpha-ketoglutarate (aKG) and carbon dioxide. IDH1 and IDH2 are homodimeric enzymes with high structural similarity (69%) that are encoded by two distinct genes, IDH1 on 2q33.3 and IDH2 on 15q26.1, respectively. The protein IDH1 is located in the cytoplasm and peroxisomes, while IDH2 is located in the mitochondria. They are both nitotinamide adenine dinucleotide phosphate (NADP+)-dependent enzymes that use NADP+ as a cofactor, producing NADPH during their enzymatic activity (27,28). IDH1 and IDH2 are the major sources of NADPH in the cytosol and mitochondria respectively (20).
IDH3 is a heterotetrameric enzyme consisting of two alpha, one beta and one gamma subunit that are encoded by the genes IDH3A on 15q25.1-2, IDH3B on 20p13 and IDH3G on Xq28, respectively. IDH3 is located in the mitochondria and is one of the most critical enzymes in the Krebs cycle. IDH3 uses NAD+ as a cofactor and produces NADH, which is necessary for energy production (29,30). Despite its important role in cellular metabolism, mutations in IDH3 genes have not as yet been linked to tumorigenesis.
IDH1 and IDH2 are necessary for the production of aKG and NADPH. NADPH is an important cofactor in many cellular functions, e.g. lipid metabolism (31), glucose metabolism (32) and oxidative stress defense, by reducing glutathione and thioredoxin, and by activating catalase (33-35). Interestingly, IDH1 is the main source of NADPH in the brain and is also thought to be a main source of NADPH in other tissuesl (36,37).
aKG is also an important molecule in diverse cellular processes, by being an essential cofactor for the family of aKG-dependent dioxygenases. This family consists of more than 60 enzymes that require aKG as a cofactor. aKG-dependent dioxygenases are present in all living organisms and affect multiple cellular functions: they catalyze hydroxylation reactions on a diverse set of substrates, including collagen, lipids, proteins, histones, transcription factors, alkylated DNA and RNA, 5-methylcytosine of genomic DNA and 6-methyladenine of RNA, as well as antibiotics. Members of the aKG-dependent dioxygenase family are propyl hydroxylases, jumonji-C domain-containing histone demethylases, ten-eleven translocation (TET) enzymes, 5-methylcytosine hydroxylases, collagen propyl-4-hydroxylases 1, 2 and 3, and many more (37,38). Many of these enzymes play a pivotal role in regulating histone and DNA demethylation in normal cells (39,40).
Biochemistry and Function of Mutant IDH
In gliomas, mutations in the IDH1 and IDH2 genes are somatic and invariably heterozygous, missense mutations that result in a single amino acid substitution. Interestingly, these mutations involve specific conserved arginine residues in the active site of the IDH enzymes that play a key role in substrate binding (4).
For the IDH1 gene, mutations are found at the arginine codon 132 (R132) and the most common mutation is the substitution of arginine by histidine (R132H), which occurs in more than 90% of all IDH1 mutants. Far less common are R132C substitutions (arginine→cysteine), R132S (arginine→serine), R132G (arginine→glycine) and R132L mutations (arginine→leucine), which occur in approximately 5% of cases. For the IDH2 gene, the homologous conserved arginine residue is at codon 172 (R172) and the reported mutations are R172K (arginine→lysine), R172M (arginine→methionine), R172G (arginine→glycine) and R172W (arginine→tryptophan) (5,41), as well as R172S (arginine→serine). All these mutations are registered in the Catalogue of Somatic Mutations in Cancer (COSMIC) database (https://cancer.sanger.ac.uk/cosmic). The incidence of mutations of each IDH gene differs among tumor types, as summarized in Table I.
The fact that IDH1/2 mutations affect the active site of the enzyme led to the initial theory that these mutations cause loss of enzymatic activity of wild-type IDH. Initial in vitro studies had shown that mutant IDH proteins impair normal IDH catalytic action in a dominant-negative fashion by heterodimerizing to the wild-type enzyme and altering its activity, leading to reduced levels of aKG and NADPH (42-44). Reduced levels of NADPH and aKG could then lead to cellular damage and genomic instability, through impairment of the normal cellular detoxification mechanisms, as well as other important cellular processes described above. This theory was supported by biochemical studies suggesting that mutant IDH1/2 was unable to catalyze the oxidative decarboxylation of isocitrate. However, subsequent studies revealed that mutant IDH enzymes were not catalytically inactive, but rather enzymes with a gain-of-function activity [summarized in (45)].
A landmark report by Dang et al. showed that the mutant IDH1 protein is not a non-functional enzyme, but rather an enzyme with a neomorphic activity, which converts aKG to 2-hydroxyglutarate (2-HG) with the simultaneous consumption of NADPH (46). 2-HG is a byproduct in normal mitochondrial metabolism that is normally found at very low levels in the cell and has two enantiomers, D-2HG and L-2HG (or, R-2HG and S-2HG, respectively). IDH1-mutant cells exclusively produce D-2HG at very high levels, up to 100-fold higher than in normal tissues. In the same manner, IDH2 mutations have been found to result in high levels of D-2HG (47). Jin et al. showed that accumulation of high levels of D-2HG takes place in vivo and in vitro only when both wild-type and mutant IDH1 alleles are expressed (48), indicating that the wild-type–mutant IDH1 heterodimer is critical for the rapid conversion of aKG to D-2HG. Indeed, loss of wild-type IDH1 expression in IDH1-mutated glioblastoma cells led to reduced levels of D-2HG. This may explain why loss of heterozygosity is rare in IDH-mutated gliomas (48,49). Interestingly, in vitro studies have additionally shown that L-2HG is increased in human neuroblastoma, pediatric glioblastoma, glioblastoma, renal cell carcinoma, and embryhonic kidney cell lines in the setting of hypoxia via enzymatic reduction of aKG (50).
It is worth noting that 2-HG accumulation in human cells has also been described in 2-HG acidurias that are rare inherited metabolic disorders characterized by abnormally high levels of hydroxyglutaric acid in almost all organ systems and body fluids (51). 2-HG aciduria is caused by inherited loss-of-function mutations in the genes encoding enantiomer specific flavin adenine dinucleotide-dependent 2-hydroxyglutarate dehydrogenases (D-2HGDH and L-2HGDH for D and L enantiomers, respectively) that normally convert 2-HG to aKG preventing its accumulation in cells (52,53). D-2HG aciduria has also been reported in association with inherited gain-of-function mutations in IDH2 and is characterized by developmental delay, epilepsy and cardiomyopathy and short life-span, while L-2HG is characterized by a variable clinical course and by increased risk of developing brain tumors in longer survivors (51,54).
The right-enantiomer, D-2HG, is structurally similar to aKG. The two molecules differ only in the presence of a C2 hydroxyl group in D-2HG instead of the C2 carbonyl group in aKG. D-2HG has been shown to act in vitro as a competitive inhibitor of aKG-dependent enzymes, including jumonji-C domain-containing histone demethylases and TET family of hydroxylases. These enzymes, as mentioned earlier, are key regulators of histone and DNA demethylation in cells and therefore their inhibition through D-2HG results in a profile of cellular hypermethylation (38,55-57). D-2HG is considered an ‘oncometabolite’ not only because it increases dramatically in cells as a consequence of IDH1/2 mutations (46), but also because it was shown to transform hematopoetic into leukemia cells (58) and promote epithelial–mesenchymal transition in colorectal cancer cells even in the absence of IDH mutation (59); by contrast, L-2HG does not appear to have such phenotype-aggravating properties.
The intracellular accumulation of 2HG in IDH-mutated gliomas has been used as a non-invasive tool for accurate diagnosis using magnetic resonance spectroscopic (MRS) technology (60).
Recently, a more specific technique has been described, Fourier-transform infrared spectroscopy, which seems promising for rapid and intraoperative glioma classification. This technique can provide prognostic information to the surgeon at the time of the surgery for optimal treatment decisions (61), but needs further development for clinical applications. In the same vein, in vivo 3D MRS may be used for patient surveillance in the context of clinical trials for assessing treatment efficiency (62).
Role of Mutant IDH in Oncogenesis
The originally proposed oncogenic effect of IDH mutations through increasing the expression of hypoxia-inducible factor-1 alpha (HIF1α) and its target genes (44) was later rejected since IDH mutations in fact result in increased degradation of HIF1α (63). The exact role of IDH mutations in tumorigenesis is still not fully understood, although much progress has been made in this direction (64).
The described gain-of-function IDH mutations giving rise to gliomas have traditionally been considered as being directly responsible for malignant transformation (13,30,65), since these are established in neural progenitors in the brain (30) before lineage-specific genomic alterations that are described in IDH-mutated gliomas (Figure 1). Corresponding knowledge on the manifestation of IDH mutations in myeloid malignancies is less concise; in leukemia, for example, the biological effects of IDH mutations, including a favorable prognostic impact of IDH2 mutations, seem to depend on coexisting genomic alterations (66); this may support a previous hypothesis that IDH mutations may develop in later steps of malignant transformation in this context (67).
Figure 1. Potential differences in the development of isocitrate dehydrogenase (IDH) mutations in the course of oncogenesis in the two main systems with IDH-mutant malignancies. A: The IDH-dependent pathway in glioma development is well characterized: IDH mutations are established in local progenitors that give rise to two glioma lineages with specific genotypic characteristics, as described in the text. B: The sequence of events with respect to IDH mutations in myeloid malignancies remains hypothetical. In cytogenetically normal (primary) acute myeloid leukemia (AML), IDH mutations occur early and are clonal. In most other cases and in secondary AML, IDH mutations may be subclonal, indicating a later establishment. MPN: Myeloproliferative neoplasm; MDS: myelodysplastic syndrome; RTK: Receptor tyrosine kinase pathway; PI3K/MAPK: phosphoinositide 3-kinase/mitogen-activated protein kinase pathway.

The early establishment of IDH mutation seems to provide the common origin and perturbed metabolic and molecular environment for the development of IDH-mutation-positive gliomas (68-70) that diverge into specific oligodendrocytic and astrocytic lineages based on additional genomic alterations aided by changes in their microenvironment (71). Thus, oligodendrogliomas acquire changes such as 1p/19q co-deletion and mutations or fusions in key genes located in 1p, such as the capicua transcriptional repressor gene (CIC) and far upstream element binding protein 1 gene (FUBP1), but also in the promoter of telomerase reverse transcriptase (TERT), the gene encoding the catalytic subunit of telomerase (72-74). In comparison, astrocytomas are characterized by the addition of mutations in tumor protein 53 (TP53) in 80% of cases and in alpha thalassemia/mental retardation syndrome X-linked gene (ATRX); the latter is involved in alternative lengthening of telomeres (75-77), while ATRX mutations are mutually exclusive from mutations in the TERT promoter. As shown with single-cell RNA sequencing, lineage determination in IDH mutation-positive gliomas is non-overlapping (71), thus justifying the WHO 2016 classification that recommends avoiding use of the histological term ‘mixed oligoastrocytoma’.
Additional genomic changes determine the malignant potential of anaplastic gliomas (Figure 1). It remains obscure however, how IDH mutations and the resulting oncometabolite D-2HG contribute to the acquisition of the changes necessary for the development of each glioma lineage, and whether the same mutations drive the transition of low-into high-grade glioma.
The main global metabolic consequence in IDH-mutated cells seems to be energy deprivation: IDH-mutated tumors have a low-energy requirement, with dozens of metabolites contributing to this pro-survival feature (68,70). The low-energy requirement of IDH mutation-positive tumors may in fact be related to the methylator phenotype that has been proposed as the oncogenic mechanism of IDH mutations. Noushmehr et al. in 2010 described a specific hypermethylation pattern in CpG islands in a subset of patients with glioma referred to as a glioma CpG island methylator phenotype (G-CIMP). This pattern is associated with altered expression of several genes through transcriptional silencing (78). Furthermore, G-CIMP has been found to be tightly associated with a specific gene-expression profile called the proneural subtype; the latter is always characterized by the presence of IDH mutations (79). Following these studies, Turcan et al. were able to replicate the G-CIMP signature in human astrocytic cells by expressing IDH1 R132H mutations, establishing in this way the attractive theory that IDH mutations constitute the molecular basis of G-CIMP (80). However, not all IDH mutation-positive gliomas exhibit the methylator phenotype (69), while the same phenotype may result from mutations in other genes.
As previously mentioned, aKG-dependent dioxygenases are directly involved in histone and DNA methylation and their inhibition through D-2HG can result in a hypermethylation pattern consistent with the G-CIMP signature. This interesting scenario is supported by multiple preclinical data. DNA demethylation is in part being regulated by TETs, enzymes that catalyze the conversion of 5-methylcytosine to its unmethylated form. It has been shown that in vitro expression of IDH mutations inhibits TET activity, resulting in DNA hypermethylation, and this inhibition can be reversed by the exogenous addition of aKG (38). In patients with AML, IDH1/2 mutations have been found to result in a DNA hypermethylation pattern similar to G-CIMP through impairment of normal TET catalytic activity. Interestingly, a subset of patients with AML harbor TET loss-of-function mutations and have the same epigenetic defects as patients with IDH mutations. It is worth noting that IDH and TET mutations are mutually exclusive (81).
Main implications of the methylator phenotype associated with IDH mutations include the altered expression of genes participating in practically all cellular functions, some of which have been studied in detail. IDH mutations have been shown to inhibit aKG-dependent histone jumonji-C demethylases that regulate histone methylation. Chowdhury et al. showed that IDH mutations inhibit members of the jumonji-C family with different potencies, with the 2-keto-3-deoxy-D-glycero-D-galacto-nononic acid (KDN) family of histone demethylases being the most sensitive to inhibition, leading to an increase in histone methylation marks (55). These findings have been replicated by other investigators (38). Lu et al. showed that this increase in histone methylation leads to a block in cellular differentiation, probably due to hypermethylation of genes associated with differentiation (82). Flavahan et al. showed that the methylator phenotype associated with IDH mutations alters normal chromosomal topology via disruption of CCCTC-binding factor (CTCF) insulator protein binding. This protein is responsible for organizing DNA into chromatin loops and boundaries. Methylation of CTCF-binding sites disrupts normal binding of the CTCF proteins, allowing aberrant gene expression. It has been shown that in IDH-mutant cells, loss of CTCF binding at a specific domain boundary results in overexpression of a prominent glioma oncogene, platelet-derived growth factor receptor A (PDGFRA). Moreover, treatment of these cells with a demethylating agent resulted in down-regulation of PDGFRA and partial restoration of the CTCF function (83). Turcan et al. also proposed that histone and chromatin modifications are the main consequences of the metabolic environment in IDH-mutant cells (84). Chesnelong et al. found that in IDH-mutant cells there is silencing of lactate dehydrogenase A gene (LDHA) via methylation of its promoter. LDHA is a subunit of LDH essential for glycolysis and silencing of LDHA in these cells results in a defective glycolytic pathway. This could perhaps explain the slow growth reported for IDH-mutant tumors compared to their wild-type IDH counterparts (85). The methylation-based silencing of genes encoding for immune checkpoint inhibitors, such as programmed cell death protein 1 (PDCD1/PD1) and programmed cell death-ligand 1 (PDL1) (86-88) is also worth mentioning in the context of the above described IDH-related methylator phenotype.
Two consequences directly attributed to the high levels of 2-HG resulting from IDH mutations include endoplasmic reticulum stress in murine glioma progenitors (89) and the stalled DNA-damage repair demonstrated in various IDH mutation-positive tumors, including gliomas (90). Compared to IDH mutation-negative gliomas, IDH mutation-positive gliomas exhibit higher apoptotic death rates that can be further increased with endoplasmic reticulum stress-inducing agents such as tunicamycin and thapsigargin (89) and with inhibition of B-cell lymphoma-extra large protein (BCL-xL) (91). IDH mutations through high 2-HG levels induce homologous recombination repair deficiency, which was accompanied by a high sensitivity of IDH-mutated tumors to poly (ADP-ribose) polymerase (PARP) inhibitors in preclinical models (90,92). The importance of the above features in IDH mutation-positive tumors lies in the fact that IDH mutations can be exploited with synthetic lethality strategies using existing drugs.
Prognostic and Predictive Role of IDH Mutations
Many studies have shown that patients with IDH mutation-positive gliomas have better survival compared to their wild-type IDH counterparts irrespective of histology and grade, making IDH mutation the most important prognostic factor for survival, followed by age, tumor grade and O6-methylguanine-DNA methyltransferase gene (MGMT) status, as summarized in (20). The favorable prognostic effect of IDH mutations compared to wild-type IDH has been reported in patients with glioblastoma (median overall survival: 31 vs. 15 months) and anaplastic astrocytoma (median overall survival: 65 vs. 38 months) (5,14,93,94) and with anaplastic oligodendroglioma (95). In line with the above findings is recent work by Yao et al. in which it was shown in glioma stem cells in vitro that the presence of IDH mutation was associated with a less aggressive phenotype compared to wild-type IDH (96).
Patients with glioma with IDH mutations have distinct clinical features. They are usually younger in age (4,5) and most commonly the lesion is found in the frontal lobes compared to patients with wild-type IDH tumors (97-101). Moreover, these tumors are associated with different characteristics on radiological imaging, such as less contrast enhancement, less necrosis and increased stiffness compared to gliomas with wild-type IDH (102,103).
As mentioned, the above findings have made it clear that histology and grading are not enough to fully characterize a glioma tumor and led to the 2016 WHO CNS classification that incorporates molecular features in addition to histology and grade. Traditionally, mutations, IDH included, have been detected with wet (polymerase chain reaction-based) molecular methods such as Sanger sequencing (Figure 2) and these are included in most next-generation sequencing panels. The development of an antibody that recognizes the most frequent IDH alteration in gliomas, namely IDH1 p.R132H (104), and its validation for diagnostic immunohistochemistry application in 2014, has greatly facilitated the integration of IDH testing in routine pathology diagnostic practice (Figure 3).
Figure 2. Example of an isocitrate dehydrogenase-2 (IDH2)-mutant grade III glioma. The patient was a 22-year-old male with a central nervous system tumor that recurred within 3 years. The case was examined before the introduction of diagnostic IDH1 R132H antibody. A: As shown in the hematoxylin- and eosin-stained section, morphologically the tumor exhibited both an oligodendrocytic and an astrocytic component (original magnification ×200). B: Sanger sequencing targeting IDH1 exon 2 and IDH2 exon 4 (CCDS 10359.1) was applied to the patient’s germline DNA from peripheral blood, on a normal brain sample, and on the indicated tumor DNA samples that were macrodissected from the same paraffin section. The pathogenic IDH2 mutation p.R172S/c.516G>T (arginine→ serine; COSM34090) was tumor-specific and was found in both the oligodendrocytic and astrocytic components. Courtesy: Department of Pathology, School of Medicine, Aristotle University of Thessaloniki, Greece (archived in 2011).

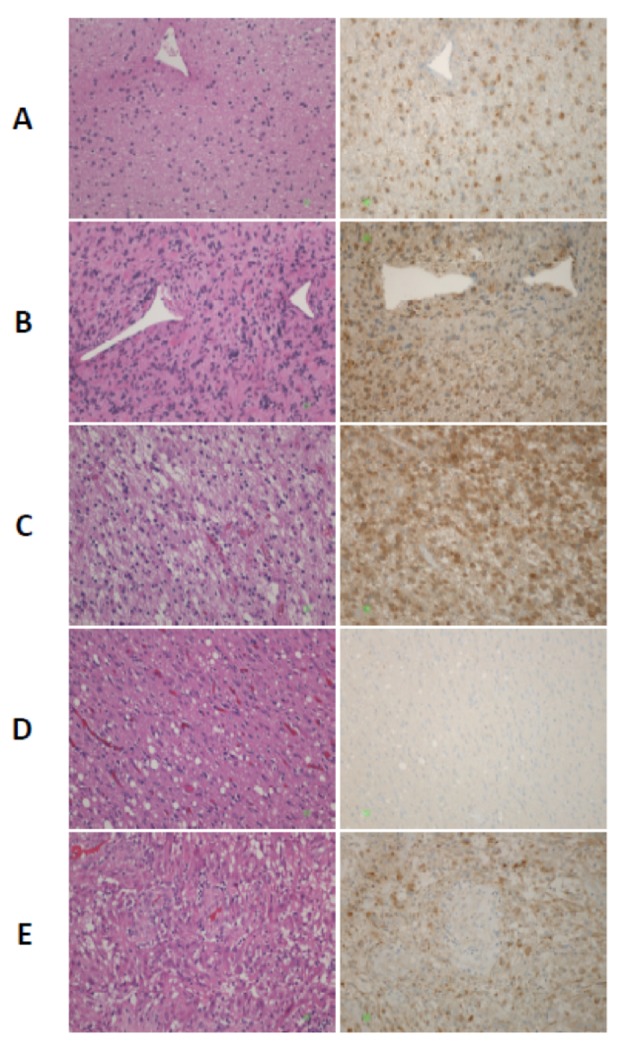
Figure 3. Isocitrate dehydrogenase-1 (IDH1)-R132H immunohistochemistry in gliomas of different histological grade. Left panel: Hematoxylin- and eosin-stained sections; Right panel: Sections stained using the diagnostic antibody to IDH1 R132H (clone DIA-H09). The antibody detects the most common IDH1 mutation, arginine→histidine at codon 132, but may also detect further amino acid changes in the same codon. Staining is cytoplasmic. A: Diffuse astrocytoma (grade II). B: Anaplastic astrocytoma (grade III). C: Anaplastic oligoastrocytoma (grade III). The area corresponds to the oligodendrocytic component. D: Same case as in C, IDH1 R132H-negative area in the periphery of the tumor. Reactive gliosis (or astrocytosis) is expected to be stain negatively with this antibody, which is of great value for distinguishing between non-tumor and tumor in areas with high cellularity in diagnostic CNS pathology. E: Glioblastoma (grade IV). Note that vascular endothelial cells are negative for this marker and serve as an internal negative control. In A, B, C and E, negative residual cells are present in the tumor bed. Original magnification ×200. Courtesy: Dr. Thomas Zaramboukas; Dept. of Pathology; School of Medicine; Aristotle University of Thessaloniki, 54124 Thessaloniki.
Similarly to patients with IDH-mutated glioma, patients with IDH-mutated cholangiocarcinoma have been shown to have better overall survival and longer time to recurrence after surgical resection (105).
In patients with AML, mutation of IDH2, but not IDH1 has been shown to be associated with better overall survival compared to patients with wild-type IDH (106). However, conflicting results have been reported (64,66) and the effect of IDH mutations in leukemia seems to depend on the genomic context and coexisting alterations rather than standing independently (66).
IDH mutations have been shown to be predictive of response to chemotherapy. This has been shown in patients with low-grade gliomas and secondary glioblastomas treated with temozolomide (107,108), as well as in patients with anaplastic oligodendroglioma treated with procarbazine, lomustine and vincristine (109).
In their study, Lu et al. showed that a possible mechanism for chemosensitivity in patients with IDH1 mutation-positive gliomas is through 2-HG production and oxidative defects, which result in impairment of PARP1-mediated DNA repair (92).
Targeted Treatment of IDH-mutant Tumors
Based on the unique characteristics of IDH-mutant tumors, it is intriguing to assume that these tumors may need to be treated differently from their wild-type IDH counterparts.
The mostly tested strategy in this context is with compounds acting as IDH inhibitors. The fact that IDH mutations occur early in gliomagenesis and that such mutations are tumor specific and are expressed uniformly in all tumor cells (13) resulted in the development of drugs targeting IDH1/2 enzymes. Data from preclinical studies showed that IDH inhibitors can reduce 2-HG production, reverse histone and DNA methylation, and induce cell differentiation (110-112). The clinical trials testing the efficiency of IDH inhibitors in gliomas and hematological malignancies are shown in Table II.
Table II. Summary of clinical trials evaluating isocitrate dehydrogenase (IDH) inhibitors, alone and in combination, in patients with glioma or myeloproliferative malignancies.

AML, Acute myeloid leukemia; MDS, myelodysplastic syndrome; mut, mutant; CT.GOV.ID, clinicaltrials.gov identifier.
Popovici-Muller et al. in 2012 were the first to develop an IDH1/2 inhibitor that lowered the 2-HG level in a glioblastoma xenograft mouse model (113). Following these findings, Chatuverdi et al. showed that the use of another IDH1 inhibitor in leukemia cells from patients with IDH1 mutations blocked colony formation without affecting normal cells (114). AGI-5198, also an IDH1 inhibitor, was shown in two in vitro studies to inhibit 2-HG production and induce cell differentiation (110,115). Similarly, AGI-6780, an IDH2 inhibitor, was shown to induce differentiation of IDH2-mutated erythroleukemia and primary human AML cell lines (111).
IDH inhibitors were first studied in patients in 2013. The first agent to be tested was AG-221 (enasidenib), a selective IDH2 inhibitor. Yen et al. showed in vitro that enasidenib suppressed 2-HG production in and induced cellular differentiation of primary human IDH2 mutation-positive AML cells ex vivo and in xenograft mouse models (116). On this basis, enasidenib was initially tested in a phase I/II clinical trials in patients with advanced hematological malignancies and a known IDH2 mutation (NCT01915498). Results from this trial were encouraging. The drug was well tolerated and resulted in 98% reduction of 2-HG levels. The objective response rate among 181 patients with relapsed or refractory AML was 41%, with 28% of patients achieving complete response and a median response duration of 5.8 months. Grade 3 or 4 drug-related side-effects were observed in 21% of patients and included indirect hyperbilirubinemia (12%) and IDH-inhibitor differentiation syndrome (7%) (117-119). Subsequent translational studies from the trial population have shown that cellular differentiation is the main mechanism of action of enasidenib (120). Interestingly, in the trial population, 23% of patients taking enasidenib at a dose of 100 mg once daily had a complete response lasting a median of 8.2 months (121).
These results led to the approval of enasidenib by the Food and Drug Administration (FDA) (August 2017) for use in patients with refractory or relapsed AML along with a companion diagnostic, the RealTime IDH2 assay to detect IDH2 mutation (USFDA Approved Drugs—FDA Granted Regular Approval to Enasidenib for the Treatment of Relapsed or Refractory AML; https://www.fda.gov/Drugs/ InformationOnDrugs/ApprovedDrugs/ucm569482.htm), which is also being tested in many clinical trials. A phase III trial evaluating enasidenib versus best supportive care in elderly patients with AML harboring IDH2 mutation is currently recruiting (NCT02577406). Enasidenib has also been evaluated in a phase I/II trial in patients with advanced solid tumors harboring an IDH2 mutation (NCT02273739).
Similarly, AG 120, an IDH1 inhibitor, has been tested in patients with advanced hematologic malignancies (NCT02074839) and solid tumors harboring IDH1 mutations (NCT02073994). Preliminary results have been presented and AG 120 has been reported to have a clinical benefit rate of 37% among all patients and 25% among those with glioma (122).
Furthermore, additional IDH inhibitors have been designed and have entered clinical trials, including IDH-305, DS-1001b and BAY-1436032, which are all IDH1-mutant inhibitors (122,123), and AG-881 (vorasidenib), a pan inhibitor of mutant IDH1 and IDH2 enzymes, which has been shown to fully penetrate the blood–brain barrier (NCT02381886, NCT03030066, NCT02746081). AG-881 is currently being tested in two clinical trials, NCT02481154 and NCT02492737, involving patients with advanced solid tumors including gliomas with IDH1/IDH2 mutations and advanced hematological malignancies, respectively (20,49).
A different treatment approach, synthetic lethality for IDH-mutant solid tumors, was recently suggested by Sulkowski et al. (90) based on the observation that 2-HG results in homologous recombination repair deficiency, rendering such cells highly sensitive to PARP inhibition. Importantly, they also observed that this sensitivity is reversed by IDH inhibitors. In the same context, Lu et al. (92) reported on synergistic effects between temozolomide and the PARP inhibitor olaparib, in patients with IDH mutation, providing the possibility to achieve improved cytotoxic effects with minimal use of alkylating agents in order to reduce bone marrow cytotoxicity (109). Based on these results, a phase I trial with the above combination in patients with relapsed glioblastoma recently completed accrual and results are awaited (NCT01390571) (124).
Another potential pathway to be targeted in IDH-mutant gliomas was identified based on the specific hypermethylation patterns associated with such tumors. As previously described, histone and DNA hypermethylation reported in IDH-mutant tumors leads to an arrest of cellular differentiation and malignant transformation. The use of decitabine and azacytidine, both DNA-demethylating agents approved by the FDA for myelodysplastic syndromes, has been shown in preclinical models to reverse the hypermethylation phenotype, induce cellular differentiation and delay tumor growth in IDH1-mutated xenograft models (125,126).
As immunotherapy constantly gains ground in the battle against cancer, many researchers have tried to use different immunotherapy approaches against IDH-mutant tumors. The fact that IDH mutations are events that occur early and steadily through time in gliomagenesis, in combination with the fact that mutant IDH enzymes are expressed only in glioma cells, have made them intriguing antigens for activating the immune system. A mutation-specific anti-IDH1(R132H)-specific peptide vaccine has been produced and been shown to generate an immune reaction in preclinical studies and to reduce tumor growth in a mouse model (127,128). Currently there are three IDH1 peptide and dendritic vaccines targeting the IDH1R132H mutation that are being evaluated in phase I clinical trials (NCT02193347, NCT02454634 and NCT02771301). Since PD1 and PDL1 genes were found to be methylated and underexpressed in IDH-mutant tumors (86-88), inhibitors of PD1 (pembrolizumab) and PDL1 (avelumab) are currently being evaluated in pretreated patients with gliomas, either alone in patients with glioma grade II-IV that harbor hypermutator phenotype, or in combination with radiotherapy in secondary glioblastoma (NCT02658279 and NCT02968940, respectively). Pretreatment with irradiation with/without temozolomide may interfere with the methylator phenotype and hence alter the expression of targets of pembrolizumab and avelumab, while temozolomide may induce a mutator phenotype in gliomas, through inactivation of DNA mismatch repair genes (129).
In conclusion, several therapeutic approaches are being tested in patients with IDH mutant tumors based on preclinical evidence (summarized in Figure 4). The first approach involves IDH inhibitors, which target the effect of IDH mutations, i.e., the oncometabolite 2-HG, and reduce its levels. The second tries to exploit the effects of the oncometabolite, i.e. deficient DNA strand breaks and apoptosis. The third is in the context of immunotherapy. Because IDH inhibitors do not kill cells but induce them to differentiate or drive them into a reversible autophagic state, these inherently counteract existing treatment modalities, such as radiotherapy, cytotoxic chemotherapeutics, as well as most molecularly targeted agents. Thus, despite approval, this class of molecules is not expected to be effectively integrated into clinical practice, for gliomas at least. In comparison, approaches of synthetic lethality appear more promising based on the rationale of combination possibilities and repurposing of existing drugs. IDH-mutant tumors are progressively being understood as an individual class of malignancies across tumors of different origins, the reason being the oncometabolite (D-)2-HG. Deepening our knowledge on how this metabolite is influenced by currently applied treatments in patients with IDH mutation-positive tumors is warranted for the efficient design of clinical trials targeting its consequences directly or after initial treatment failure, particularly in patients with glioma.
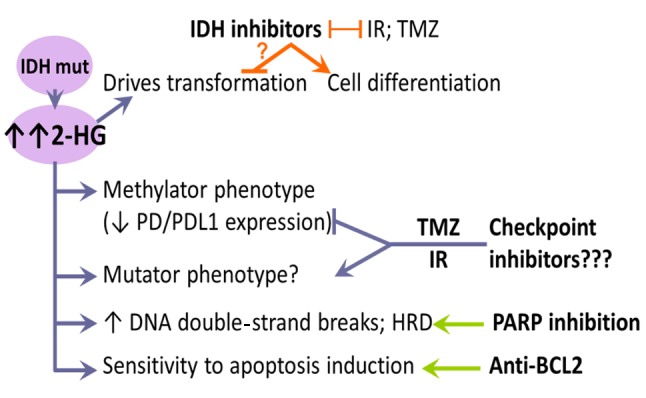
Figure 4. Features of isocitrate dehydrogenase (IDH) mutations in gliomas and potential treatment approaches. IDH inhibitors effectively reduce the level of 2-hydroxyglutarate (the oncometabolite in IDHmutated tumors) and induce cell differentiation, but are incompatible with major treatment modalities for gliomas. In addition, these inherently counteract any of the molecular treatment options presented (right panel). In the clinical setting, when tumors are diagnosed, transformation has been accomplished and tumors have already acquired additional molecular/genomic characteristics, questioning the value of targeting IDH for effective tumor growth inhibition. The three drug classes on the right target the effects of 2-HG and not 2-HG itself. Checkpoint inhibitors are under trial, but these need their target molecules programmed cell death protein 1 (PD1) and programmed death ligand 1 (PDL1) to be expressed or overexpressed in order to be effective. These molecules are underexpressed in IDH-mutant tumors due to promoter hypermethylation. Temozolomide (TMZ) may reverse the low expression of checkpoint molecules in IDH-mutant gliomas and induce the hyper-mutator phenotype, but this has still to be proven in the clinical setting. The straightforward options seem to be poly-ADPribose polymerase (PARP) targeting in the context of synthetic lethality, and apoptosis inhibition with already available drugs. Combinations of the three classes of drugs on the right may be considered. BCL2: B-cell lymphoma 2 protein; IR: irradiation; HRD: homologous recombination repair deficiency.
Acknowledgements
This work was supported by a Hesmo (Hellenic Society of Medical Oncology) Grant.
References
- 1.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016;131(6):803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 2.Wesseling P, Capper D. Who 2016 classification of gliomas. Neuropathol Appl Neurobiol. 2018;44(2):139–150. doi: 10.1111/nan.12432. [DOI] [PubMed] [Google Scholar]
- 3.Iuchi T, Sugiyama T, Ohira M, Kageyama H, Yokoi S, Sakaida T, Hasegawa Y, Setoguchi T, Itami M. Clinical significance of the 2016 who classification in japanese patients with gliomas. Brain Tumor Pathol. 2018;35(2):71–80. doi: 10.1007/s10014-018-0309-0. [DOI] [PubMed] [Google Scholar]
- 4.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr., Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. Idh1 and idh2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the idh1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008;116(6):597–602. doi: 10.1007/s00401-008-0455-2. [DOI] [PubMed] [Google Scholar]
- 7.Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, Frattini M, Molinari F, Knowles M, Cerrato A, Rodolfo M, Scarpa A, Felicioni L, Buttitta F, Malatesta S, Marchetti A, Bardelli A. Idh1 mutations at residue p.R132 (idh1(r132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat. 2009;30(1):7–11. doi: 10.1002/humu.20937. [DOI] [PubMed] [Google Scholar]
- 8.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O'Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, Collins VP. Idh1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 2009;11(4):341–347. doi: 10.1215/15228517-2009-025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nobusawa S, Watanabe T, Kleihues P, Ohgaki H. Idh1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res. 2009;15(19):6002–6007. doi: 10.1158/1078-0432.CCR-09-0715. [DOI] [PubMed] [Google Scholar]
- 11.Qi ST, Yu L, Lu YT, Ou YH, Li ZY, Wu LX, Yao F. Idh mutations occur frequently in chinese glioma patients and predict longer survival but not response to concomitant chemoradiotherapy in anaplastic gliomas. Oncol Rep. 2011;26(6):1479–1485. doi: 10.3892/or.2011.1428. [DOI] [PubMed] [Google Scholar]
- 12.Shaban ZM, Al-Aubaidy SR, Hameedi AD. Idh1 mutation in gliomas in baghdad by immunohistochemical study. International Journal of Genetics and Genomics. 2018;6(1):1–7. [Google Scholar]
- 13.Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. Idh1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. 2009;174(4):1149–1153. doi: 10.2353/ajpath.2009.080958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartmann C, Hentschel B, Wick W, Capper D, Felsberg J, Simon M, Westphal M, Schackert G, Meyermann R, Pietsch T, Reifenberger G, Weller M, Loeffler M, von Deimling A. Patients with idh1 wild type anaplastic astrocytomas exhibit worse prognosis than idh1-mutated glioblastomas, and idh1 mutation status accounts for the unfavorable prognostic effect of higher age: Implications for classification of gliomas. Acta Neuropathol. 2010;120(6):707–718. doi: 10.1007/s00401-010-0781-z. [DOI] [PubMed] [Google Scholar]
- 15.Olar A, Wani KM, Alfaro-Munoz KD, Heathcock LE, van Thuijl HF, Gilbert MR, Armstrong TS, Sulman EP, Cahill DP, Vera-Bolanos E, Yuan Y, Reijneveld JC, Ylstra B, Wesseling P, Aldape KD. Idh mutation status and role of who grade and mitotic index in overall survival in grade ii-iii diffuse gliomas. Acta Neuropathol. 2015;129(4):585–596. doi: 10.1007/s00401-015-1398-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, Pollock R, O'Donnell P, Grigoriadis A, Diss T, Eskandarpour M, Presneau N, Hogendoorn PC, Futreal A, Tirabosco R, Flanagan AM. Idh1 and idh2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224(3):334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 17.Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, Schenkein DP, Hezel AF, Ancukiewicz M, Liebman HM, Kwak EL, Clark JW, Ryan DP, Deshpande V, Dias-Santagata D, Ellisen LW, Zhu AX, Iafrate AJ. Frequent mutation of isocitrate dehydrogenase (idh)1 and idh2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17(1):72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kosmider O, Gelsi-Boyer V, Slama L, Dreyfus F, Beyne-Rauzy O, Quesnel B, Hunault-Berger M, Slama B, Vey N, Lacombe C, Solary E, Birnbaum D, Bernard OA, Fontenay M. Mutations of idh1 and idh2 genes in early and accelerated phases of myelodysplastic syndromes and mds/myelo-proliferative neoplasms. Leukemia. 2010;24(5):1094–1096. doi: 10.1038/leu.2010.52. [DOI] [PubMed] [Google Scholar]
- 19.Lopez GY, Reitman ZJ, Solomon D, Waldman T, Bigner DD, McLendon RE, Rosenberg SA, Samuels Y, Yan H. Idh1(r132) mutation identified in one human melanoma metastasis, but not correlated with metastases to the brain. Biochem Biophys Res Commun. 2010;398(3):585–587. doi: 10.1016/j.bbrc.2010.06.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madala HR, Punganuru SR, Arutla V, Misra S, Thomas TJ, Srivenugopal KS. Beyond brooding on oncometabolic havoc in idh-mutant gliomas and aml: Current and future therapeutic strategies. Cancers (Basel) 2018;10(2):49. doi: 10.3390/cancers10020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller JJ, Shih HA, Andronesi OC, Cahill DP. Isocitrate dehydrogenase-mutant glioma: Evolving clinical and therapeutic implications. Cancer. 2017;123(23):4535–4546. doi: 10.1002/cncr.31039. [DOI] [PubMed] [Google Scholar]
- 22.Molenaar RJ, Maciejewski JP, Wilmink JW, van Noorden CJF. Wild-type and mutated idh1/2 enzymes and therapy responses. Oncogene. 2018;37(15):1949–1960. doi: 10.1038/s41388-017-0077-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Kronke J, Bullinger L, Spath D, Kayser S, Zucknick M, Gotze K, Horst HA, Germing U, Dohner H, Dohner K. Idh1 and idh2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with npm1 mutation without flt3 internal tandem duplication. J Clin Oncol. 2010;28(22):3636–3643. doi: 10.1200/JCO.2010.28.3762. [DOI] [PubMed] [Google Scholar]
- 24.Shibata T, Kokubu A, Miyamoto M, Sasajima Y, Yamazaki N. Mutant idh1 confers an in vivo growth in a melanoma cell line with braf mutation. Am J Pathol. 2011;178(3):1395–1402. doi: 10.1016/j.ajpath.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders JE, Zeilemaker A, van Putten WJ, Rijneveld AW, Lowenberg B, Valk PJ. Acquired mutations in the genes encoding idh1 and idh2 both are recurrent aberrations in acute myeloid leukemia: Prevalence and prognostic value. Blood. 2010;116(12):2122–2126. doi: 10.1182/blood-2009-11-250878. [DOI] [PubMed] [Google Scholar]
- 26.Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, Niknafs N, Guthrie VB, Maitra A, Argani P, Offerhaus GJA, Roa JC, Roberts LR, Gores GJ, Popescu I, Alexandrescu ST, Dima S, Fassan M, Simbolo M, Mafficini A, Capelli P, Lawlor RT, Ruzzenente A, Guglielmi A, Tortora G, de Braud F, Scarpa A, Jarnagin W, Klimstra D, Karchin R, Velculescu VE, Hruban RH, Vogelstein B, Kinzler KW, Papadopoulos N, Wood LD. Exome sequencing identifies frequent inactivating mutations in bap1, arid1a and pbrm1 in intrahepatic cholangiocarcinomas. Nat Genet. 2013;45(12):1470–1473. doi: 10.1038/ng.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X, Zhao J, Xu Z, Peng B, Huang Q, Arnold E, Ding J. Structures of human cytosolic nadp-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J Biol Chem. 2004;279(32):33946–33957. doi: 10.1074/jbc.M404298200. [DOI] [PubMed] [Google Scholar]
- 28.Zhang C, Moore LM, Li X, Yung WK, Zhang W. Idh1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro Oncol. 2013;15(9):1114–1126. doi: 10.1093/neuonc/not087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dang L, Yen K, Attar EC. Idh mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 2016;27(4):599–608. doi: 10.1093/annonc/mdw013. [DOI] [PubMed] [Google Scholar]
- 30.Weller M, Wick W, von Deimling A. Isocitrate dehydrogenase mutations: A challenge to traditional views on the genesis and malignant progression of gliomas. Glia. 2011;59(8):1200–1204. doi: 10.1002/glia.21130. [DOI] [PubMed] [Google Scholar]
- 31.Koh HJ, Lee SM, Son BG, Lee SH, Ryoo ZY, Chang KT, Park JW, Park DC, Song BJ, Veech RL, Song H, Huh TL. Cytosolic nadp+-dependent isocitrate dehydrogenase plays a key role in lipid metabolism. J Biol Chem. 2004;279(38):39968–39974. doi: 10.1074/jbc.M402260200. [DOI] [PubMed] [Google Scholar]
- 32.Ronnebaum SM, Ilkayeva O, Burgess SC, Joseph JW, Lu D, Stevens RD, Becker TC, Sherry AD, Newgard CB, Jensen MV. A pyruvate cycling pathway involving cytosolic nadp-dependent isocitrate dehydrogenase regulates glucose-stimulated insulin secretion. J Biol Chem. 2006;281(41):30593–30602. doi: 10.1074/jbc.M511908200. [DOI] [PubMed] [Google Scholar]
- 33.Biaglow JE, Miller RA. The thioredoxin reductase/ thioredoxin system: Novel redox targets for cancer therapy. Cancer Biol Ther. 2005;4(1):6–13. doi: 10.4161/cbt.4.1.1434. [DOI] [PubMed] [Google Scholar]
- 34.Dikalov S. Cross talk between mitochondria and nadph oxidases. Free Radic Biol Med. 2011;51(7):1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee SM, Koh HJ, Park DC, Song BJ, Huh TL, Park JW. Cytosolic nadp(+)-dependent isocitrate dehydrogenase status modulates oxidative damage to cells. Free Radic Biol Med. 2002;32(11):1185–1196. doi: 10.1016/s0891-5849(02)00815-8. [DOI] [PubMed] [Google Scholar]
- 36.Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, Tigchelaar W, Troost D, Vandertop WP, Bardelli A, Van Noorden CJ. The prognostic idh1( r132 ) mutation is associated with reduced nadp+-dependent idh activity in glioblastoma. Acta Neuropathol. 2010;119(4):487–494. doi: 10.1007/s00401-010-0645-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waitkus MS, Diplas BH, Yan H. Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol. 2016;18(1):16–26. doi: 10.1093/neuonc/nov136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shih AH, Levine RL. Idh1 mutations disrupt blood, brain, and barriers. Cancer Cell. 2012;22(3):285–287. doi: 10.1016/j.ccr.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 40.Yang H, Ye D, Guan KL, Xiong Y. Idh1 and idh2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin Cancer Res. 2012;18(20):5562–5571. doi: 10.1158/1078-0432.CCR-12-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, Weller M, Herold-Mende C, Unterberg A, Jeuken JW, Wesseling P, Reifenberger G, von Deimling A. Type and frequency of idh1 and idh2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118(4):469–474. doi: 10.1007/s00401-009-0561-9. [DOI] [PubMed] [Google Scholar]
- 42.Yang B, Zhong C, Peng Y, Lai Z, Ding J. Molecular mechanisms of "off-on switch" of activities of human idh1 by tumor-associated mutation r132h. Cell Res. 2010;20(11):1188–1200. doi: 10.1038/cr.2010.145. [DOI] [PubMed] [Google Scholar]
- 43.Zhao S, Guan KL. Idh1 mutant structures reveal a mechanism of dominant inhibition. Cell Res. 2010;20(12):1279–1281. doi: 10.1038/cr.2010.160. [DOI] [PubMed] [Google Scholar]
- 44.Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, Xiong Y. Glioma-derived mutations in idh1 dominantly inhibit idh1 catalytic activity and induce hif-1alpha. Science. 2009;324(5924):261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yen KE, Bittinger MA, Su SM, Fantin VR. Cancer-associated idh mutations: Biomarker and therapeutic opportunities. Oncogene. 2010;29(49):6409–6417. doi: 10.1038/onc.2010.444. [DOI] [PubMed] [Google Scholar]
- 46.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Cancer-associated idh1 mutations produce 2-hydroxyglutarate. Nature. 2010;465(7300):966. doi: 10.1038/nature09132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB. The common feature of leukemia-associated idh1 and idh2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin G, Reitman ZJ, Duncan CG, Spasojevic I, Gooden DM, Rasheed BA, Yang R, Lopez GY, He Y, McLendon RE, Bigner DD, Yan H. Disruption of wild-type idh1 suppresses d-2-hydroxyglutarate production in idh1-mutated gliomas. Cancer Res. 2013;73(2):496–501. doi: 10.1158/0008-5472.CAN-12-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han CH, Batchelor TT. Isocitrate dehydrogenase mutation as a therapeutic target in gliomas. Chin Clin Oncol. 2017;6(3):33. doi: 10.21037/cco.2017.06.11. [DOI] [PubMed] [Google Scholar]
- 50.Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, Rustenburg AS, Grinaway PB, Chodera JD, Cross JR, Thompson CB. Hypoxia induces production of l-2-hydroxyglutarate. Cell Metab. 2015;22(2):304–311. doi: 10.1016/j.cmet.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kranendijk M, Struys EA, van Schaftingen E, Gibson KM, Kanhai WA, van der Knaap MS, Amiel J, Buist NR, Das AM, de Klerk JB, Feigenbaum AS, Grange DK, Hofstede FC, Holme E, Kirk EP, Korman SH, Morava E, Morris A, Smeitink J, Sukhai RN, Vallance H, Jakobs C, Salomons GS. Idh2 mutations in patients with d-2-hydroxyglutaric aciduria. Science. 2010;330(6002):336. doi: 10.1126/science.1192632. [DOI] [PubMed] [Google Scholar]
- 52.Rzem R, Vincent MF, Van Schaftingen E, Veiga-da-Cunha M. L-2-hydroxyglutaric aciduria, a defect of metabolite repair. J Inherit Metab Dis. 2007;30(5):681–689. doi: 10.1007/s10545-007-0487-0. [DOI] [PubMed] [Google Scholar]
- 53.Struys EA. D-2-hydroxyglutaric aciduria: Unravelling the biochemical pathway and the genetic defect. J Inherit Metab Dis. 2006;29(1):21–29. doi: 10.1007/s10545-006-0317-9. [DOI] [PubMed] [Google Scholar]
- 54.Moroni I, Bugiani M, D'Incerti L, Maccagnano C, Rimoldi M, Bissola L, Pollo B, Finocchiaro G, Uziel G. L-2-hydroxyglutaric aciduria and brain malignant tumors: A predisposing condition. Neurology. 2004;62(10):1882–1884. doi: 10.1212/01.wnl.0000125335.21381.87. [DOI] [PubMed] [Google Scholar]
- 55.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, McDonough MA, King ON, Clifton IJ, Klose RJ, Claridge TD, Ratcliffe PJ, Schofield CJ, Kawamura A. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12(5):463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yan H, Bigner DD, Velculescu V, Parsons DW. Mutant metabolic enzymes are at the origin of gliomas. Cancer Res. 2009;69(24):9157–9159. doi: 10.1158/0008-5472.CAN-09-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ye D, Xiong Y, Guan KL. The mechanisms of idh mutations in tumorigenesis. Cell Res. 2012;22(7):1102–1104. doi: 10.1038/cr.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WGJr. (r)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339(6127):1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Colvin H, Nishida N, Konno M, Haraguchi N, Takahashi H, Nishimura J, Hata T, Kawamoto K, Asai A, Tsunekuni K, Koseki J, Mizushima T, Satoh T, Doki Y, Mori M, Ishii H. Oncometabolite d-2-hydroxyglurate directly induces epithelial-mesenchymal transition and is associated with distant metastasis in colorectal cancer. Sci Rep. 2016;6:36289. doi: 10.1038/srep36289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leather T, Jenkinson MD, Das K, Poptani H. Magnetic resonance spectroscopy for detection of 2-hydroxyglutarate as a biomarker for idh mutation in gliomas. Metabolites. 2017;7(2) doi: 10.3390/metabo7020029. doi: 10.3390/metabo7020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hollon TC, Orringer DA. Shedding light on idh1 mutation in gliomas. Clin Cancer Res. 2018;24(11):2467–2469. doi: 10.1158/1078-0432.CCR-18-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Andronesi OC, Arrillaga-Romany IC, Ly KI, Bogner W, Ratai EM, Reitz K, Iafrate AJ, Dietrich J, Gerstner ER, Chi AS, Rosen BR, Wen PY, Cahill DP, Batchelor TT. Pharmacodynamics of mutant-idh1 inhibitors in glioma patients probed by in vivo 3d mrs imaging of 2-hydroxyglutarate. Nat Commun. 2018;9(1):1474. doi: 10.1038/s41467-018-03905-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Travins J, Weiss S, Looper R, Ligon KL, Verhaak RG, Yan H, Kaelin WG Jr. Transformation by the (r)-enantiomer of 2-hydroxyglutarate linked to egln activation. Nature. 2012;483(7390):484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dang L, Su SM. Isocitrate dehydrogenase mutation and (r)-2-hydroxyglutarate: From basic discovery to therapeutics development. Annu Rev Biochem. 2017;86:305–331. doi: 10.1146/annurev-biochem-061516-044732. [DOI] [PubMed] [Google Scholar]
- 65.Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, Asthana S, Jalbert LE, Nelson SJ, Bollen AW, Gustafson WC, Charron E, Weiss WA, Smirnov IV, Song JS, Olshen AB, Cha S, Zhao Y, Moore RA, Mungall AJ, Jones SJM, Hirst M, Marra MA, Saito N, Aburatani H, Mukasa A, Berger MS, Chang SM, Taylor BS, Costello JF. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343(6167):189–193. doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Medeiros BC, Fathi AT, DiNardo CD, Pollyea DA, Chan SM, Swords R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. 2017;31(2):272–281. doi: 10.1038/leu.2016.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cazzola M. Idh1 and idh2 mutations in myeloid neoplasms--novel paradigms and clinical implications. Haematologica. 2010;95(10):1623–1627. doi: 10.3324/haematol.2010.030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Parker SJ, Metallo CM. Metabolic consequences of oncogenic idh mutations. Pharmacol Ther. 2015;152:54–62. doi: 10.1016/j.pharmthera.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Malta TM, de Souza CF, Sabedot TS, Silva TC, Mosella MS, Kalkanis SN, Snyder J, Castro AVB, Noushmehr H. Glioma cpg island methylator phenotype (g-cimp): Biological and clinical implications. Neuro Oncol. 2018;20(5):608–620. doi: 10.1093/neuonc/nox183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fack F, Tardito S, Hochart G, Oudin A, Zheng L, Fritah S, Golebiewska A, Nazarov PV, Bernard A, Hau AC, Keunen O, Leenders W, Lund-Johansen M, Stauber J, Gottlieb E, Bjerkvig R, Niclou SP. Altered metabolic landscape in idh-mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol Med. 2017;9(12):1681–1695. doi: 10.15252/emmm.201707729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman C, Gillespie SM, Dionne D, Luo CC, Ravichandran H, Mylvaganam R, Mount C, Onozato ML, Nahed BV, Wakimoto H, Curry WT, Iafrate AJ, Rivera MN, Frosch MP, Golub TR, Brastianos PK, Getz G, Patel AP, Monje M, Cahill DP, Rozenblatt-Rosen O, Louis DN, Bernstein BE, Regev A, Suvà ML. Decoupling genetics, lineages, and microenvironment in idh-mutant gliomas by single-cell rna-seq. Science. 2017;355(6332) doi: 10.1126/science.aai8478. doi: 10.1126/science.aai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duncan R, Bazar L, Michelotti G, Tomonaga T, Krutzsch H, Avigan M, Levens D. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 1994;8(4):465–480. doi: 10.1101/gad.8.4.465. [DOI] [PubMed] [Google Scholar]
- 73.Hsiao HH, Nath A, Lin CY, Folta-Stogniew EJ, Rhoades E, Braddock DT. Quantitative characterization of the interactions among c-myc transcriptional regulators fuse, fbp, and fir. Biochemistry. 2010;49(22):4620–4634. doi: 10.1021/bi9021445. [DOI] [PubMed] [Google Scholar]
- 74.Jimenez G, Guichet A, Ephrussi A, Casanova J. Relief of gene repression by torso rtk signaling: Role of capicua in drosophila terminal and dorsoventral patterning. Genes Dev. 2000;14(2):224–231. [PMC free article] [PubMed] [Google Scholar]
- 75.Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr., Friedman AH, Friedman H, Gallia GL, Giovanella BC, Grollman AP, He TC, He Y, Hruban RH, Jallo GI, Mandahl N, Meeker AK, Mertens F, Netto GJ, Rasheed BA, Riggins GJ, Rosenquist TA, Schiffman M, Shih Ie M, Theodorescu D, Torbenson MS, Velculescu VE, Wang TL, Wentzensen N, Wood LD, Zhang M, McLendon RE, Bigner DD, Kinzler KW, Vogelstein B, Papadopoulos N, Yan H. Tert promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. 2013;110(15):6021–6026. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M, Rice T, Kosel ML, Smirnov IV, Sarkar G, Caron AA, Kollmeyer TM, Praska CE, Chada AR, Halder C, Hansen HM, McCoy LS, Bracci PM, Marshall R, Zheng S, Reis GF, Pico AR, O'Neill BP, Buckner JC, Giannini C, Huse JT, Perry A, Tihan T, Berger MS, Chang SM, Prados MD, Wiemels J, Wiencke JK, Wrensch MR, Jenkins RB. Glioma groups based on 1p/19q, idh, and tert promoter mutations in tumors. N Engl J Med. 2015;372(26):2499–2508. doi: 10.1056/NEJMoa1407279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, Offerhaus GJ, McLendon R, Rasheed BA, He Y, Yan H, Bigner DD, Oba-Shinjo SM, Marie SK, Riggins GJ, Kinzler KW, Vogelstein B, Hruban RH, Maitra A, Papadopoulos N, Meeker AK. Altered telomeres in tumors with atrx and daxx mutations. Science. 2011;333(6041):425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K. Identification of a cpg island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O'Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in pdgfra, idh1, egfr, and nf1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, Levine R, Heguy A, Viale A, Morris LG, Huse JT, Mellinghoff IK, Chan TA. Idh1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A. Leukemic idh1 and idh2 mutations result in a hypermethylation phenotype, disrupt tet2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O'Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB. Idh mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, Bernstein BE. Insulator dysfunction and oncogene activation in idh mutant gliomas. Nature. 2016;529(7584):110–114. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Turcan S, Makarov V, Taranda J, Wang Y, Fabius AWM, Wu W, Zheng Y, El-Amine N, Haddock S, Nanjangud G, LeKaye HC, Brennan C, Cross J, Huse JT, Kelleher NL, Osten P, Thompson CB, Chan TA. Mutant-idh1-dependent chromatin state reprogramming, reversibility, and persistence. Nat Genet. 2018;50(1):62–72. doi: 10.1038/s41588-017-0001-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chesnelong C, Chaumeil MM, Blough MD, Al-Najjar M, Stechishin OD, Chan JA, Pieper RO, Ronen SM, Weiss S, Luchman HA, Cairncross JG. Lactate dehydrogenase a silencing in idh mutant gliomas. Neuro Oncol. 2014;16(5):686–695. doi: 10.1093/neuonc/not243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mu L, Long Y, Yang C, Jin L, Tao H, Ge H, Chang YE, Karachi A, Kubilis PS, De Leon G, Qi J, Sayour EJ, Mitchell DA, Lin Z, Huang J. The idh1 mutation-induced oncometabolite, 2-hydroxyglutarate, may affect DNA methylation and expression of pd-l1 in gliomas. Front Mol Neurosci. 2018;11:82. doi: 10.3389/fnmol.2018.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rover LK, Gevensleben H, Dietrich J, Bootz F, Landsberg J, Goltz D, Dietrich D. Pd-1 (pdcd1) promoter methylation is a prognostic factor in patients with diffuse lower-grade gliomas harboring isocitrate dehydrogenase (idh) mutations. EBioMedicine. 2018;28:97–104. doi: 10.1016/j.ebiom.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Qian Z, Li Y, Fan X, Zhang C, Wang Y, Jiang T, Liu X. Molecular and clinical characterization of idh associated immune signature in lower-grade gliomas. OncoImmunology. 2018;20 (suppl 1):e1434466. doi: 10.1080/2162402X.2018.1434466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Y, Ningning L, Lau J, Richard-londt A, von Deimling A, Pusch S, Brandner S. Idh1 mutant glioma initiating cells are predisposed to apoptosis under endoplasmic reticulum (er) stress. Neuro-Oncology. 2018;20(suppl 1):i13–i13. [Google Scholar]
- 90.Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, Liu Y, Sundaram RK, Hegan DC, Fons NR, Breuer GA, Song Y, Mishra-Gorur K, De Feyter HM, de Graaf RA, Surovtseva YV, Kachman M, Halene S, Gunel M, Glazer PM, Bindra RS. 2-hydroxyglutarate produced by neomorphic idh mutations suppresses homologous recombination and induces parp inhibitor sensitivity. Med. 2017;9(375) doi: 10.1126/scitranslmed.aal2463. doi: 10.1126/scitranslmed.aal2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Karpel-Massler G, Ishida CT, Bianchetti E, Zhang Y, Shu C, Tsujiuchi T, Banu MA, Garcia F, Roth KA, Bruce JN, Canoll P, Siegelin MD. Induction of synthetic lethality in idh1-mutated gliomas through inhibition of bcl-xl. Nat Commun. 2017;8(1):1067. doi: 10.1038/s41467-017-00984-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lu Y, Kwintkiewicz J, Liu Y, Tech K, Frady LN, Su YT, Bautista W, Moon SI, MacDonald J, Ewend MG, Gilbert MR, Yang C, Wu J. Chemosensitivity of idh1-mutated gliomas due to an impairment in parp1-mediated DNA repair. Cancer Res. 2017;77(7):1709–1718. doi: 10.1158/0008-5472.CAN-16-2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, El Hallani S, Boisselier B, Mokhtari K, Hoang-Xuan K, Delattre JY. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009;27(25):4150–4154. doi: 10.1200/JCO.2009.21.9832. [DOI] [PubMed] [Google Scholar]
- 94.Labussiere M, Sanson M, Idbaih A, Delattre JY. Idh1 gene mutations: A new paradigm in glioma prognosis and therapy. Oncologist. 2010;15(2):196–199. doi: 10.1634/theoncologist.2009-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van den Bent MJ, Dubbink HJ, Marie Y, Brandes AA, Taphoorn MJ, Wesseling P, Frenay M, Tijssen CC, Lacombe D, Idbaih A, van Marion R, Kros JM, Dinjens WN, Gorlia T, Sanson M. Idh1 and idh2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: A report of the european organization for research and treatment of cancer brain tumor group. Clin Cancer Res. 2010;16(5):1597–1604. doi: 10.1158/1078-0432.CCR-09-2902. [DOI] [PubMed] [Google Scholar]
- 96.Yao Q, Cai G, Yu Q, Shen J, Gu Z, Chen J, Shi W, Shi J. Idh1 mutation diminishes aggressive phenotype in glioma stem cells. Int J Oncol. 2018;52(1):270–278. doi: 10.3892/ijo.2017.4186. [DOI] [PubMed] [Google Scholar]
- 97.Beiko J, Suki D, Hess KR, Fox BD, Cheung V, Cabral M, Shonka N, Gilbert MR, Sawaya R, Prabhu SS, Weinberg J, Lang FF, Aldape KD, Sulman EP, Rao G, McCutcheon IE, Cahill DP. Idh1 mutant malignant astrocytomas are more amenable to surgical resection and have a survival benefit associated with maximal surgical resection. Neuro Oncol. 2014;16(1):81–91. doi: 10.1093/neuonc/not159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, Robertson AG, Noushmehr H, Laird PW, Cherniack AD, Akbani R, Huse JT, Ciriello G, Poisson LM, Barnholtz-Sloan JS, Berger MS, Brennan C, Colen RR, Colman H, Flanders AE, Giannini C, Grifford M, Iavarone A, Jain R, Joseph I, Kim J, Kasaian K, Mikkelsen T, Murray BA, O'Neill BP, Pachter L, Parsons DW, Sougnez C, Sulman EP, Vandenberg SR, Van Meir EG, von Deimling A, Zhang H, Crain D, Lau K, Mallery D, Morris S, Paulauskis J, Penny R, Shelton T, Sherman M, Yena P, Black A, Bowen J, Dicostanzo K, Gastier-Foster J, Leraas KM, Lichtenberg TM, Pierson CR, Ramirez NC, Taylor C, Weaver S, Wise L, Zmuda E, Davidsen T, Demchok JA, Eley G, Ferguson ML, Hutter CM, Mills Shaw KR, Ozenberger BA, Sheth M, Sofia HJ, Tarnuzzer R, Wang Z, Yang L, Zenklusen JC, Ayala B, Baboud J, Chudamani S, Jensen MA, Liu J, Pihl T, Raman R, Wan Y, Wu Y, Ally A, Auman JT, Balasundaram M, Balu S, Baylin SB, Beroukhim R, Bootwalla MS, Bowlby R, Bristow CA, Brooks D, Butterfield Y, Carlsen R, Carter S, Chin L, Chu A, Chuah E, Cibulskis K, Clarke A, Coetzee SG, Dhalla N, Fennell T, Fisher S, Gabriel S, Getz G, Gibbs R, Guin R, Hadjipanayis A, Hayes DN, Hinoue T, Hoadley K, Holt RA, Hoyle AP, Jefferys SR, Jones S, Jones CD, Kucherlapati R, Lai PH, Lander E, Lee S, Lichtenstein L, Ma Y, Maglinte DT, Mahadeshwar HS, Marra MA, Mayo M, Meng S, Meyerson ML, Mieczkowski PA, Moore RA, Mose LE, Mungall AJ, Pantazi A, Parfenov M, Park PJ, Parker JS, Perou CM, Protopopov A, Ren X, Roach J, Sabedot TS, Schein J, Schumacher SE, Seidman JG, Seth S, Shen H, Simons JV, Sipahimalani P, Soloway MG, Song X, Sun H, Tabak B, Tam A, Tan D, Tang J, Thiessen N, Triche T Jr, Van Den Berg DJ, Veluvolu U, Waring S, Weisenberger DJ, Wilkerson MD, Wong T, Wu J, Xi L, Xu AW, Yang L, Zack TI, Zhang J, Aksoy BA, Arachchi H, Benz C, Bernard B, Carlin D, Cho J, DiCara D, Frazer S, Fuller GN, Gao J, Gehlenborg N, Haussler D, Heiman DI, Iype L, Jacobsen A, Ju Z, Katzman S, Kim H, Knijnenburg T, Kreisberg RB, Lawrence MS, Lee W, Leinonen K, Lin P, Ling S, Liu W, Liu Y, Liu Y, Lu Y, Mills G, Ng S, Noble MS, Paull E, Rao A, Reynolds S, Saksena G, Sanborn Z, Sander C, Schultz N, Senbabaoglu Y, Shen R, Shmulevich I, Sinha R, Stuart J, Sumer SO, Sun Y, Tasman N, Taylor BS, Voet D, Weinhold N, Weinstein JN, Yang D, Yoshihara K, Zheng S, Zhang W, Zou L, Abel T, Sadeghi S, Cohen ML, Eschbacher J, Hattab EM, Raghunathan A, Schniederjan MJ, Aziz D, Barnett G, Barrett W, Bigner DD, Boice L, Brewer C, Calatozzolo C, Campos B, Carlotti CG Jr, Chan TA, Cuppini L, Curley E, Cuzzubbo S, Devine K, DiMeco F, Duell R, Elder JB, Fehrenbach A, Finocchiaro G, Friedman W, Fulop J, Gardner J, Hermes B, Herold-Mende C, Jungk C, Kendler A, Lehman NL, Lipp E, Liu O, Mandt R, McGraw M, McLendon R, McPherson C, Neder L, Nguyen P, Noss A, Nunziata R, Ostrom QT, Palmer C, Perin A, Pollo B, Potapov A, Potapova O, Rathmell WK, Rotin D, Scarpace L, Schilero C, Senecal K, Shimmel K, Shurkhay V, Sifri S, Singh R, Sloan AE, Smolenski K, Staugaitis SM, Steele R, Thorne L, Tirapelli DP, Unterberg A, Vallurupalli M, Wang Y, Warnick R, Williams F, Wolinsky Y, Bell S, Rosenberg M, Stewart C, Huang F, Grimsby JL, Radenbaugh AJ, Zhang J. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372(26):2481–2498. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Qi S, Yu L, Li H, Ou Y, Qiu X, Ding Y, Han H, Zhang X. Isocitrate dehydrogenase mutation is associated with tumor location and magnetic resonance imaging characteristics in astrocytic neoplasms. Oncol Lett. 2014;7(6):1895–1902. doi: 10.3892/ol.2014.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jaiswal S, Chaudhary N, Prasad P, Khatri D, Das KK, Mehrotra A, Jaiswal AK, Pal L, Behari S. Expression of isocitrate dehydrogenase-1 (idh-1) mutant protein in gliomas. Tech Neurosurg Neurol 1(3), 2018. TNN.000514.2018. Last accessed. Neurosurg Neurol. 2018;1 (3) TNN.000514.2018. Last accessed on 24-7-2018. [Google Scholar]
- 101.Chen JR, Yao Y, Xu HZ, Qin ZY. Isocitrate dehydrogenase (idh)1/2 mutations as prognostic markers in patients with glioblastomas. Medicine (Baltimore) 2016;95(9):e2583. doi: 10.1097/MD.0000000000002583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Villanueva-Meyer JE, Wood MD, Choi BS, Mabray MC, Butowski NA, Tihan T, Cha S. Mri features and idh mutational status of grade ii diffuse gliomas: Impact on diagnosis and prognosis. AJR Am J Roentgenol. 2018;210(3):621–628. doi: 10.2214/AJR.17.18457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pepin KM, McGee KP, Arani A, Lake DS, Glaser KJ, Manduca A, Parney IF, Ehman RL, Huston J 3rd. Mr elastography analysis of glioma stiffness and idh1-mutation status. AJNR Am J Neuroradiol. 2018;39(1):31–36. doi: 10.3174/ajnr.A5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Capper D, Weissert S, Balss J, Habel A, Meyer J, Jager D, Ackermann U, Tessmer C, Korshunov A, Zentgraf H, Hartmann C, von Deimling A. Characterization of r132h mutation-specific idh1 antibody binding in brain tumors. Brain Pathol. 2010;20(1):245–254. doi: 10.1111/j.1750-3639.2009.00352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang P, Dong Q, Zhang C, Kuan PF, Liu Y, Jeck WR, Andersen JB, Jiang W, Savich GL, Tan TX, Auman JT, Hoskins JM, Misher AD, Moser CD, Yourstone SM, Kim JW, Cibulskis K, Getz G, Hunt HV, Thorgeirsson SS, Roberts LR, Ye D, Guan KL, Xiong Y, Qin LX, Chiang DY. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32(25):3091–3100. doi: 10.1038/onc.2012.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chou WC, Lei WC, Ko BS, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Wu SJ, Huang SY, Hsu SC, Chen YC, Chang YC, Kuo KT, Lee FY, Liu MC, Liu CW, Tseng MH, Huang CF, Tien HF. The prognostic impact and stability of isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia. 2011;25(2):246–253. doi: 10.1038/leu.2010.267. [DOI] [PubMed] [Google Scholar]
- 107.Houillier C, Wang X, Kaloshi G, Mokhtari K, Guillevin R, Laffaire J, Paris S, Boisselier B, Idbaih A, Laigle-Donadey F, Hoang-Xuan K, Sanson M, Delattre JY. Idh1 or idh2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. 2010;75(17):1560–1566. doi: 10.1212/WNL.0b013e3181f96282. [DOI] [PubMed] [Google Scholar]
- 108.SongTao Q, Lei Y, Si G, YanQing D, HuiXia H, XueLin Z, LanXiao W, Fei Y. Idh mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012;103(2):269–273. doi: 10.1111/j.1349-7006.2011.02134.x. [DOI] [PubMed] [Google Scholar]
- 109.Cairncross JG, Wang M, Jenkins RB, Shaw EG, Giannini C, Brachman DG, Buckner JC, Fink KL, Souhami L, Laperriere NJ, Huse JT, Mehta MP, Curran WJ Jr. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of idh. J Clin Oncol. 2014;32(8):783–790. doi: 10.1200/JCO.2013.49.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, Kunii K, Pedraza A, Schalm S, Silverman L, Miller A, Wang F, Yang H, Chen Y, Kernytsky A, Rosenblum MK, Liu W, Biller SA, Su SM, Brennan CW, Chan TA, Graeber TG, Yen KE, Mellinghoff IK. An inhibitor of mutant idh1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, Yang H, Gross S, Artin E, Saada V, Mylonas E, Quivoron C, Popovici-Muller J, Saunders JO, Salituro FG, Yan S, Murray S, Wei W, Gao Y, Dang L, Dorsch M, Agresta S, Schenkein DP, Biller SA, Su SM, de Botton S, Yen KE. Targeted inhibition of mutant idh2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 112.Kernytsky A, Wang F, Hansen E, Schalm S, Straley K, Gliser C, Yang H, Travins J, Murray S, Dorsch M, Agresta S, Schenkein DP, Biller SA, Su SM, Liu W, Yen KE. Idh2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood. 2015;125(2):296–303. doi: 10.1182/blood-2013-10-533604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Popovici-Muller J, Saunders JO, Salituro FG, Travins JM, Yan S, Zhao F, Gross S, Dang L, Yen KE, Yang H, Straley KS, Jin S, Kunii K, Fantin VR, Zhang S, Pan Q, Shi D, Biller SA, Su SM. Discovery of the first potent inhibitors of mutant idh1 that lower tumor 2-hg in vivo. ACS Med Chem Lett. 2012;3(10):850–855. doi: 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Gorlich K, Wichmann M, Schwarzer A, Preller M, Thol F, Meyer J, Haemmerle R, Struys EA, Jansen EE, Modlich U, Li Z, Sly LM, Geffers R, Lindner R, Manstein DJ, Lehmann U, Krauter J, Ganser A, Heuser M. Mutant idh1 promotes leukemogenesis in vivo and can be specifically targeted in human aml. Blood. 2013;122(16):2877–2887. doi: 10.1182/blood-2013-03-491571. [DOI] [PubMed] [Google Scholar]
- 115.Li L, Paz AC, Wilky BA, Johnson B, Galoian K, Rosenberg A, Hu G, Tinoco G, Bodamer O, Trent JC. Treatment with a small molecule mutant idh1 inhibitor suppresses tumorigenic activity and decreases production of the oncometabolite 2-hydroxyglutarate in human chondrosarcoma cells. PLoS One. 2015;10(9):e0133813. doi: 10.1371/journal.pone.0133813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yen K, Travins J, Wang F, David MD, Artin E, Straley K, Padyana A, Gross S, DeLaBarre B, Tobin E, Chen Y, Nagaraja R, Choe S, Jin L, Konteatis Z, Cianchetta G, Saunders JO, Salituro FG, Quivoron C, Opolon P, Bawa O, Saada V, Paci A, Broutin S, Bernard OA, de Botton S, Marteyn BS, Pilichowska M, Xu Y, Fang C, Jiang F, Wei W, Jin S, Silverman L, Liu W, Yang H, Dang L, Dorsch M, Penard-Lacronique V, Biller SA, Su SM. Ag-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic idh2 mutations. Cancer Discov. 2017;7(5):478–493. doi: 10.1158/2159-8290.CD-16-1034. [DOI] [PubMed] [Google Scholar]
- 117.Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, Stone RM, DeAngelo DJ, Levine RL, Flinn IW, Kantarjian HM, Collins R, Patel MR, Frankel AE, Stein A, Sekeres MA, Swords RT, Medeiros BC, Willekens C, Vyas P, Tosolini A, Xu Q, Knight RD, Yen KE, Agresta S, de Botton S, Tallman MS. Enasidenib in mutant idh2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–731. doi: 10.1182/blood-2017-04-779405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stein EM. Enasidenib, a targeted inhibitor of mutant idh2 proteins for treatment of relapsed or refractory acute myeloid leukemia. Future Oncol. 2018;14(1):23–40. doi: 10.2217/fon-2017-0392. [DOI] [PubMed] [Google Scholar]
- 119.Stein EM, DiNardo C, Altman JK, Collins R, DeAngelo DJ, Kantarjian HM, Sekeres MA, Fathi AT, Flinn IW, Frankel AE, Levine RL, Medeiros BC, Patel MR, Pollyea D, Roboz GJ, Stone RM, Swords RT, Tallman MS, Yen K, Attar EC, Xu Q, Tosolini A, Mei JM, Thakurta A, Knight RD, De Botton S. Safety and efficacy of ag-221, a potent inhibitor of mutant idh2 that promotes differentiation of myeloid cells in patients with advanced hematologic malignancies: Results of a phase 1/2 trial. Blood. 2015;126(23):323–323. [Google Scholar]
- 120.Amatangelo MD, Quek L, Shih A, Stein EM, Roshal M, David MD, Marteyn B, Farnoud NR, de Botton S, Bernard OA, Wu B, Yen KE, Tallman MS, Papaemmanuil E, Penard-Lacronique V, Thakurta A, Vyas P, Levine RL. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood. 2017;130(6):732–741. doi: 10.1182/blood-2017-04-779447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim ES. Enasidenib: First global approval. Drugs. 2017;77(15):1705–1711. doi: 10.1007/s40265-017-0813-2. [DOI] [PubMed] [Google Scholar]
- 122.Burris H, Mellinghoff I, Maher E, Wen P, Beeram M, Touat M, Faris J, Azad N, Cloughesy T, Gore L, Trent J, Hoff DV, Goldwasser M, Fan B, Agresta S. Abstract pl04-05: The first reported results of ag-120, a first-in-class, potent inhibitor of the idh1 mutant protein, in a phase i study of patients with advanced idh1-mutant solid tumors, including gliomas. Molecular Cancer Therapeutics. 2015;14(12 Suppl 2):PL04–05-PL04-05. [Google Scholar]
- 123.Chaturvedi A, Herbst L, Pusch S, Klett L, Goparaju R, Stichel D, Kaulfuss S, Panknin O, Zimmermann K, Toschi L, Neuhaus R, Haegebarth A, Rehwinkel H, Hess-Stumpp H, Bauser M, Bochtler T, Struys EA, Sharma A, Bakkali A, Geffers R, Araujo-Cruz MM, Thol F, Gabdoulline R, Ganser A, Ho AD, von Deimling A, Rippe K, Heuser M, Kramer A. Pan-mutant-idh1 inhibitor bay1436032 is highly effective against human idh1 mutant acute myeloid leukemia in vivo. Leukemia. 2017;31(10):2020–2028. doi: 10.1038/leu.2017.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Picca A, Berzero G, Sanson M. Current therapeutic approaches to diffuse grade ii and iii gliomas. Ther Adv Neurol Disord. 2018;11 doi: 10.1177/1756285617752039. https://doi.org/10.1177%2F175628561 7752039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Turcan S, Fabius AW, Borodovsky A, Pedraza A, Brennan C, Huse J, Viale A, Riggins GJ, Chan TA. Efficient induction of differentiation and growth inhibition in idh1 mutant glioma cells by the dnmt inhibitor decitabine. Oncotarget. 2013;4(10):1729–1736. doi: 10.18632/oncotarget.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Borodovsky A, Salmasi V, Turcan S, Fabius AW, Baia GS, Eberhart CG, Weingart JD, Gallia GL, Baylin SB, Chan TA, Riggins GJ. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived idh1 mutant glioma xenograft. Oncotarget. 2013;4(10):1737–1747. doi: 10.18632/oncotarget.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, Menn O, Osswald M, Oezen I, Ott M, Keil M, Balss J, Rauschenbach K, Grabowska AK, Vogler I, Diekmann J, Trautwein N, Eichmuller SB, Okun J, Stevanovic S, Riemer AB, Sahin U, Friese MA, Beckhove P, von Deimling A, Wick W, Platten M. A vaccine targeting mutant idh1 induces antitumour immunity. Nature. 2014;512(7514):324–327. doi: 10.1038/nature13387. [DOI] [PubMed] [Google Scholar]
- 128.Pellegatta S, Valletta L, Corbetta C, Patane M, Zucca I, Riccardi Sirtori F, Bruzzone MG, Fogliatto G, Isacchi A, Pollo B, Finocchiaro G. Effective immuno-targeting of the idh1 mutation r132h in a murine model of intracranial glioma. Acta Neuropathol Commun. 2015;3:4. doi: 10.1186/s40478-014-0180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Saito K, Suzuki K, Shimizu S, Kobayashi K, Shimada D, Kume S, Iijima S, Chiba T, Shibahara J, Shiokawa Y, Nagane M. Gene-61. Temozolomide-induced mismatch repair insufficiency and hypermethylation of mgmt promoter with hypermutation in malignant gliomas. Neuro-Oncology. 2017;19(suppl_6):vi106–vi106. [Google Scholar]
- 130.Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, Shimamura T, Niida A, Motomura K, Ohka F, Yamamoto T, Tanahashi K, Ranjit M, Wakabayashi T, Yoshizato T, Kataoka K, Yoshida K, Nagata Y, Sato-Otsubo A, Tanaka H, Sanada M, Kondo Y, Nakamura H, Mizoguchi M, Abe T, Muragaki Y, Watanabe R, Ito I, Miyano S, Natsume A, Ogawa S. Mutational landscape and clonal architecture in grade ii and iii gliomas. Nat Genet. 2015;47(5):458–468. doi: 10.1038/ng.3273. [DOI] [PubMed] [Google Scholar]