

Abstract
Up-regulation of thrombospondin-4 (TSP4) or voltage-gated calcium channel subunit α2δ1 (Cavα2δ1) proteins in the spinal cord contributes to neuropathic pain development through an unidentified mechanism. We have previously shown that TSP4 interacts with Cavα2δ1 to promote excitatory synaptogenesis and the development of chronic pain states. However, the TSP4 determinants responsible for these changes are not known. Here, we tested the hypothesis that the Cavα2δ1-binding domains of TSP4 are synaptogenic and pronociceptive. We mapped the major Cavα2δ1-binding domains of TSP4 within the coiled-coil and epidermal growth factor (EGF)-like domains in vitro. Intrathecal injection of TSP4 fragment proteins containing the EGF-like domain (EGF-LIKE) into naïve rodents was sufficient for inducing behavioral hypersensitivity similar to that produced by an equal molar dose of full-length TSP4. Gabapentin, a drug that binds to Cavα2δ1, blocked EGF-LIKE–induced behavioral hypersensitivity in a dose-dependent manner, supporting the notion that EGF-LIKE interacts with Cavα2δ1 and thereby mediates behavioral hypersensitivity. This notion was further supported by our findings that a peptide within EGF-LIKE (EGFD355–369) could block TSP4- or Cavα2δ1-induced behavioral hypersensitivity after intrathecal injections. Furthermore, only TSP4 proteins that contained EGF-LIKE could promote excitatory synaptogenesis between sensory and spinal cord neurons, which could be blocked by peptide EGFD355–369. Together, these findings indicate that EGF-LIKE is the molecular determinant that mediates aberrant excitatory synaptogenesis and chronic pain development. Blocking interactions between EGF-LIKE and Cavα2δ1 could be an alternative approach in designing target-specific pain medications.
Keywords: thrombospondin, pain, synaptic plasticity, calcium channel, extracellular matrix protein, allodynia, neuropathic pain, pro-nociceptive domain, synaptogenesis, TSP4
Introduction
Neuropathic pain, or pain states derived from injuries to the peripheral or central nervous systems, includes spontaneous pain and evoked pain such as tactile allodynia (exaggerated response to otherwise innocuous tactile stimuli) and hyperalgesia (exaggerated pain sensations to mildly noxious stimuli) (1, 2). Most current pain medications are not efficacious against neuropathic pain and are often associated with intolerable side effects. Identifying new targets and pathways involved in neuropathic pain processing is an unmet medical need that can provide mechanistic insights leading to development of target-specific pain medication. In the search for new targets/pathways critical in pain state development, our group and others have reported that peripheral nerve injury induces a parallel up-regulation of the astrocyte-secreted extracellular matrix protein thrombospondin-4 (TSP4)3 and voltage-gated calcium channel α2δ1 subunit (Cavα2δ1) in the dorsal root ganglia (DRG) and dorsal spinal cord that correlates with the development of behavioral hypersensitivities (3–9).
TSPs are large oligomeric, multidomain, extracellular matrix proteins that mediate cell/cell and cell/matrix interactions through binding to other extracellular matrix proteins, membrane proteins, and cytokines (10, 11). The TSP family consists of two subfamilies, subgroup A (TSP1 and -2) and subgroup B (TSP3–5), which are distinguished by their oligomerization states and domain structures. TSP4 has been shown to play a role in the development of neuropathic pain states. Intrathecal injection of TSP4 recombinant proteins into naïve rodent animals causes exaggerated presynaptic excitatory input into dorsal spinal cord and behavioral hypersensitivity (9, 12). TSP4 blockade by intrathecal treatments with antisense oligodeoxynucleotides, antibodies, or genetic ablation of the TSP4 gene reverses or prevents behavioral hypersensitivity induced by injuries to the peripheral and central nervous systems or intrathecal injection of TSP4 recombinant proteins (9, 13, 14).
Interestingly, TSP4-induced spinal cord neuron sensitization and behavioral hypersensitivity can be blocked by gabapentin, a gabapentinoid drug that binds to Cavα2δ1 (15, 16), suggesting that TSP4 induces spinal cord sensitization and behavioral hypersensitivity through its interactions with Cavα2δ1 (12). Multiple lines of evidence also support this notion. Transgenic (TG) mice overexpressing Cavα2δ1 in neuronal tissues exhibit dorsal horn neuron hyperexcitability (17–19) and behavioral hypersensitivity (20) without nerve injury that can be blocked by TSP4 antibodies or viral or genetic ablation of TSP4 (12). In addition, genetic ablation of Cavα2δ1 or gabapentin treatment can block TSP4-induced spinal neuron sensitization and behavioral hypersensitivity (12, 21). Furthermore, TSP4 proteins interact with Cavα2δ1 to promote excitatory synaptogenesis in dorsal spinal cord that can be blocked by gabapentin or genetic ablation of Cavα2δ1 (12, 21). All these findings support that activation of a TSP4/Cavα2δ1 pathway is critical in promoting central sensitization and pain states, at least partially through aberrant excitatory synaptogenesis (12, 21). Thus, blocking the activated TSP4/Cavα2δ1 pathway could be an alternative approach in developing specific interventions for pain relief. However, both TSP4 and Cavα2δ1 are large proteins, so it is difficult to design small molecules or other drugs to block their interactions for pain relief without knowing the Cavα2δ1-interacting and nociceptive domains of TSP4. Accordingly, we tested the hypothesis in this study that the Cavα2δ1-interacting domains of TSP4 play a critical role in promoting behavioral hypersensitivity, at least partially through enhancing spinal cord excitatory synaptogenesis.
Results
Mapping Cavα2δ1-binding motifs of TSP4
We have shown previously that injury-induced spinal TSP4 contributes to the development of neuropathic pain states (9) through a direct interaction with Cavα2δ1 proteins (12). To identify the TSP4-binding domain to Cavα2δ1, we created five recombinant truncation constructs of TSP4: the N-terminal domain deletion construct (ND) encoding the EGF-LIKE domains, type-3 calcium-binding domain, and C-terminal domain; the EGF-like domain construct (EGF-LIKE) encoding EGF-LIKE; the C-terminal domain construct (CD) encoding the type-3 calcium-binding domain and C-terminal domain; the N-terminal domain alone construct (NT); and the N-terminal domain plus the coiled-coil domain construct (NT+CC) (Fig. 1A, top). TSP4 mutant proteins from these constructs were expressed in the human embryonic kidney (HEK) cell line 293-EBNA and purified through His-tag columns (Fig. 1A, bottom). Binding of the recombinant TSP4 proteins to recombinant Cavα2δ1 proteins was tested in an ELISA-based ligand binding assay. Our data indicated that domain proteins containing EGF-LIKE (ND and EGF-LIKE) and coiled-coil domain of TSP4 bound to Cavα2δ1 with a similar or over 70% capacity as the full-length TSP4, whereas binding of domain proteins containing the N-terminal domain, type-3 calcium-binding domain, and C-terminal domain to Cavα2δ1 was about 35% of that observed for full-length TSP4 (Fig. 1B). Thus, the coiled-coil and EGF-LIKE domains of TSP4 likely contained the major Cavα2δ1-binding sites.
Figure 1.

Identification of Cavα2δ1-binding domains of TSP4. A, top, domain structure of full-length TSP4 and recombinant TSP4 truncation proteins. Shown are the N-terminal domain (blue), coiled-coil oligomerization domain (gray), four type II EGF-like domain repeats (orange), seven type-3 calcium-binding repeats (yellow), and the C-terminal domain (green). Bottom, representative Western blots under reducing conditions showing each purified recombinant TSP4 truncated protein detected by anti-His antibodies. The approximate molecular mass (kDa) is shown on the left. B, binding of FLAG-Cavα2δ1 to immobilized TSP4 or truncated TSP4 proteins at equal molar concentrations to TSP4 (80 μg/ml) in an ELISA-based ligand binding assay. Data presented in the box-and-whisker plot are from six to nine independent determinations; ****, p < 0.0001 compared with full-length TSP4 by one-way ANOVA with Bonferroni's multiple comparisons test. C, summary of normalized binding of Cavα2δ1 lysates to 15-mer peptides (overlapping by 12 amino acids) encompassing the entire TSP4 amino acid sequence. Summarized data are normalized to peptide 78.
We further examined the Cavα2δ1-binding motifs of TSP4 with SPOT peptide array analysis by measuring binding of recombinant Cavα2δ1 purified from transfected HEK 293 cells to immobilized overlapping 15-mer peptides that covered the entire length of TSP4. Similar to data from protein binding assays, TSP4 linear peptides with high affinity to Cavα2δ1 were clustered in the coiled-coil and EGF-LIKE domains but not in the type-3 calcium-binding domain (Fig. 1C). A few peptides from the N-terminal and C-terminal domains also bound to Cavα2δ1, which might contribute to the low binding affinity to Cavα2δ1 observed from the NT and CD truncated proteins (Fig. 1, B and C).
EGF-LIKE domain of TSP4 is pronociceptive
Previous findings have shown that increased spinal TSP4 protein alone is sufficient to induce tactile allodynia and thermal hyperalgesia (9) through interactions with Cavα2δ1 (12). To identify the pronociceptive domain of TSP4, we injected the recombinant truncated TSP4 proteins into L5/6 spinal region of naïve adult rats and tested their hind paw sensitivity to mechanical and thermal stimuli. Intrathecal (i.t.) injection of EGF-LIKE–containing proteins (EGF-like and ND), at an equal molar dose to the pronociceptive dose of full-length TSP4 (45 μg/rat) (9), induced a reduction in paw withdrawal thresholds to von Frey filaments (tactile allodynia) (Fig. 2A) and paw withdrawal latency to radiant heat (thermal hyperalgesia) (Fig. 2B). The pronociceptive effects of EGF-LIKE–containing proteins started within 2 days and lasted for about 1 week post-i.t. injection, similar to that induced by full-length TSP4 proteins (9). In contrast, neither lowering the dose of EGF-LIKE proteins to a molar dose (5 μg/rat) equivalent to the sub-pronociceptive dose of full-length TSP4 (20 μg/rat) (Fig. 3A) (9) nor injecting recombinant truncated proteins without EGF-LIKE (CD, NT, and NT+CC) at a molar dose equivalent to the pronociceptive dose of full-length TSP4 (45 μg/rat) (Fig. 2) (9) cause behavioral hypersensitivity. Similar to its anti-nociceptive effects post-TSP4 injection (12), Cavα2δ1 ligand gabapentin could dose-dependently reverse tactile allodynia induced by EGF-LIKE within an hour post-i.t. injection (Fig. 3, A and B). Together, these findings support that EGF-LIKE is the functional determinant of TSP4's pronociceptive effects, and its interaction with the Cavα2δ1 proteins is likely required for TSP4's pronociceptive effects.
Figure 2.

Induction of tactile allodynia and thermal hyperalgesia in naïve rats after bolus intrathecal injection of TSP4 truncation proteins containing the EGF-LIKE domains. PWTs to mechanical stimulation (A) were tested daily, and paw withdrawal latencies (PWL) to thermal stimulation (B) were tested at days 4 and 10 after bolus i.t. injection of TSP4 truncated proteins at day 0 following baseline tests. A, data presented are the means from at least five rats in each group with error bars representing S.E. B, data presented in the box-and-whisker plot are from at least five rats in each group. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 compared with pretreatment level in each group by two-way ANOVA with Tukey's multiple comparisons test in A and one-way ANOVA with Sidak's multiple comparisons test in B.
Figure 3.
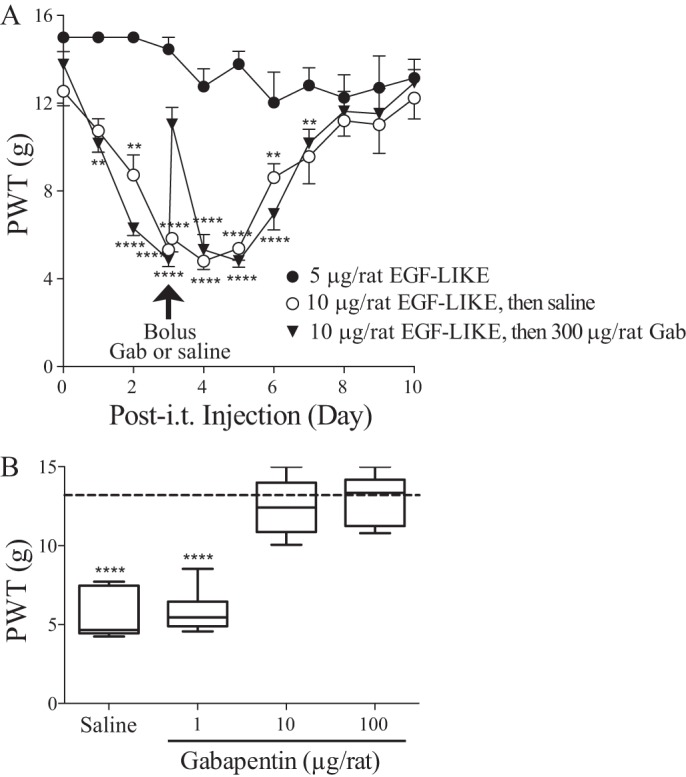
Gabapentin could block tactile allodynia induced by intrathecal injection of EGF-LIKE domain proteins of TSP4 into naïve rats. A, daily PWTs to von Frey filament stimulation were tested blindly after bolus i.t. injection of EGF-LIKE proteins into naïve rats at time 0. Bolus intrathecal gabapentin (Gab) (300 μg/rat) or saline was administered at day 3 postinjection of EGF-LIKE (indicated by the arrow) followed by behavioral tests after 1 h and then daily. Data presented are the means from six rats in each group with error bars representing S.E. **, p < 0.01; ****, p < 0.0001 compared with pretreatment level by repeated measures one-way ANOVA with Bonferroni's multiple comparisons test. B, dose-dependent effects of gabapentin 1 h after intrathecal injections in reversal of EGF-LIKE–induced tactile allodynia described in A. The dotted line represents the baseline level of PWT (before i.t. injections of EGF-LIKE). Data presented in the box-and-whisker plot are from five to seven rats in each group. ****, p < 0.0001 compared with pretreatment level by one-way ANOVA with Bonferroni's multiple comparisons test.
Effects of intrathecal injection of peptide EGFD355–369 on behavioral hypersensitivities induced by elevated TSP4 and/or Cavα2δ1
Because EGF-LIKE is the pronociceptive domain, we tested whether a peptide within the EGF-LIKE domain of TSP4 could interfere with behavioral hypersensitivity induced by increased TSP4 and/or Cavα2δ1 in vivo. Based on the SPOT peptide array analysis (Fig. 1C), we identified peptide EGFD355–369 within EGF-LIKE as highly conserved in rodents and having high binding affinity to Cavα2δ1. This peptide was synthesized and injected i.t. into the L5/6 spinal region of TSP4-injected rats when the animals had severe allodynia (day 3 or 4 after bolus TSP4 injection). A blind behavioral sensitivity test to von Frey filament stimulation was performed daily after the TSP4 injection before and every 2 h for up to 6 h after each peptide injection. Our data indicated that peptide EGFD355–369, but not its scrambled peptide control, blocked TSP4-induced tactile allodynia dose-dependently. This effect lasted for >6 h (Fig. 4A). Bolus injection of peptide EGFD355–369 alone into naïve rats at the highest dose tested (2.1 nmol/rat) did not cause significant changes in baseline behavioral sensitivity acutely (Fig. 4A) or chronically (Fig. 4B). These data support that peptide EGFD355–369 can block TSP4-induced tactile allodynia.
Figure 4.

TSP4-induced tactile allodynia could be attenuated by intrathecal injection of peptide EGFD355–369 from the EGF-LIKE domain of TSP4. A, TSP4 proteins (45 μg/rat; ∼1.6 nm considering averaged volume of rat cerebrospinal fluid of 290 μl (56)) or sterile saline were injected into the L5/6 spinal region of naïve rats at day 0 after baseline testing followed by daily behavioral tests using von Frey filaments. As shown previously (9), tactile allodynia peaked between 3 and 4 days post-TSP4 injection. Bolus peptide EGFD355–369 or its scrambled control peptide was administered at day 3 or 4 post-TSP4 injection as indicated followed by behavioral tests every 2 h for up to 6 h after peptide treatment. The peptide doses used were at 5× molar excess (2.1 nmol/rat), molar equivalent (0.4 nmol/rat), or molar dose (0.08 nmol/rat) of the pronociceptive dose of TSP4 (45 μg/rat). Data presented are the means from six to seven rats in each group with error bars representing S.E. *, p < 0.05; #, p < 0.01; &, p < 0.001; $, p < 0.0001 compared with pretreatment level in each group by repeated measures two-way ANOVA with Dunnett's multiple comparisons test. B, PWTs to von Frey filament stimuli were tested blindly before, at day 4, and at day 10 after bolus i.t. injection of peptide EGFD355–369 (2.1 nmol/rat) into naïve rats. Data presented in the box-and-whisker plot are from seven rats in each group. 15 g was the maximum value assigned to a rat as described under “Experimental procedures.”
To confirm that the anti-nociceptive effects of peptide EGFD355–369 were mediated by interfering with the activation of a pathway due to TSP4/Cavα2δ1 interactions, we examined its effect on mechanical allodynia in a Cavα2δ1 TG mouse line with increased neuronal Cavα2δ1 overexpression (20). This model has been shown to enhance excitatory synaptic transmission in dorsal spinal cord that contributes to behavioral hypersensitivity (12, 17–20). Our data indicated that intrathecal injection of the effective dose (0.047 nmol/mouse) of peptide EGFD355–369, but not its scrambled peptide, could reverse allodynia in the Cavα2δ1 TG mice with an onset time of 2 h and a duration <6 h (Fig. 5). These data indicate that peptide EGFD355–369 can attenuate tactile allodynia induced by Cavα2δ1, most likely through its interactions with TSP4 (12).
Figure 5.
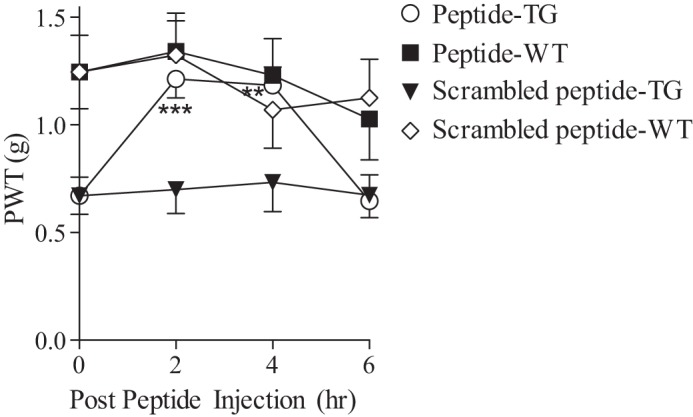
Cavα2δ1-induced tactile allodynia could be attenuated by intrathecal injection of peptide EGFD355–369. A bolus i.t. injection of peptide EGFD355–369 or its scrambled control peptide (0.047 nmol/mouse) at an equal molar dose to the pronociceptive dose of TSP4 (5 μg/mouse) was administered to Cavα2δ1-overexpressing TG mice and their age- and sex-matched WT littermates at time 0 followed by a blind behavioral test for PWTs to von Frey filament stimuli at 2, 4, and 6 h after bolus peptide treatments. Data presented are the means from 10 mice in each group with error bars representing S.E. **, p < 0.01; ***, p < 0.001 compared with pretreatment level by repeated measures one-way ANOVA with Bonferroni's multiple comparisons test.
EGF-LIKE domain of TSP4 is synaptogenic
To determine whether EGF-LIKE induces excitatory synaptogenesis that may underlie its nociceptive effects, we examined the synaptogenic effects of TSP4 domain proteins derived from the constructs shown in Fig. 1 in cocultures of DRG and spinal cord neurons. Treatments of cocultures with an equal molar concentration of recombinant proteins containing EGF-LIKE, such as that encoded by full-length TSP4, ND, and EGF-LIKE domain constructs, resulted in significant increases of excitatory synapse numbers compared with control (PBS treatment). There was no significant difference in synaptic numbers among treatments with these EGF-LIKE–containing proteins (Fig. 6). In contrast, treatments with proteins lacking EGF-LIKE, such as those encoded by the CD and NT+CC constructs, did not induce significant excitatory synapse formation compared with control (Fig. 6). These data indicate that EGF-LIKE is synaptogenic and promotes excitatory synapse formation between sensory neurons and spinal cord neurons.
Figure 6.

TSP4 EGF-LIKE domain proteins promote synapse formation between sensory neurons and spinal cord neurons. Synaptogenesis analysis was performed in cocultures of DRG and spinal cord neurons after treatment for 4 days with PBS (CTL) or an equal molar concentration of either full-length or different domain proteins of TSP4 in multiple-well plates of independent experiments. A, representative confocal images showing immunoreactivities to antibodies of synaptic markers (Vglut2, red; PSD95, green) along spinal neuron dendrites (MAP2, blue). Puncta of colocalized synaptic marker immunoreactivity were quantified as synapse numbers. B, summarized synapse numbers in cocultures after treatment with TSP4 or its domain proteins. Data presented are the means from 20 neurons per group of three independent experiments with error bars representing 95% confidence interval. ***, p < 0.001 compared with control by repeated measures one-way ANOVA with a Dunnett's post hoc test.
EGFD355–369 can block synaptogenesis induced by EGF-LIKE domain of TSP4
To determine whether peptide EGFD355–369 could block excitatory synaptogenesis induced by EGF-LIKE, we examined the effects of peptide EGFD355–369 on excitatory synaptogenesis induced by EGF-LIKE in the cocultures. Similar to that shown in Fig. 6, treatments with EGF-LIKE resulted in a significant increase of excitatory synapses compared with the control. Concurrent treatments with peptide EGFD355–369, but not the scrambled control peptide, blocked the increase of synapse formation induced by EGF-LIKE (Fig. 7). These data support that peptide EGFD355–369 can block the synaptogenic effects of EGF-LIKE.
Figure 7.
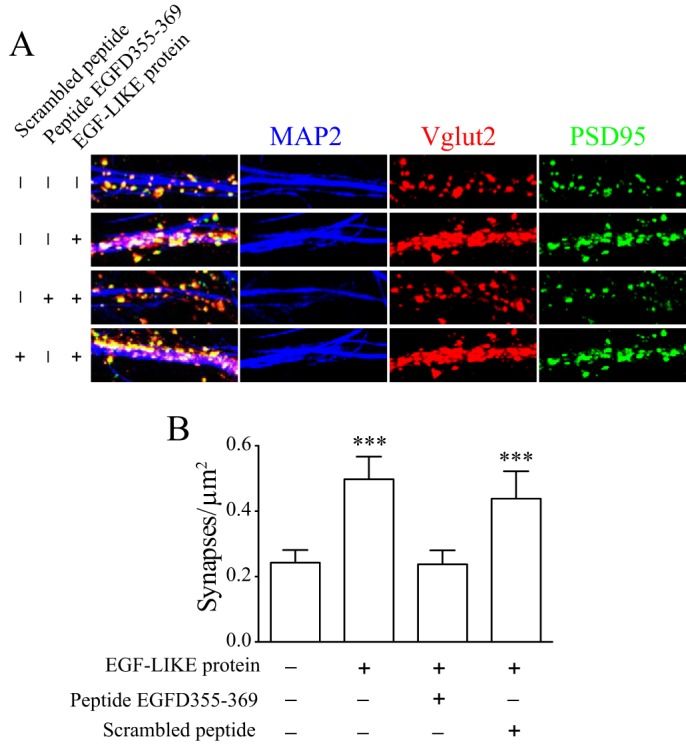
Synaptogenic effects of the TSP4 EGF-LIKE domain protein were blocked by peptide EGFD355–369. Synaptogenesis analysis was performed in cocultures of DRG and spinal cord neurons after treatment for 4 days with PBS (CTL) or 20 nm EGF-LIKE proteins in the absence or presence of an equal molar concentration of scrambled control peptide or peptide EGFD355–369 in multiple-well plates of independent experiments. A, representative confocal images showing immunoreactivities to antibodies of synaptic markers (Vglut2, red; PSD95, green) along spinal neuron dendrites (MAP2, blue). Puncta of colocalized synaptic marker immunoreactivity were quantified as synapse numbers. B, summarized synapse numbers in cocultures after each treatment. Data presented are the means from 13–17 neurons per group in each of three independent experiments with error bars representing 95% confidence interval. ***, p < 0.001 compared with control by repeated measures one-way ANOVA with a Dunnett's post hoc test.
Discussion
Peripheral nerve injury induces concurrent up-regulation of TSP4 and Cavα2δ1 in dorsal spinal cord (5, 9), and TSP4/Cavα2δ1 interaction promotes excitatory synaptogenesis and chronic pain states (12). To gain molecular insight into these processes, here we have identified the Cavα2δ1-binding motifs of TSP4 and that EGF-LIKE is prosynaptogenic and nociceptive. Administration of gabapentin blocks behavioral hypersensitivity induced by EGF-LIKE. Administration of the EGFD355–369 peptide from within the EGF-LIKE motif blocks excitatory synaptogenesis induced by EGF-LIKE as well as tactile allodynia induced by elevated TSP4 or Cavα2δ1. Together, our findings support that EGF-LIKE is a functional determinant in promoting aberrant excitatory synaptogenesis and sensory hypersensitivity resulting from activation of a TSP4/Cavα2δ1 pathway.
Multidomain interactions between TSP4 and Cavα2δ1
We used SPOT peptide synthesis method and an ELISA-based binding assay to map the linear and discontinuous binding sites of TSP4 to Cavα2δ1. Our data indicate that most TSP4 linear peptides with relatively high binding affinity to Cavα2δ1 are located within the coiled-coil and EGF-LIKE domains, but not the type-3 calcium-binding domain, of TSP4. However, a few discontinuous peptides from the TSP4 N-terminal and C-terminal domains also bind to Cavα2δ1 with high affinity and may be responsible for the low-affinity binding of TSP4 truncated N- or C-terminal domain proteins to Cavα2δ1. Thus, the binding interaction between TSP4 and Cavα2δ1 is likely mediated through multidomain interactions, which may lead to subsequent conformational changes of the binding proteins. Recent data from a cotransfection study suggest that TSP4/Cavα2δ1 interactions occur intracellularly (22). Therefore, the exact location of TSP4/Cavα2δ1 interactions in vivo remains to be established.
Specific interactions between the TSP4 EGF-LIKE domain and Cavα2δ1 are critical in TSP4-induced behavioral hypersensitivity
Functionally, we show that only truncated TSP4 proteins containing EGF-LIKE promote behavioral hypersensitivities like full-length TSP4 proteins. Intriguingly, those containing the N-terminal, coiled-coil, calcium-binding, and C-terminal domains fail to elicit behavioral hypersensitivity even though some of these proteins, such as that encoded by the NT+CC construct, have similar Cavα2δ1 binding affinity as full-length TSP4. Thus, multidomain-binding interactions between TSP4 and Cavα2δ1 may facilitate specific interactions between EGF-LIKE and Cavα2δ1, which are critical in mediating behavioral hypersensitivity. It is reported that EGF-LIKE interacts with von Willebrand factor A (VWF-A) domain of Cavα2δ1 in promoting central nervous system excitatory synaptogenesis (23); a similar mechanism in spinal cord can lead to behavioral hypersensitivity. Because gabapentin binds to a site upstream of the VWF-A domain of Cavα2δ1 (24), it is less likely that the inhibitory effects of gabapentin on EGF-LIKE–induced behavioral hypersensitivity are mediated through direct blockage of TSP4 binding to Cavα2δ1. Instead, binding of small molecules such as gabapentin to Cavα2δ1 may cause conformational changes of the protein that may interfere with TSP4/Cavα2δ1 interactions. This is supported by recent findings that TSP4 binding to Cavα2δ1 can reduce gabapentin binding to Cavα2δ1, but this effect is diminished with mutations in the VWF-A domain of Cavα2δ1 (22).
We hypothesized that a peptide from the EGF-LIKE sequence might interfere with EGF-LIKE/Cavα2δ1 interactions, thereby blocking TSP4/Cavα2δ1 pathway activation and attenuating behavioral hypersensitivity. To test this, we designed peptide EGFD355–369 and tested its effects in blocking behavioral hypersensitivity induced by elevated TSP4 and/or Cavα2δ1. Intrathecal injection of this peptide can block tactile allodynia induced by intrathecal TSP4 injection in rats and Cavα2δ1 overexpression in transgenic mice without affecting baseline behavioral sensitivity in naïve animals. Therefore, peptide EGFD355–369 can block behavioral hypersensitivity induced by elevated TSP4 or Cavα2δ1 alone. Its anti-nociceptive effects are specific because neither a high-affinity binding peptide from the coiled-coil domain (data not shown) nor the scrambled control peptide has any functional effect in blocking nociception. Because intrathecal injection of EGF-LIKE–containing proteins (TSP4, ND, and EGF-LIKE) in naïve animals is pronociceptive but that of EGF-LIKE linear peptides alone is not, our data suggest that the nociceptive effect of TSP4 may require multiple binding sites and an optimal protein interface upon its interactions with Cavα2δ1.
Potential mechanisms underlying TSP4/Cavα2δ1-induced pain transduction and drug actions
Bolus injection of peptide EGFD355–369 blocks Cavα2δ1-induced tactile allodynia, confirming the requirement of a basal level of TSP4 for the pronociceptive effects of elevated Cavα2δ1 (12). Enhanced excitatory transmission and behavioral hypersensitivity induced by Cavα2δ1 overexpression in neuronal cells can be attenuated by gabapentin (20) through inhibiting Cavα2δ1 and voltage-gated calcium channel trafficking to presynaptic axon terminals of nerve-injured DRG neurons in the dorsal horn (25, 26). Future studies to explore whether peptide EGFD355–369 could modulate excitatory neurotransmission and reduce Cavα2δ1 trafficking to the presynaptic sites in dorsal spinal cord would shed some light on the mechanistic contribution of TSP4 to behavioral hypersensitivity associated with Cavα2δ1.
Conversely, the ability of gabapentin to inhibit TSP4- (12) or EGF-LIKE–induced central sensitization and behavioral hypersensitivity highlights a critical role of the TSP4/Cavα2δ1 pathway in mediating chronic pain states. Because gabapentin can block TSP-induced excitatory synapse formation (21, 23), dorsal horn neuron sensitization, and behavioral hypersensitivities (12), it is likely that activation of the TSP4/Cavα2δ1 pathway by EGF-LIKE contributes to aberrant excitatory synaptogenesis, central sensitization, and pain states. This is supported by our recent findings that trigeminal nerve injury causes increased expression of Cavα2δ1 and TSP4 in dorsal spinal cord that correlates with aberrant excitatory synaptogenesis and enhances presynaptic excitatory input and neuropathic pain states (14, 27). Our findings reveal that only TSP4 proteins containing EGF-LIKE are synaptogenic and pronociceptive, but NT+CC domain proteins lacking EGF-LIKE are not even though they have similar Cavα2δ1 binding affinity as full-length TSP4. Together, these data support that specific interactions between EGF-LIKE and Cavα2δ1, but not multidomain-binding interactions between TSP4 and Cavα2δ1, are critical in mediating synaptogenesis and behavioral hypersensitivity.
Our findings reveal that gabapentin and peptide EGFD355–369 can block excitatory synaptogenesis and behavioral hypersensitivity induced by EGF-LIKE without affecting basal level values, supporting that aberrant synapse formation induced by interactions of EGF-LIKE with Cavα2δ1 contributes to behavioral hypersensitivity. Interestingly, both gabapentin and peptide EGFD355–369 have a rapid onset time (within 2 h) and short duration (<4 h) in blocking pain states. This time frame coincides with that of new excitatory synapse formation in vitro and in vivo. For example, new excitatory synapses can be assembled in vitro within 1–2 h of initial axodendritic contact (28, 29), which correlates with the time for new synaptic spine formation after high frequency stimulation (30–32). In vivo data indicate that activity-dependent synaptic stabilization and elimination are dynamic processes that can occur as rapidly as <30 min to 2 h poststimulation (33, 34). These rapidly assembled excitatory synapses are initiated by presynaptic remodeling through recruitment and stabilization of presynaptic packets containing calcium channel subunits (28). Thus, it is possible that binding of TSP4, or EGF-LIKE, proteins to Cavα2δ1 promotes the assembly/recruitment of presynaptic protein precursors and/or stabilization of presynaptic remodeling after nerve injury that allows a rapid initiation of new excitatory synapse formation. Supportive findings include that injury-induced Cavα2δ1 proteins are translocated from injured DRG neurons to their presynaptic terminals in dorsal spinal cord (5, 26) and play a role in axonal trafficking and presynaptic assembly (26, 35). This in turn can lead to increased presynaptic excitatory input into dorsal horn neurons (18, 19, 35) that can accelerate stimulation-dependent aberrant synapse formation (30–32). Gabapentin or peptide EGFD355–369 can bind to Cavα2δ1, likely induce conformational changes of target proteins, and prevent rapid aberrant synaptogenesis initiated by TSP4/Cavα2δ1 interactions, leading to pain state reversal. This is supported by our recent findings that only early gabapentin treatment can prevent TSP4-induced excitatory synaptogenesis and behavioral hypersensitivity (21).
The transient effects of gabapentin and EGFD355–369 in reversing pain state may be due to the short half-life of these agents in vivo because gabapentin has a mean elimination half-life about 2 h in rats (36), and the half-life of peptides in rats can range from minutes to 2 h (37, 38). Modifications to improve the stability and pharmacokinetics of these agents may provide better therapeutic values. However, we cannot rule out the possibility that the effects of gabapentin and the peptide are independent from its binding to Cavα2δ1–TSP4 complexes. Detailed studies using mutational analyses across Cavα2δ1 and/or EGF-LIKE should provide further mechanistic insights. A recent study reports that gabapentin's anti-nociceptive effects could be mediated by its selective inhibition of Cavα2δ1-bound NMDA receptors on the cell membrane surface (39). Because TSP4 and Cavα2δ1 also form complexes in spinal cord (12), it will be interesting to investigate whether TSP4 plays a contributory role to events associated with Cavα2δ1–NMDA receptor complex formation.
Summary and conclusion
In summary, our findings demonstrate that EGF-LIKE is the molecular determinant in mediating excitatory synaptogenesis and behavioral hypersensitivity and requires its interaction with Cavα2δ1. Blocking EGF-LIKE/Cavα2δ1 interaction can be an alternative approach in designing target-specific medications for neuropathic pain management.
Experimental procedures
Construction of recombinant TSP4 truncated cDNA
The recombinant truncated TSP4 constructs were prepared by the PCR-driven overlap extension technique (40) using full-length rat TSP4 cDNA (GenBankTM accession number X89963) in a pCEP-Pu vector as template (a gift from Dr. Frank Zaucke, University of Cologne). Segments of the TSP4 cDNA were amplified using two flanking master primers and internal primers that introduce the mutation for truncation and create an overlapping nucleotide sequence. The construct with the N-terminal domain of TSP4 deletion was created with the internal primers CACGCGCTAGTCTCTCTGTTCCAG (forward), CTGGAACAGAGAGACTAGCGCGTG (reverse) and master primers CGCAGAACTGGTAGGTATGG (forward) and CTTATCATGTCTGGATCCGGC (reverse). The construct containing only the EGF-like domains of TSP4 (EGF-LIKE) was obtained with internal primers CGCGCTAGTCCCAACACGTC (forward) and GCTTCTTGGTTTATCCACAGACATAG (reverse) and CTATGTCTGTGGATAAACCAAGAAGC (forward) and GACGTGTTGGGACTAGCGCG (reverse) and master primers CGGGACTTTCCTACTTGGCAG (forward) and CCCGACACCCGCCAACACC (reverse). The construct containing the type-3 calcium-binding domain and C-terminal domain of TSP4 was created with the internal primers CACGCGCTAGTCAAGGATGTGGAC (forward) and GTCCACATCCTTGACTAGCGCGTG (reverse) and master primers CGCAGAACTGGTAGGTATGG (forward) and CTTATCATGTCTGGATCCGGC (reverse). The N-terminal domain-only construct was created with the internal primers GTGGTGAGGGGCTAAACCAAGAAG (forward) and CTTCTTGGTTTAGCCCCTCACCAC (reverse) and master primers CTAGAAGCTGGGTACCTTAAGGC (forward) and CAAGCTGTGACCGTCTCCG (reverse). The construct containing the N-terminal domain and coiled-coil domain of TSP4 was obtained with the internal primers CTCCTCCAGCATAAACCAAGAAGC (forward) and GCTTCTTGGTTTATGCTGGAGGAG (reverse). The same master primers for the N-terminal domain-only construct were used. The constructs were inserted back into the pCEP-Pu vector containing the BM40 signal peptide (41, 42) and N-terminal His6 tag using restriction sites KpnI and BamHI. The constructs were confirmed by sequencing (Eton Biosciences, San Diego, CA).
Expression and purification of recombinant TSP4 proteins
The recombinant rat TSP4 cDNA (GenBank accession number X89963) with a His6 tag at the N-terminal was transfected into human embryonic kidney cell line 293-EBNA (Invitrogen) using the calcium phosphate transfection method. The transfected cells were selected with 0.5 μg/ml puromycin and grown to confluence. Secretion of the full-length TSP4-His and truncated TSP4-His proteins into Dulbecco's modified Eagle's medium (DMEM)/F-12 medium (Mediatech, Manassas, VA) was confirmed by Western blotting using anti-penta-His monoclonal antibodies (Qiagen, Valencia, CA). The recombinant TSP4-His proteins were purified using a nickel-nitrilotriacetic acid column based on the manufacturer's instructions (Invitrogen), concentrated with an Amicon Ultra-4 centrifugal filter unit based on the cutoff molecular weight (Millipore, Billerica, MA), verified using Western blot analysis, aliquoted, and stored at −80 °C until use.
Western blot analysis
Protein samples were denatured in the presence of DTT, heated to 70 °C for 10 min, and then loaded onto a 4–12% NuPAGE Bis-Tris gel (Invitrogen) for gel electrophoresis. Proteins were transferred onto polyvinylidene difluoride membrane and blocked with 5% skim milk powder in PBS-T (137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, 1.4 mm KH2PO4, 0.1% Tween 20, pH 7.4) at room temperature. The immunoblot was probed with anti-penta-His monoclonal antibodies (Qiagen; 1:1000) overnight at 4 °C and then incubated with secondary anti-mouse IgG antibody conjugated with horseradish peroxidase (HRP) (Cell Signaling Technology; 1:2000) for 1 h at room temperature followed by enhanced chemiluminescence detection of the protein–antibody complexes in an Eastman Kodak Co. image station (2000MM).
Purification of FLAG-Cavα2δ1
FLAG-Cavα2δ1 cDNA was transiently transfected into HEK 293 cells using Lipofectamine 2000 (Invitrogen). Transfected cells were washed twice with PBS and then extracted in protein extraction buffer (50 mm Tris, 150 mm NaCl, 1 mm EDTA, 0.1% Triton X-100, pH 7.4) 2–3 days after the transfection. The cell lysate was incubated on ice for 15 min followed by centrifugation at 13,000 × g for 20 min at 4 °C. The supernatant was incubated with anti-FLAG M2 agarose affinity resin (Sigma-Aldrich) on a rotating mixer for 2 h at 4 °C and then washed three times with protein extraction buffer. FLAG-Cavα2δ1 was eluted in elution buffer (0.1 m glycine, pH 3.5) and stored at −20 °C for future use.
ELISA-based binding assay
The reagents for solid-phase binding were from Invitrogen (catalog number CNB0011). Recombinant TSP4 proteins were immobilized onto a 96-well polystyrene plate (Thermo Fisher Scientific) overnight at 4 °C in coating buffer A (10 mm phosphate buffer, 0.1% azide, pH 7.4). All further incubations were carried out at room temperature for 1 h, and proteins or antibodies were diluted in assay buffer containing bovine serum albumin (BSA). After blocking, the plates were incubated with affinity-purified FLAG-Cavα2δ1 for 1 h, washed, and then incubated with mouse monoclonal anti-FLAG antibodies (1:1000; Sigma-Aldrich) followed by HRP-conjugated secondary antibodies. The bound FLAG-Cavα2δ1 complexes were detected by measuring a color reaction (yellow product) at 450 nm after adding tetramethylbenzidine for 15 min followed by adding sulfuric acid to stop the reaction.
SPOT peptide array and far-Western blotting
Peptide arrays containing overlapping peptides (15-mers; overlapping by 12 amino acids) covering the entire length of rat TSP4 protein were synthesized and immobilized onto a cellulose membrane (Sigma-Aldrich custom SPOT service). The peptide array was rinsed with methanol for 5 min, washed with TBS-T (50 mm Tris, 137 mm NaCl, 2.7 mm KCl, 0.05% Tween 20, pH 8.0), and blocked with 5% dry milk in TBS-T for 1 h at room temperature. Plasma membranes from tsA-201 cells stably expressing Cav2.2e (Δ24a,31a), Cavβ3, and Cavα2δ1 (43) (a gift from Dr. Diane Lipscombe at Brown University) were collected in Tris buffer (5 mm Tris/HCl, 5 mm EDTA, pH 7.4, containing PMSF, leupeptin, and pepstatin A), incubated on ice for 15 min, sonicated, and then centrifuged at 1000 × g for 10 min. The supernatant was collected and centrifuged at 50,000 × g for 30 min at 4 °C. The resulting pellet was resuspended in Tris buffer (0.1 μg/ml) and incubated with the peptide array in 5% dry milk for 1 h at room temperature. Binding of Cavα2δ1 to the peptide array was detected by incubating the peptide array for 2 h at room temperature with primary antibodies against Cavα2δ1 (1:1000; mouse; Sigma-Aldrich) and then HRP-conjugated secondary antibodies against mouse IgG for 1 h at room temperature (1:2000; Cell Signaling Technology) followed by enhanced chemiluminescence detection.
Peptides
The following peptides were synthesized and verified by MS and HPLC analysis by Genscript (Piscataway, NJ): EGF-LIKE domain peptide of TSP4 (PGVRCTNLAPGFRCD) (EGFD355–369; GenBank accession number CAA62002) and its scrambled control peptide (GLAFVNCPRRDTGCP). The lyophilized peptides were dissolved in sterile water before use.
Animals
Male adult Harlan Sprague-Dawley rats (<150 g) were from Harlan Sprague-Dawley Industries (Indianapolis, IN). Adult male and female mice (20–30 g) with 129sv background were from Charles River Laboratories (Wilmington, MA). They were bred internally to obtain pregnant females for spinal cord cell collection for cell cultures. The Cavα2δ1-overexpressing transgenic mice were generated and characterized as described in our previous publication (20). Briefly, a neuronal specific thy-1 promoter (44) was used to drive overexpression of the mouse brain Cavα2δ1 cDNA (GenBank accession number U73484) in the transgenic mice. These genetically modified mice were fertile and showed normal growth, grooming, social interactions, and feeding. They were backcrossed to the 129sv background for over 10 generations before use. Mouse genotyping was performed by TransnetYX, Inc. (Cordova, TN). All animals were housed in separate cages and exposed to a 12-h light/dark cycle with food and water ad libitum. All animal care and experiments were performed according to protocols approved by the Institutional Animal Care Committees of the University of California, Irvine.
Tactile allodynia
The animals were placed in Plexiglass chambers on a wire mesh–bottomed cage for acclimatization (at least 60 min). von Frey filaments (Stoelting, Wood Dale, IL) were used to determine the 50% paw withdrawal threshold (PWT) using the up-down method of Dixon (45). A series of von Frey filaments were applied to both plantar surfaces of the hind paws, starting with a buckling weight of 2.0 or 0.41 g for a rat or mouse, respectively, with sufficient force to cause the filament to buckle. A positive response was defined as a rapid withdrawal and/or licking of the paw upon application of the stimulus, which prompted the use of the next weaker filament. Absence of a paw withdrawal response after 5 s prompted the use of the next filament of increasing weight. This paradigm was continued until four more measurements have been made after the initial change of the behavioral response or until five consecutive negative (assigned a score of 15 g for rats or 2 g for mice) or four consecutive positive (assigned a score of 0.25 g for rats or 0.01 g for mice) responses had occurred. The resulting scores of six (starting from the one before the first change in response) were used to calculate the 50% response threshold as described previously (3).
Thermal hyperalgesia (Hargreaves method)
The animals were placed in Plexiglass chambers on a glass panel that was maintained at 30 °C for acclimatization (at least 60 min). A heat stimulus projecting through a small aperture below the glass panel from a high intensity light bulb was applied to both plantar surfaces of the hind paws. When the animals moved their paw away from the thermal stimulus, the light beam was shut off automatically. The length of time between the start of the light beam and the hind paw withdrawal was defined as the paw withdrawal latency. A cutoff time of 20 s was used to avoid tissue damage to the hind paw.
Cell culture
A DRG/spinal cord primary neuron coculture system derived from published protocols (46–49) with minor modifications (26, 50–53) was used for in vitro synaptogenesis analysis. Briefly, at least six E14–E19 mouse embryos (for spinal cord neurons) and three adult mice (for DRG neurons) were used for each multiwell culture plate in independent experiments. Spinal cord neurons were dissociated with 0.25% trypsin in DMEM, plated onto 0.1 mg/ml poly-d-lysine– and 0.04 mg/ml laminin-coated glass coverslips, and grown in DMEM supplemented with 10% horse serum and 10% fetal bovine serum. After 24 h, the media were replaced with neurobasal medium supplemented with B27 supplement (NB/B27) to allow spinal cord neuron maturation and neurite sprouting for 3 days. Adult DRG (T9–L5) were harvested after hydraulic extrusion of spinal cord and laminectomy from Advillin-Cre/Rosa-tdTomato mice, which expressed red fluorescent proteins driven by the Advillin promoter in about 90% of DRG neurons (54, 55). Neurons were dissociated in DMEM containing 1.25 mg/ml collagenase (Sigma) and added to the spinal cord neuron cultures at the time of culture media change at day 4. The cocultures were maintained in NB/B27 supplement with 100 nm uridine and 20 nm 5-fluorodeoxyuridine to control nonneuronal cell proliferation. The culture cell treatments started about 2–3 days after DRG cell addition.
Immunohistochemistry
Excitatory synapse numbers in cultured cells were analyzed as described previously (12, 21, 51). Because a single investigator performed all the immunohistochemical experiments, data analyses were not double blinded. Briefly, after heat-based antigen retrieval as described previously, cell samples were stained with primary antibodies against vesicular glutamate transporter 2 (VGlut2; guinea pig; Synaptic Systems), postsynaptic density protein 95 (PSD95; rabbit; Thermo Fisher Scientific), and microtubule-associated protein 2 (MAP2) (chicken; Abcam) at 4 °C overnight. Our laboratory and others have done validation of these antibodies as reported previously (12). After washing, samples were incubated with species-specific secondary antibodies conjugated to unique fluorophores for 2 h at room temperature and washed again. Fluorescence images were acquired with a confocal microscope (Zeiss LSM700, University of California Irvine Optical Biology Core) in 0.3-μm-thick Z stacks. DRG axons could be traced by tdTomato red fluorescence. Because VGlut2 immunoreactivity is mainly in presynaptic vesicles, but not axons, and tdTomato fluorescence is mainly expressed along DRG neuron axons, but not in presynaptic vesicles, it was highly likely that VGlut2 immunoreactivity detected within the dendritic spinelike structure along MAP2 immunoreactivity was from VGlut2 immunostaining and not from tdTomato fluorescence (21). This allowed measurement of excitatory synapses between sensory axons and individual dendrites of each spinal cord neuron with overlapping VGlut2, PSD95, and MAP2 immunoreactivity along with or surrounded by red DRG axons. Corrections for variations in dendrite size and numbers among individual spinal cord neurons were done by calculating synapse count/μm2 area of MAP2-immunoreactive dendrites of that neuron. Data analyses were performed on merged seven consecutive Z stacks with the best signal using Volocity 6.0 (PerkinElmer Life Sciences).
Author contributions
J. F. P. data curation; J. F. P., Y. P. Y., and N. G. formal analysis; J. F. P., Y. P. Y., N. G., and V. N. T. investigation; J. F. P. writing-original draft; Z. D. L. conceptualization; Z. D. L. supervision; Z. D. L. funding acquisition; Z. D. L. validation; Z. D. L. writing-review and editing.
Acknowledgments
We thank Dr. Frank Zaucke (University of Cologne) for the TSP4 cDNA vector and Dr. Diane Lipscombe (Brown University) for the tsA-201 stable cell lines.
This work supported in part by National Institutes of Health Grants DE021847 and NS064341 (to Z. D. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- TSP4
- thrombospondin-4
- EGF
- epidermal growth factor
- Cav
- voltage-gated calcium channel
- EGF-LIKE
- EGF-like domain
- DRG
- dorsal root ganglia
- TG
- transgenic
- HEK
- human embryonic kidney
- i.t.
- intrathecal
- VWF-A
- von Willebrand factor A
- NMDA
- N-methyl-d-aspartate
- DMEM
- Dulbecco's modified Eagle's medium
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- HRP
- horseradish peroxidase
- PWT
- paw withdrawal threshold
- VGlut2
- vesicular glutamate transporter 2
- PSD95
- postsynaptic density protein 95
- MAP2
- microtubule-associated protein 2
- ANOVA
- analysis of variance.
References
- 1. Woolf C. J., and Mannion R. J. (1999) Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 353, 1959–1964 10.1016/S0140-6736(99)01307-0 [DOI] [PubMed] [Google Scholar]
- 2. Costigan M., Scholz J., and Woolf C. J. (2009) Neuropathic pain: a maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 32, 1–32 10.1146/annurev.neuro.051508.135531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Luo Z. D., Chaplan S. R., Higuera E. S., Sorkin L. S., Stauderman K. A., Williams M. E., and Yaksh T. L. (2001) Upregulation of dorsal root ganglion α2δ calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J. Neurosci. 21, 1868–1875 10.1523/JNEUROSCI.21-06-01868.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luo Z. D., Calcutt N. A., Higuera E. S., Valder C. R., Song Y. H., Svensson C. I., and Myers R. R. (2002) Injury type-specific calcium channel α2δ-1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J. Pharmacol. Exp. Ther. 303, 1199–1205 10.1124/jpet.102.041574 [DOI] [PubMed] [Google Scholar]
- 5. Li C. Y., Song Y. H., Higuera E. S., and Luo Z. D. (2004) Spinal dorsal horn calcium channel α2δ-1 subunit upregulation contributes to peripheral nerve injury-induced tactile allodynia. J. Neurosci. 24, 8494–8499 10.1523/JNEUROSCI.2982-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim D. S., Figueroa K. W., Li K. W., Boroujerdi A., Yolo T., and Luo Z. D. (2009) Profiling of dynamically changed gene expression in dorsal root ganglia post peripheral nerve injury and a critical role of injury-induced glial fibrillary acidic protein in maintenance of pain behaviors [corrected]. Pain 143, 114–122 10.1016/j.pain.2009.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Valder C. R., Liu J. J., Song Y. H., and Luo Z. D. (2003) Coupling gene chip analyses and rat genetic variances in identifying potential target genes that may contribute to neuropathic allodynia development. J. Neurochem. 87, 560–573 10.1046/j.1471-4159.2003.02016.x [DOI] [PubMed] [Google Scholar]
- 8. Wang H., Sun H., Della Penna K., Benz R. J., Xu J., Gerhold D. L., Holder D. J., and Koblan K. S. (2002) Chronic neuropathic pain is accompanied by global changes in gene expression and shares pathobiology with neurodegenerative diseases. Neuroscience 114, 529–546 10.1016/S0306-4522(02)00341-X [DOI] [PubMed] [Google Scholar]
- 9. Kim D. S., Li K. W., Boroujerdi A., Peter Yu Y., Zhou C. Y., Deng P., Park J., Zhang X., Lee J., Corpe M., Sharp K., Steward O., Eroglu C., Barres B., Zaucke F., et al. (2012) Thrombospondin-4 contributes to spinal sensitization and neuropathic pain states. J. Neurosci. 32, 8977–8987 10.1523/JNEUROSCI.6494-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Adams J. C., and Lawler J. (2004) The thrombospondins. Int. J. Biochem. Cell Biol. 36, 961–968 10.1016/j.biocel.2004.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bornstein P. (2001) Thrombospondins as matricellular modulators of cell function. J. Clin. Investig. 107, 929–934 10.1172/JCI12749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Park J., Yu Y. P., Zhou C. Y., Li K. W., Wang D., Chang E., Kim D. S., Vo B., Zhang X., Gong N., Sharp K., Steward O., Vitko I., Perez-Reyes E., Eroglu C., et al. (2016) Central mechanisms mediating thrombospondin-4-induced pain states. J. Biol. Chem. 291, 13335–13348 10.1074/jbc.M116.723478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zeng J., Kim D., Li K. W., Sharp K., Steward O., Zaucke F., and Luo Z. D. (2013) Thrombospondin-4 contributes to spinal cord injury-induced changes in nociception. Eur. J. Pain 17, 1458–1464 10.1002/j.1532-2149.2013.00326.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li K. W., Kim D. S., Zaucke F., and Luo Z. D. (2014) Trigeminal nerve injury-induced thrombospondin-4 up-regulation contributes to orofacial neuropathic pain states in a rat model. Eur. J. Pain 18, 489–495 10.1002/j.1532-2149.2013.00396.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gee N. S., Brown J. P., Dissanayake V. U., Offord J., Thurlow R., and Woodruff G. N. (1996) The novel anticonvulsant drug, gabapentin (Neurontin), binds to the α2δ subunit of a calcium channel. J. Biol. Chem. 271, 5768–5776 10.1074/jbc.271.10.5768 [DOI] [PubMed] [Google Scholar]
- 16. Marais E., Klugbauer N., and Hofmann F. (2001) Calcium channel α2δ subunits-structure and gabapentin binding. Mol. Pharmacol. 59, 1243–1248 10.1124/mol.59.5.1243 [DOI] [PubMed] [Google Scholar]
- 17. Nguyen D., Deng P., Matthews E. A., Kim D. S., Feng G., Dickenson A. H., Xu Z. C., and Luo Z. D. (2009) Enhanced pre-synaptic glutamate release in deep-dorsal horn contributes to calcium channel α-2-δ-1 protein-mediated spinal sensitization and behavioral hypersensitivity. Mol. Pain 5, 6 10.1186/1744-8069-5-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou C., and Luo Z. D. (2014) Electrophysiological characterization of spinal neuron sensitization by elevated calcium channel α-2-δ-1 subunit protein. Eur. J. Pain 18, 649–658 10.1002/j.1532-2149.2013.00416.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou C., and Luo Z. D. (2015) Nerve injury-induced calcium channel α-2-δ-1 protein dysregulation leads to increased pre-synaptic excitatory input into deep dorsal horn neurons and neuropathic allodynia. Eur. J. Pain 19, 1267–1276 10.1002/ejp.656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li C. Y., Zhang X. L., Matthews E. A., Li K. W., Kurwa A., Boroujerdi A., Gross J., Gold M. S., Dickenson A. H., Feng G., and Luo Z. D. (2006) Calcium channel α2δ1 subunit mediates spinal hyperexcitability in pain modulation. Pain 125, 20–34 10.1016/j.pain.2006.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu Y. P., Gong N., Kweon T. D., Vo B., and Luo Z. D. (2018) Gabapentin prevents synaptogenesis between sensory and spinal cord neurons induced by thrombospondin-4 acting on pre-synaptic Cav α2 δ1 subunits and involving T-type Ca2+ channels. Br. J. Pharmacol. 175, 2348–2361 10.1111/bph.14149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lana B., Page K. M., Kadurin I., Ho S., Nieto-Rostro M., and Dolphin A. C. (2016) Thrombospondin-4 reduces binding affinity of [3H]-gabapentin to calcium-channel α2δ-1-subunit but does not interact with α2δ-1 on the cell-surface when co-expressed. Sci. Rep. 6, 24531 10.1038/srep24531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eroglu C., Allen N. J., Susman M. W., O'Rourke N. A., Park C. Y., Ozkan E., Chakraborty C., Mulinyawe S. B., Annis D. S., Huberman A. D., Green E. M., Lawler J., Dolmetsch R., Garcia K. C., Smith S. J., Luo Z. D., et al. (2009) Gabapentin receptor α2δ-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392 10.1016/j.cell.2009.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang M., Offord J., Oxender D. L., and Su T. Z. (1999) Structural requirement of the calcium-channel subunit α2δ for gabapentin binding. Biochem. J. 342, 313–320 [PMC free article] [PubMed] [Google Scholar]
- 25. Hendrich J., Van Minh A. T., Heblich F., Nieto-Rostro M., Watschinger K., Striessnig J., Wratten J., Davies A., and Dolphin A. C. (2008) Pharmacological disruption of calcium channel trafficking by the α2δ ligand gabapentin. Proc. Natl. Acad. Sci. U.S.A. 105, 3628–3633 10.1073/pnas.0708930105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bauer C. S., Nieto-Rostro M., Rahman W., Tran-Van-Minh A., Ferron L., Douglas L., Kadurin I., Sri Ranjan Y., Fernandez-Alacid L., Millar N. S., Dickenson A. H., Lujan R., and Dolphin A. C. (2009) The increased trafficking of the calcium channel subunit α2δ-1 to presynaptic terminals in neuropathic pain is inhibited by the α2δ ligand pregabalin. J. Neurosci. 29, 4076–4088 10.1523/JNEUROSCI.0356-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li K. W., Yu Y. P., Zhou C., Kim D. S., Lin B., Sharp K., Steward O., and Luo Z. D. (2014) Calcium channel α2δ1 proteins mediate trigeminal neuropathic pain states associated with aberrant excitatory synaptogenesis. J. Biol. Chem. 289, 7025–7037 10.1074/jbc.M114.548990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ahmari S. E., Buchanan J., and Smith S. J. (2000) Assembly of presynaptic active zones from cytoplasmic transport packets. Nat. Neurosci. 3, 445–451 10.1038/74814 [DOI] [PubMed] [Google Scholar]
- 29. Friedman H. V., Bresler T., Garner C. C., and Ziv N. E. (2000) Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron 27, 57–69 10.1016/S0896-6273(00)00009-X [DOI] [PubMed] [Google Scholar]
- 30. Engert F., and Bonhoeffer T. (1999) Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399, 66–70 10.1038/19978 [DOI] [PubMed] [Google Scholar]
- 31. Maletic-Savatic M., Malinow R., and Svoboda K. (1999) Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science 283, 1923–1927 10.1126/science.283.5409.1923 [DOI] [PubMed] [Google Scholar]
- 32. Toni N., Buchs P. A., Nikonenko I., Bron C. R., and Muller D. (1999) LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature 402, 421–425 10.1038/46574 [DOI] [PubMed] [Google Scholar]
- 33. Ruthazer E. S., Li J., and Cline H. T. (2006) Stabilization of axon branch dynamics by synaptic maturation. J. Neurosci. 26, 3594–3603 10.1523/JNEUROSCI.0069-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alsina B., Vu T., and Cohen-Cory S. (2001) Visualizing synapse formation in arborizing optic axons in vivo: dynamics and modulation by BDNF. Nat. Neurosci. 4, 1093–1101 10.1038/nn735 [DOI] [PubMed] [Google Scholar]
- 35. Hoppa M. B., Lana B., Margas W., Dolphin A. C., and Ryan T. A. (2012) α2δ expression sets presynaptic calcium channel abundance and release probability. Nature 486, 122–125 10.1038/nature11033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Radulovic L. L., Türck D., von Hodenberg A., Vollmer K. O., McNally W. P., DeHart P. D., Hanson B. J., Bockbrader H. N., and Chang T. (1995) Disposition of gabapentin (Neurontin) in mice, rats, dogs, and monkeys. Drug Metab. Dispos. 23, 441–448 [PubMed] [Google Scholar]
- 37. Milligan E. D., Langer S. J., Sloane E. M., He L., Wieseler-Frank J., O'Connor K., Martin D., Forsayeth J. R., Maier S. F., Johnson K., Chavez R. A., Leinwand L. A., and Watkins L. R. (2005) Controlling pathological pain by adenovirally driven spinal production of the anti-inflammatory cytokine, interleukin-10. Eur. J. Neurosci. 21, 2136–2148 10.1111/j.1460-9568.2005.04057.x [DOI] [PubMed] [Google Scholar]
- 38. Ruzza C., Rizzi A., Malfacini D., Molinari S., Giuliano C., Lovati E., Pietra C., and Calo' G. (2015) In vitro and in vivo pharmacological characterization of Pronetupitant, a prodrug of the neurokinin 1 receptor antagonist Netupitant. Peptides 69, 26–32 10.1016/j.peptides.2015.03.021 [DOI] [PubMed] [Google Scholar]
- 39. Chen J., Li L., Chen S. R., Chen H., Xie J. D., Sirrieh R. E., MacLean D. M., Zhang Y., Zhou M. H., Jayaraman V., and Pan H. L. (2018) The α2δ-1-NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Rep. 22, 2307–2321 10.1016/j.celrep.2018.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heckman K. L., and Pease L. R. (2007) Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2, 924–932 10.1038/nprot.2007.132 [DOI] [PubMed] [Google Scholar]
- 41. Kohfeldt E., Maurer P., Vannahme C., and Timpl R. (1997) Properties of the extracellular calcium binding module of the proteoglycan testican. FEBS Lett. 414, 557–561 10.1016/S0014-5793(97)01070-3 [DOI] [PubMed] [Google Scholar]
- 42. Narouz-Ott L., Maurer P., Nitsche D. P., Smyth N., and Paulsson M. (2000) Thrombospondin-4 binds specifically to both collagenous and non-collagenous extracellular matrix proteins via its C-terminal domains. J. Biol. Chem. 275, 37110–37117 10.1074/jbc.M007223200 [DOI] [PubMed] [Google Scholar]
- 43. Lin Y., McDonough S. I., and Lipscombe D. (2004) Alternative splicing in the voltage-sensing region of N-type CaV2.2 channels modulates channel kinetics. J. Neurophysiol. 92, 2820–2830 10.1152/jn.00048.2004 [DOI] [PubMed] [Google Scholar]
- 44. Vidal M., Morris R., Grosveld F., and Spanopoulou E. (1990) Tissue-specific control elements of the Thy-1 gene. EMBO J. 9, 833–840 10.1002/j.1460-2075.1990.tb08180.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dixon W. J. (1980) Efficient analysis of experimental observations. Annu. Rev. Pharmacol. Toxicol. 20, 441–462 10.1146/annurev.pa.20.040180.002301 [DOI] [PubMed] [Google Scholar]
- 46. Albuquerque C., Joseph D. J., Choudhury P., and MacDermott A. B. (2009) Dissection, plating, and maintenance of dorsal root ganglion neurons for monoculture and for coculture with dorsal horn neurons. Cold Spring Harb. Protoc. 2009, pdb.prot5275 10.1101/pdb.prot5275 [DOI] [PubMed] [Google Scholar]
- 47. Albuquerque C., Joseph D. J., Choudhury P., and MacDermott A. B. (2009) Dissection, plating, and maintenance of dorsal horn neuron cultures. Cold Spring Harb. Protoc. 2009, pdb.prot5274 10.1101/pdb.prot5274 [DOI] [PubMed] [Google Scholar]
- 48. Burkey T. H., Hingtgen C. M., and Vasko M. R. (2004) Isolation and culture of sensory neurons from the dorsal-root ganglia of embryonic or adult rats. Methods Mol. Med. 99, 189–202 10.1385/1-59259-770-X:189 [DOI] [PubMed] [Google Scholar]
- 49. Seybold V. S., and Abrahams L. G. (2004) Primary cultures of neonatal rat spinal cord. Methods Mol. Med. 99, 203–213 10.1385/1-59259-770-X:203 [DOI] [PubMed] [Google Scholar]
- 50. Delree P., Leprince P., Schoenen J., and Moonen G. (1989) Purification and culture of adult rat dorsal root ganglia neurons. J. Neurosci. Res. 23, 198–206 10.1002/jnr.490230210 [DOI] [PubMed] [Google Scholar]
- 51. Joseph D. J., Choudhury P., and Macdermott A. B. (2010) An in vitro assay system for studying synapse formation between nociceptive dorsal root ganglion and dorsal horn neurons. J. Neurosci. Methods 189, 197–204 10.1016/j.jneumeth.2010.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ohshiro H., Ogawa S., and Shinjo K. (2007) Visualizing sensory transmission between dorsal root ganglion and dorsal horn neurons in co-culture with calcium imaging. J. Neurosci. Methods 165, 49–54 10.1016/j.jneumeth.2007.05.018 [DOI] [PubMed] [Google Scholar]
- 53. Varon S., and Raiborn C. (1971) Excitability and conduction in neurons of dissociated ganglionic cell cultures. Brain Res. 30, 83–98 10.1016/0006-8993(71)90007-2 [DOI] [PubMed] [Google Scholar]
- 54. Hasegawa H., Abbott S., Han B. X., Qi Y., and Wang F. (2007) Analyzing somatosensory axon projections with the sensory neuron-specific Advillin gene. J. Neurosci. 27, 14404–14414 10.1523/JNEUROSCI.4908-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zurborg S., Piszczek A., Martínez C., Hublitz P., Al Banchaabouchi M., Moreira P., Perlas E., and Heppenstall P. A. (2011) Generation and characterization of an Advillin-Cre driver mouse line. Mol. Pain 7, 66 10.1186/1744-8069-7-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Artru A. (1999) Spinal cerebrospinal fluid chemistry and physiology, in Spinal Drug Delivery (Yaksh T. L., ed) pp. 176–237, Elsevier Sciences, Amsterdam [Google Scholar]