

Abstract
Rosuvastatin has cardiac protective effects through its anti-inflammatory effects. The nuclear protein high-mobility group box 1 (HMGB1) can activate inflammatory pathways when released from dying cells. The present study aimed to investigate the effects of rosuvastatin in adriamycin (ADR)-treated rats. Adult male rats were randomized to three groups: i) Control group, ii) ADR group, and iii) ADR+rosuvastatin group. Serum biochemical indices were measured using an enzyme-linked immunosorbent assay. Cardiac function was assessed by echocardiography. The expression of HMGB1 and receptors for advanced glycation end products (RAGE) were assessed by reverse transcription-quantitative polymerase chain reaction analysis, western blot analysis, and immunohistochemistry. Cytokines were measured using flow cytometry. Rosuvastatin improved the biochemical indices and cardiac morphology and alleviated the pathological lesions. In the ADR+rosuvastatin group, the mRNA and protein levels of HMGB1 and RAGE in the myocardium were significantly lower compared with those in the ADR group (both P<0.05). The results showed that rosuvastatin significantly reduced the levels of HMGB1 and RAGE in the myocardium of the ADR-treated rats. These results suggest that the protective effects of rosuvastatin may be associated with attenuation of the HMGB1/RAGE-mediated inflammatory response in ADR-treated rats. Despite this protective effect of rosuvastatin in the present study, it did not improve cardiac function in terms of the diastolic left ventricular internal dimension, systolic left ventricular internal dimension, left ventricular ejection fraction and left ventricular fractional shortening; this may be due the observation duration being insufficient.
Keywords: rosuvastatin, myocardium, adriamycin, inflammatory response
Introduction
Chronic heart failure (CHF) is the advanced stage or end-stage of various heart diseases. The pathogenesis of CHF involves the renin-angiotensin-aldosterone system and excessive neurohormonal activation. Immune system activation and a number of inflammatory cytokines are involved in CHF (1–4).
High mobility group box-1 (HMGB1) is a conserved nuclear DNA-binding protein involved in the maintenance of nucleosome structure and in DNA recombination, replication, and gene transcription (5–8). HMGB1 is released passively from necrotic or damaged cells and is actively secreted by immune cells, including monocytes, macrophages, and dendritic cells (8). HMGB1 is a potent extracellular cytokine involved in cellular activation and triggers a rigorous inflammatory response through interactions with its receptors (8,9). Extracellular HMGB1 binds to a variety of cell surface receptors, including receptors for advanced glycation end products (RAGE) (10), toll-like receptor (TLR)2, and TLR4 (11), leading to the activation of downstream physiologic and pathologic responses (12). The binding of HMGB1 to RAGE receptors results in the activation of mitogen-activated protein kinases (MAPKs) and of the nuclear factor-κB (NF-κB) transcription factor, which induces the production of various pro-inflammatory cytokines (13). In addition, previous studies have demonstrated that HMGB1 is pivotal in cardiovascular diseases, including atherosclerosis, myocardial ischemia/reperfusion injury, CHF, and myocardial infarction (14). In particular, CHF is considered to be an inflammatory disease and HMGB-1 is important in its progression (10,15,16). A high level of HMGB-1 is often found in patients with CHF, where it is released by activated macrophages and, in turn, induces the expression of other inflammatory cytokines that are able to amplify macrophage recruitment, starting a vicious circle (17).
Statins are inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A reductase and are used extensively for treatment of coronary heart diseases due to their cholesterol-lowering effects (18). Furthermore, statins exert beneficial pleiotropic effects, including plaque stabilization, anti-inflammatory effects, and the prevention of endothelial dysfunction (19). In addition, statins protect endothelial cells against ischemia reperfusion injury through the HMGB1/TLR4 pathway (20,21). However, whether statins can influence the HMGB1/RAGE/NF-κB pathway and the production of inflammatory agents remains to be fully elucidated.
Therefore, in the present study, it was hypothesized that rosuvastatin may attenuate myocardial injury by inhibiting the expression of HMGB1 and RAGE. To test this hypothesis, adriamycin (ADR)-treated rats were used. The aim of the present study was to assess whether rosuvastatin attenuates myocardial injury in ADR-treated rats and whether the activation of HMGB1/RAGE is involved.
Materials and methods
Animals
Male Sprague-Dawley rats aged 6–8 weeks (220–260 g) were obtained from the Animal Center of the Chinese Academy of Sciences (Shanghai, China). The rats were housed under a 12-h dark-light cycle at 20–25°C and 40–60% humidity, with five rats/cage, and free access to food and water. The experiments were performed following 1 week of adaptive feeding. The experiments were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Animal Care and Use Committee of Nanjing Medical University (Nanjing, China). During the experiment, the following criteria were used to indicate end of life of the experimental animals: i) significant weight loss (>30% weight loss) or cachexia; ii) complete loss of appetite for >24 h or poor appetite (<50% of normal diet) for >3 days; ii) unable to eat or drink water under no anesthesia or sedation; iv) depression or hypothermia (<37°C) under no anesthesia or sedation; v) clinical symptoms of severe functional loss of organs and treatment failure; vi) expiratory dyspnea; vii) severe diarrhea and peritonitis; viii) tremors and paralysis; or ix) severe ulcerations of the skin.
Grouping
The rats were randomized into the following groups (n=10/group): i) Control group, rats administered with 0.9% saline solution without ADR at 1 mg/kg/day for 14 days via intraperitoneal injection and with 0.9% saline without rosuvastatin at a dosage of 1 mg/kg/day for 6 weeks by gavage administration; ii) ADR group, rats injected with equal volumes of ADR solution (ADR dissolved in 0.9% saline solution, 10 mg/ml) for 2 weeks plus administration of equal volumes of 0.9% saline solution without rosuvastatin; iii) ADR+rosuvastatin group, rats administered with equal volumes of ADR for 2 weeks and rosuvastatin (1 mg/kg/day) (22) solution for 6 weeks (rosuvastatin was dissolved in 0.9% saline solution, 10 mg/ml). All rats received the drug at 10 a.m. and the intraperitoneal injection at 9 a.m. (1 h prior to gavage). ADR was purchased from Haizheng Chemical Co., Ltd. (Zhejiang, China). Rosuvastatin was provided by AstraZeneca (London, UK). All examinations and sample collection (echocardiography, blood collection, and myocardial tissue collection) were performed at 12 weeks.
Enzyme-linked immunosorbent assay (ELISA)
Blood samples were collected from the ophthalmic artery and the serum was separated by centrifugation at 1,500 × g for 15 min at 4°C. The levels of blood urea nitrogen (BUN; cat. no. R0119), creatinine (Cr; cat. no. R0120), alanine aminotransferase (ALT; cat. no. R0116), aspartate aminotransferase (AST; cat. no. R0117), lactate dehydrogenase (LDH; cat. no. R0042), creatine kinase isoenzyme-MB (CK-MB; cat. no. R0043), triglycerides (TG; cat. no. R0794), total cholesterol (CHO; cat. no. R0794), low-density lipoprotein cholesterol (LDL-C; cat. no. R0794), and high density lipoprotein cholesterol (HDL-C; cat. no. R0794) in serum samples were determined using ELISA kits (Nanjing Senbeijia Biological Technology Co., Ltd., Nanjing, China), according to the manufacturer’s protocol.
Echocardiography measurements
Echocardiography was performed to assess the heart function of the rats 12 weeks following the first treatment, according to a previously published method (23). The rats were anesthetized using chloral hydrate (5%, 0.7 ml/100 g, equivalent to 350 mg/kg) via intraperitoneal injection. Images were captured using a 12-MHz linear transducer connected to a Vivid 7 echocardiography machine (GE Healthcare Life Sciences, Chalfont, UK). A two-dimensional short-axis view of the left ventricle was obtained at the level of the papillary muscle and two-dimensional targeted M-mode tracings were recorded. The detection indices were as follows: Systolic left ventricular internal dimension (LVIDs), diastolic left ventricular internal dimension (LVIDd), left ventricular ejection fraction (LVEF), left ventricular end-diastolic pressure (LVEDP), and left ventricular fractional shortening (LVFS). All these parameters were measured over three consecutive cardiac cycles.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis
The mRNA expression levels of HMGB1 and RAGE were determined by RT-qPCR analysis. Total RNA was extracted with TRIzol (Promega Corporation, Madison, WI, USA), according to the manufacturer’s protocol. Total cDNA was synthesized using the HiScript II 1st Strand cDNA Synthesis kit (+gDNA wiper), purchased from Vazyme Biotech Co., Ltd. (Nanjing, China). The cDNA was then amplified by PCR. The forward and reverse primers were as follows: HMGB1 forward, 5′-CCT GAG AAT GTA TCC CCA AAA GC-3′ and reverse, 5′-CAG TCA AGT TTC CTG AGC AA TCC-3′ (product size: 149 bp); RAGE forward, 5′-TAGCCA TGG ACC TCA GGA AAG-3′ and reverse, 5′-CCA ATG AGC AGA GCG GCT AT-3′ (product size: 159 bp); and GAPDH forward, 5′-GGT GGA CCT CAT GGC CTA CA-3′ and reverse, 5′-TTG TGA GGG AGA TGC TCA GTG T-3′ (product size: 246 bp). PCR was performed with 12.5 μl Maxima SYBR Green/ROX qPCR Master Mix (2X; Thermo Scientific, Inc., K0221, Waltham, MA, USA), 0.75 μl forward primers (10 μM), 0.75 μl reverse primer (10 μM), 2 μl cDNA and 9 μl distilled water, at 95°C for 15 sec, 60°C for 30 sec, and 30 sec at 72°C for 40 cycles, according to the manufacturer’s protocol. The comparative Ct method was used to calculate the relative abundance of the mRNA and the results for target gene expression were compared with those for GAPDH. The results were obtained from three independent experiments. The method (24) was calculated to represent the relative mRNA expression of target genes.
Western blot analysis
Myocardial tissues from the left ventricle were completely homogenized and centrifuged at 13,000 × g for 15 min at 4°C. The supernatant was collected. An equal volume of 5X SDS sample buffer was added, and the samples were boiled for 10 min. Protein concentration was measured using the BCA method. The samples (50 μg of protein per lane) were subjected to electrophoresis on 10% SDS-polyacrylamide gels for 30 min at 80 V followed by 80 min at 120 V. The proteins were transferred onto polyvinylidene fluoride membranes (Immobilon-PSQ; EMD Millipore, Billerica, MA, USA) for 1 h at 100 V and 300 mA. The membranes were blocked with 5% skimmed milk for 2 h at room temperature and incubated overnight at 4°C with rabbit anti-HMGB1 monoclonal antibody (1:1,000; cat. no. ab79823; Abcam, Cambridge, MA, USA), rabbit anti-RAGE polyclonal antibody (1:1,000; cat. no. ab3611; Abcam), and rabbit GAPDH monoclonal antibody (1:1,000; cat. no. M20006; Abmart, Berkeley Heights, NJ, USA). The membranes were washed three times for 10 min each time in PBS with 0.1% Tween-20 (PBST) and were incubated with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G secondary antibody (1:1,000; cat. no. 7074; Cell Signaling Technology, Inc., Danvers, MA, USA) at room temperature for 2 h. The blotted protein bands were visualized by enhanced chemiluminescence (Amersham; GE Healthcare Life Sciences), and exposed to X-ray films. The developed films were digitized using an Epson Perfection 2480 scanner (Seiko Co., Ltd., Nagano, Japan). The optical densities were obtained using Glyko Bandscan software version 4.5 (Glyko Inc., Novato, CA, USA).
Histology and immunohistochemistry
The rats were sacrificed following 12 weeks of treatment and cardiac tissues were harvested. The tissue samples were formalin-fixed, embedded in paraffin, and sectioned at 3–5 μm. The sections were stained with hematoxylin and eosin (H&E). The histological examination of all sections was performed with an optical microscope in a blinded-manner.
Immunohistochemistry was performed to detect HMGB1 and RAGE. Briefly, the paraffin sections were dewaxed in xylene and rehydrated through graded ethanol to water. Antigens were retrieved by boiling in 10 mM citrate buffer (pH 6.0) and endogenous peroxidase activity was quenched in methanol containing 3% hydrogen peroxide. The sections were incubated in phosphate-buffered saline (PBS) containing 3% bovine serum albumin (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) to block nonspecific binding, and with anti-HMGB1 monoclonal antibody (1:1,000; Abcam) and anti-RAGE polyclonal antibody (1:800; Abcam) overnight at 4°C, followed by a 15-min period of washing in PBS. The sections were then incubated with HRP-conjugated IgG (1:500; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) for 60 min at room temperature. Those cells in which HMGB1 and RAGE were expressed in the extranuclear space were considered to be HMGB1-positive and RAGE-positive cells. The positive cells were identified, counted and analyzed with an optical microscope by two pathologists.
Flow cytometry
Tumor necrosis factor (TNF)-α, interferon (IFN)-γ, interleukin (IL)-4, and IL-10 were measured using the CBA Rat Soluble Protein Detection kit (Bio-Rad Laboratories, Inc.). The samples and standards (50 μl each) were incubated in Falcon tubes with capture beads for 1 h at room temperature in the dark. The phycoerythrin detection reagent was added to each tube for an additional 2 h of incubation at room temperature in the dark. The samples were washed, and the bead pellets were re-suspended in washing buffer. The re-suspended samples were run on a flow cytometer (FACS Canto II; BD Biosciences, Franklin Lake, NJ, USA) equipped with the BD FACSDiva software version 6.1.3. A total of 2,000 events in the gated bead population were collected, and the 5-parameter data were saved for subsequent analysis using BD FCAP Array software version 3.0. Serum concentrations were derived using the standard curve and are expressed in pg/ml.
Statistical analysis
SPSS 18.0 (SPSS, Inc., Chicago, IL, USA) was used for statistical analysis. Data are expressed as the mean ± standard error of the mean and were analyzed using one-way analysis of variance followed by the Tukey’s post hoc test. P<0.05 (two-sided) was considered to indicate a statistically significant difference.
Results
Characteristics of the rats
Three weeks prior to drug administration, the rats in each group had normal water intake, their hair was soft, and they were active. At the beginning of week 4, the rats in the ADR group became lethargic, had decreased food and water intake, slow movement, loss of hair luster, decreased activity, and symptoms of eyeball hyperemia and diarrhea. One rat died on each of days 35, 47, and 70 of the experiment in the ADR group, and the survival rate was 70%. The rats in the ADR+rosuvastatin group showed similar symptoms to rats in the ADR group, although they occurred later and were milder. One rat died on day 72 in this group; the survival rate was 90%. The rats in the control group were generally in good conditions and none of the rats died. All deceased rats were found during routine rounds, and none of the rats met the criteria for end of life as all symptoms were observed to be mild to moderate. The maximum weight loss observed in rats was ~25%, and the mortality rate was 30%, which was similar to a previous study (25).
The weights of the rats were measured prior to model establishment (0 weeks) and at 4, 8, and 12 weeks following model establishment. The weight of the rats in the control group increased during each subsequent time period. At 4 weeks prior to model establishment, the weights of the rats in the ADR and ADR+rosuvastatin groups maintained the same trend of growth as those in the control group. At 4 weeks post-modeling, the body weight of the rats in the ADR group did not increase or decrease. At 12 weeks post-model establishment, the weight of the rats in the ADR group was significantly decreased compared with that in the control group (P<0.01). The weight of the rats in the ADR+rosuvastatin group was significantly higher than that in the ADR group (P<0.05) (Fig. 1).
Figure 1.
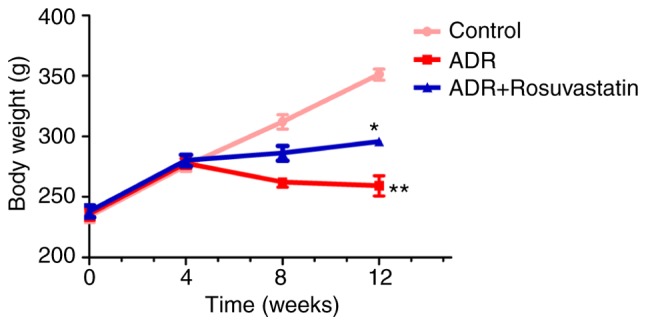
Body weight changes. Weights of the rats were measured prior to model establishment (week 0) and at 4, 8, and 12 weeks following model establishment. **P<0.01, vs. control; *P<0.05, vs. ADR. ADR, adriamycin.
At the end of study, no significant differences in BUN, Cr, ALT, LDH, TG, or HDL-C were observed among the three groups. The serum levels of CHO and LDL-C were significantly lower in the ADR+rosuvastatin group compared with those in the other groups (P<0.05). The serum levels of AST and CK-MB were elevated in the ADR group compared with those in the control group and were significantly lower in the ADR+rosuvastatin group than in the ADR group (P<0.05; Table I).
Table I.
Biochemical indices in each group at the end of the study.
| Index | Control | ADR | ADR+rosuvastatin |
|---|---|---|---|
| Bun | 5.46±0.10 | 6.29±0.43 | 6.42±0.68 |
| Cr | 24.92±3.00 | 26.24±3.13 | 24.56±1.92 |
| ALT | 42.00±6.18 | 47.20±8.28 | 35.80±3.68 |
| AST | 78.7±4.44 | 246.6±63.10b | 117.0±30.5c |
| CK-MB | 420±72 | 2,848±1,443b | 491±72d |
| LDH | 1,218±214 | 1,049±521 | 347±73.9 |
| CHO | 3.13±0.38 | 3.10±0.30 | 1.44±0.12a,c |
| TG | 1.26±0.45 | 1.07±0.30 | 0.55±0.06 |
| HDL | 1.36±0.20 | 0.90±0.03 | 1.17±0.22 |
| LDL | 0.61±0.07 | 0.60±0.08 | 0.25±0.05a,c |
P<0.05 and
P<0.01, vs. control group;
P<0.05 and
P<0.01, vs. ADR group. ADR, adriamycin; Bun, blood urea nitrogen; Cr, creatinine; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CK-MB, creatine kinase isoenzyme-MB; LDH, lactate dehydrogenase; CHO, total cholesterol; TG, triglycerides; HDL, high density lipoprotein; LDL, low-density lipoprotein.
In order to determine whether ADR affected the cardiac function of the rats, echocardiography was performed at 12 weeks following the first treatment. The echocardiography revealed that, compared with the control group, the rats in the ADR and ADR+rosuvastatin groups exhibited significant left ventricular dilation and systolic dysfunction. The LVIDd, and LVIDs in these two groups were significantly higher than those in the control group (P<0.05). The LVEF and LVFS in these two groups were significantly lower than those in the control group (P<0.05), whereas the LVEDP was higher (P<0.05; Fig. 2A-E). Rosuvastatin itself did not improve the heart function (LVIDd, LVIDs, LVEF and LVFS) of the ADR-treated rats.
Figure 2.

Cardiac function in control, ADR, and ADR+rosuvastatin groups of rats. (A) LVIDd; (B) LVIDs; (C) LVEF; (D) FS; (E) LVEDP. Data are presented as the mean ± standard error of the mean. ADR, adriamycin; LVIDd, diastolic left ventricular internal dimension diastolic; LVIDs, systolic left ventricular internal dimension; LVEF, left ventricular ejection fraction; FS, left ventricular fractional shortening; LVEDP, left ventricular end-diastolic pressure.
The H&E-stained sections of left ventricular myocardial tissue are shown in Fig. 3A-C. The tissues from the ADR group showed enhancement and loose arrangement of the myocardial fibers, loss of myocytes, and vacuolar degeneration. The administration of rosuvastatin improved cardiac morphology and alleviated pathological lesions.
Figure 3.

H&E staining of cardiomyocytes of rats in the control, ADR, and ADR+rosuvastatin groups. Representative left ventricular sections of hearts from the (A) control, (B) ADR, and (C) ADR+rosuvastatin experimental groups, stained with H&E (magnification, ×200). The arrows point to regions of myocardial fibrosis. ADR, adriamycin; H&E, hematoxylin and eosin.
Effect of rosuvastatin on the expression of HMGB1 and RAGE
The mRNA expression levels of HMGB1 and RAGE in the myocardium were significantly higher in the ADR group compared with those in the control group (P<0.01 and P<0.01, respectively). In the ADR+rosuvastatin group, the mRNA levels of HMGB1 and RAGE in the myocardium were signifi-cantly lower compared with those in the ADR group (P<0.05 and P<0.05, respectively; Fig. 4).
Figure 4.

Effects of rosuvastatin on the mRNA expression levels of HMGB1 and RAGE in the three groups. In the control, ADR, and ADR+rosuvastatin groups, mRNA expression levels of HMGB1 and RAGE were determined by reverse transcription-quantitative polymerase chain reaction analysis. Data are presented as the mean ± standard error of the mean. ADR, adriamycin; HMGB1, high-mobility group box 1; RAGE, receptors for advanced glycation end products.
To determine the effect of rosuvastatin on the expression of HMGB1 and RAGE in the ADR-treated rats, western blot analysis was performed to detect changes in the protein levels of HMGB1 and RAGE. The results showed low protein levels of HMGB1 and RAGE in the control group. The protein levels of HMGB1 and RAGE were significantly higher in the myocardium of the ADR group compared with those of the control group (P<0.01 and P<0.001, respectively). The increased levels of HMGB1 and RAGE were markedly suppressed in the ADR+rosuvastatin group (P<0.05 and P<0.001, respectively; Fig. 5A and B).
Figure 5.

Effect of rosuvastatin on the protein expression of HMGB1 and RAGE. In the controls, ADR, and ADR+rosuvastatin groups, protein levels of HMGB1 and RAGE were measured by western blot analysis. (A) Original representative western blots are presented. (B) Data are presented as the mean ± standard error of the mean. ADR, adriamycin; HMGB1, high-mobility group box 1; RAGE, receptors for advanced glycation end products.
Rosuvastatin decreases the proportion of HMGB1- and RAGE-positive cells in the myocardium of ADR-treated rats
Immunohistochemistry was performed to investigate the localization and expression of HMGB1 and RAGE in the rat myocardial tissue. Those myocardial cells in which HMGB1 was found in the cytoplasm were considered to be HMGB1-positive cells. Few HMGB1 and RAGE-positive cells were observed in the control group. Higher numbers of HMGB1 and RAGE-positive cells were observed in the ADR group. Rosuvastatin significantly decreased HMGB1- and RAGE-positivity. These results showed that rosuvastatin significantly reduced HMGB1 and RAGE immunoreactivity in the myocardium of the ADR-treated rats (Fig. 6A-D).
Figure 6.

Immunohistochemistry showing the protein expression of HMGB1 and RAGE in the myocardial tissue of the control, ADR, and ADR+rosuvastatin groups. The cells with cytoplasm or extranuclear HMGB1 and RAGE staining were considered HMGB1-positive and RAGE-positive cells. (A) HMGB1 (magnification, ×200). (B) HMGB1 (magnification, ×400). (C) RAGE (magnification, ×200). (D) RAGE (magnification, ×400). ADR, adriamycin; HMGB1, high-mobility group box 1; RAGE, receptors for advanced glycation end products.
Levels of pro-inflammatory and anti-inflammatory cytokines in the serum
As shown in Fig. 7, the serum levels of TNF-α and IFN-γ were low, and the levels of IL-4 and IL-10 were high in the control group. In the ADR group, the serum levels of TNF-α and IFN-γ were increased, whereas the levels of IL-4 and IL-10 were decreased. Rosuvastatin significantly decreased the serum levels of TNF-α and IFN-γ and elevated the serum levels of IL-4 and IL-10 in the ADR+rosuvastatin group (Fig. 7).
Figure 7.

Levels of proinflammatory (TNF-α and IFN-γ) and anti-inflammatory (IL-4 and IL-10) cytokines in the three groups. In the control, ADR, and ADR+rosuvastatin groups, cytokines were measured by flow cytometry. Data are presented as the mean ± standard error of the mean. ADR, adriamycin; TNF, tumor necrosis factor; IFN, interferon; IL, interleukin.
Discussion
Statins have been shown to mediate HMGB1 and its different receptors, including RAGE or TLR2/TLR4, to exert cytoprotective actions and anti-inflammatory effects by inhibiting the expression of various proinflammatory cytokines (26–28). HMGB1 can activate inflammatory pathways when released from dying cells (14). In the heart, pre- and post-conditioning with rosuvastatin has been shown to reduce ischemia/reperfusion myocardial injury through the inhibition of HMGB1 (29,30). Therefore, the present study aimed to investigate the effects of rosuvastatin in ADR-treated rats. The results suggested that the protective effects of rosuvastatin may be associated with a reduction of the HMGB1/RAGE-mediated inflammatory response in rats treated with ADR.
Rosuvastatin is one of the most potent statins and has well-established lipid-lowering effects in addition to pleiotropic effects, including anti-inflammatory and endothelial protective effects (31–33). Accordingly, the present study showed improved biochemical indices and histopathology following rosuvastatin treatment. These results are supported by previous studies; Fei et al (34) showed that rosuvastatin had protective effects on the risk of acute myocardial injury through lowering of vascular endothelial growth factor A. In addition, Kim et al (35) showed that rosuvastatin prevented the long-term detrimental effects of ADR on left ventricular function. Ke et al (30) and Du et al (29) showed that pre- and post-conditioning with rosuvastatin reduces ischemia/reperfusion myocardial injury through the inhibition of HMGB1. However, the direct effects of rosuvastatin on cardiac function were not observed in the present study. This may be due to the model itself or to the short treatment time. In addition, the main effect of statins is to lower the blood cholesterol levels, whereas their pleiotropic effects are secondary effects.
The present study revealed that rosuvastatin significantly inhibited the expression of HMGB1 and RAGE. Xu et al (36) showed that atorvastatin inhibited the expression of RAGE in rat aortas, Yang et al (20) showed that statins decreased the activation of HMGB1, and Jin et al (37) showed that atorvastatin decreased the serum levels of HMGB1. HMGB1 binding to RAGE results in the upregulation of proinflammatory cytokines, including TNF-α and IFN-γ, following ADR injury (38). These results are supported by Gao et al (39), who showed that HMGB1 and RAGE mediated the overexpression of TNF-α, and by a number of previous studies (40–42). Similar results have been obtained for IFN-γ (43–45).
A previous study demonstrated that HMGB1-haptoglobin β complexes binding to CD163 result in the release of anti-inflammatory cytokines, including IL-10 in sterile and infectious inflammation (38). In addition, HMGB1 binding to RAGE results in anti-inflammatory cytokines, including IL-4 and IL-10, being upregulated following inflammatory injury (40,46,47). Du et al (29) showed that post-conditioning with rosuvastatin decreased markers of oxidative stress in rats following ischemia/reperfusion injury. In addition, Ke et al (30) showed that preconditioning with rosuvastatin decreased ischemia/reperfusion injury by reducing the accumulation of inflammatory cells and regulatory T cells in the heart, which is associated with increased production of inflammatory cytokines, including TNF-α and IFN-γ, and dysregulated anti-inflammatory cytokines, including IL-4 and IL-10 (48). In the present study, it was found that the administration of rosuvastatin in the ADR-treated rats inhibited the expression of HMGB1 and RAGE, in addition to the decrease of TNF-α and IFN-γ and increase of IL-4 and IL-10. Together, these results suggest that the activation of HMGB1/RAGE by rosuvastatin may result in improved functional recovery. However, additional investigations are necessary to examine the effects of rosuvastatin on inflammatory cells, regulatory T cells, and a comprehensive cytokine panel in ADR heart injury. In addition, previous studies have shown that HMGB1 can activate the phosphoinositide 3-kinase/Akt and TLR4/NF-κB pathways during heart ischemia/reperfusion injury, high-lighting the role of HMGB1 in heart injury (20,49–51). The effects of rosuvastatin on pathways including PI3K/Akt and TLR4/NF-κB in the context of heart injury also require investigation in the future.
The present study did not show that rosuvastatin significantly improved left ventricular structure and function or LVEF. This may be due to a number of reasons, including administration time and dose of statin. It had been reported that the improvement of LVEF by statins is time-dependent, and that long-term treatment may be more beneficial in improving LVEF (52). It was hypothesized that changes in serological parameters and the expression of inflammatory cytokines by rosuvastatin may occur ahead of changes in myocardial tissue. Additional investigations are necessary to address this issue.
The present study had certain limitations. There was no statin-only group. However, previous studies have suggested that treatment with statin alone did not differ significantly from the control group in terms of the baseline blood biochemical indicators (AST, ALT, and ALP), heart structure indicators (LVEF, LVFS, LVIDd, and LVIDs), injury to other organs and tissues, and oxidative stress indicators, including superoxide dismutase, malondialdehyde, and catalase (53,54). Therefore, considering animal protection, a statin-only group was not included in the present study. In addition, only one dose of rosuvastatin was used, however, preliminary experiments showed that 1.0 mg/kg/day led to more significant improvement for myocardial dysfunction in the ADR-treated rats compared to the other doses (0.1 and 5.0 mg/kg/day).
In conclusion, the present study suggested that ADR can induce the expression of HMGB1 and RAGE in the myocardium of ADR-treated rats, which can be inhibited by rosuvastatin. The protective effects of rosuvastatin in ADR-treated rats may be associated with the inhibition of HMGB1/RAGE pathway activation. Despite this protective effect of rosuvastatin in the present study, it did not improve cardiac function (LVIDd, LVIDs, LVEF, and LVFS). Further investigation on the effects of rosuvastatin may provide novel modalities for myocardial injury induced by ADR.
Acknowledgments
The authors gratefully acknowledge Mrs. Ping Zhou from the Department of Geriatrics (The Second Affiliated Hospital of Nanjing Medical University, Nanjing, China) for her technical assistance.
Funding
This study was supported by grants from the National Natural Science Foundation of China (grant nos. 81270428, 81300999, and 81470501).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors’ contributions
XL and HZ conceived the study and designed the experiments. HZ, ZL and KD performed the experiments. HZ and ZL collected and analyzed the experimental results. HZ wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The experiments were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Animal Care and Use Committee of Nanjing Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Christensen HM, Schou M, Goetze JP, Faber J, Frystyk J, Flyvbjerg A, Kistorp C. Body mass index in chronic heart failure: Association with biomarkers of neurohormonal activation, inflammation and endothelial dysfunction. BMC Cardiovasc Disord. 2013;13:80. doi: 10.1186/1471-2261-13-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kazymyrko VK, Kutovyi VV, Ivanyts’ka LM, Dubkova AG, Silant’ieva TS. Biomarkers of iron metabolism and inflammation in patients with chronic heart failure and various types of left ventricular dysfunction (In Ukrainian) Lik Sprava. 2013:19–22. [PubMed] [Google Scholar]
- 3.Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: Critical importance of the cardiosplenic axis. Circ Res. 2014;114:266–282. doi: 10.1161/CIRCRESAHA.113.301720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zykov KA, Tatenkulova SN, Masenko VP, Kuznetsova TV, Rvacheva AV, Belenkov IuN. Characteristics of autoimmune reactions in chronic cardiac failure of different etiology (In Russian) Ter Arkh. 2009;81:22–28. [PubMed] [Google Scholar]
- 5.Yamada S, Maruyama I. HMGB1, a novel inflammatory cytokine. Clin Chim Acta. 2007;375:36–42. doi: 10.1016/j.cca.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 6.Bustin M, Reeves R. High-mobility-group chromosomal proteins: Architectural components that facilitate chromatin function. Prog Nucleic Acid Res Mol Biol. 1996;54:35–100. doi: 10.1016/S0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- 7.Park JS, Arcaroli J, Yum HK, Yang H, Wang H, Yang KY, Choe KH, Strassheim D, Pitts TM, Tracey KJ, et al. Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am J Physiol Cell Physiol. 2003;284:C870–C879. doi: 10.1152/ajpcell.00322.2002. [DOI] [PubMed] [Google Scholar]
- 8.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 9.Kostova N, Zlateva S, Ugrinova I, Pasheva E. The expression of HMGB1 protein and its receptor RAGE in human malignant tumors. Mol Cell Biochem. 2010;337:251–258. doi: 10.1007/s11010-009-0305-0. [DOI] [PubMed] [Google Scholar]
- 10.Volz HC, Kaya Z, Katus HA, Andrassy M. The role of HMGB1/RAGE in inflammatory cardiomyopathy. Semin Thromb Hemost. 2010;36:185–194. doi: 10.1055/s-0030-1251503. [DOI] [PubMed] [Google Scholar]
- 11.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q, O’Hearn S, Kavalukas SL, Barbul A. Role of high mobility group box 1 (HMGB1) in wound healing. J Surg Res. 2012;176:343–347. doi: 10.1016/j.jss.2011.06.069. [DOI] [PubMed] [Google Scholar]
- 13.Li W, Sama AE, Wang H. Role of HMGB1 in cardiovascular diseases. Curr Opin Pharmacol. 2006;6:130–135. doi: 10.1016/j.coph.2005.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding HS, Yang J. High mobility group box-1 and cardiovascular diseases. Saudi Med J. 2010;31:486–489. [PubMed] [Google Scholar]
- 15.Liu T, Zhang DY, Zhou YH, Han QF, Wang LH, Wu L, Yao HC. Increased serum HMGB1 level may predict the fatal outcomes in patients with chronic heart failure. Int J Cardiol. 2015;184:318–320. doi: 10.1016/j.ijcard.2015.02.088. [DOI] [PubMed] [Google Scholar]
- 16.Volz HC, Laohachewin D, Schellberg D, Wienbrandt AR, Nelles M, Zugck C, Kaya Z, Katus HA, Andrassy M. HMGB1 is an independent predictor of death and heart transplantation in heart failure. Clin Res Cardiol. 2012;101:427–435. doi: 10.1007/s00392-011-0409-x. [DOI] [PubMed] [Google Scholar]
- 17.Volz HC, Seidel C, Laohachewin D, Kaya Z, Muller OJ, Pleger ST, Lasitschka F, Bianchi ME, Remppis A, Bierhaus A, et al. HMGB1: The missing link between diabetes mellitus and heart failure. Basic Res Cardiol. 2010;105:805–820. doi: 10.1007/s00395-010-0114-3. [DOI] [PubMed] [Google Scholar]
- 18.Blumenthal RS. Statins: Effective antiatherosclerotic therapy. Am Heart J. 2000;139:577–583. doi: 10.1016/S0002-8703(00)90033-4. [DOI] [PubMed] [Google Scholar]
- 19.Stalker TJ, Lefer AM, Scalia R. A new HMG-CoA reductase inhibitor, rosuvastatin, exerts anti-inflammatory effects on the microvascular endothelium: The role of mevalonic acid. Br J Pharmacol. 2001;133:406–412. doi: 10.1038/sj.bjp.0704070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J, Huang C, Yang J, Jiang H, Ding J. Statins attenuate high mobility group box-1 protein induced vascular endothelial activation: A key role for TLR4/NF-kappaB signaling pathway. Mol Cell Biochem. 2010;345:189–195. doi: 10.1007/s11010-010-0572-9. [DOI] [PubMed] [Google Scholar]
- 21.Han QF, Wu L, Zhou YH, Wang LH, Zhang DY, Liu T, Yao HC. Simvastatin protects the heart against ischemia reperfusion injury via inhibiting HMGB1 expression through PI3K/Akt signal pathways. Int J Cardiol. 2015;201:568–569. doi: 10.1016/j.ijcard.2015.08.180. [DOI] [PubMed] [Google Scholar]
- 22.Jones SP, Gibson MF, Rimmer DM, III, Gibson TM, Sharp BR, Lefer DJ. Direct vascular and cardioprotective effects of rosuvastatin, a new HMG-CoA reductase inhibitor. J Am Coll Cardiol. 2002;40:1172–1178. doi: 10.1016/S0735-1097(02)02115-0. [DOI] [PubMed] [Google Scholar]
- 23.Hauck L, Stanley-Hasnain S, Fung A, Grothe D, Rao V, Mak TW, Billia F. Cardiac-specific ablation of the E3 ubiquitin ligase Mdm2 leads to oxidative stress, broad mitochondrial deficiency and early death. PLoS One. 2017;12:e0189861. doi: 10.1371/journal.pone.0189861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Rabelo E, De Angelis K, Bock P, Gatelli Fernandes T, Cervo F, Belló Klein A, Clausell N, Cláudia Irigoyen M. Baroreflex sensitivity and oxidative stress in adriamycin-induced heart failure. Hypertension. 2001;38:576–580. doi: 10.1161/hy09t1.096185. [DOI] [PubMed] [Google Scholar]
- 26.Zhu Z, Fang Z. Statin protects endothelial cell against ischemia reperfusion injury through HMGB1/TLR4 pathway. Int J Cardiol. 2016;203:74. doi: 10.1016/j.ijcard.2015.10.095. [DOI] [PubMed] [Google Scholar]
- 27.Haraba R, Suica VI, Uyy E, Ivan L, Antohe F. Hyperlipidemia stimulates the extracellular release of the nuclear high mobility group box 1 protein. Cell Tissue Res. 2011;346:361–368. doi: 10.1007/s00441-011-1277-4. [DOI] [PubMed] [Google Scholar]
- 28.Prasad K. Do statins have a role in reduction/prevention of post-PCI restenosis. Cardiovasc Ther. 2013;31:12–26. doi: 10.1111/j.1755-5922.2011.00302.x. [DOI] [PubMed] [Google Scholar]
- 29.Du X, Hu X, Wei J. Postconditioning with rosuvastatin reduces myocardial ischemia-reperfusion injury by inhibiting high mobility group box 1 protein expression. Exp Ther Med. 2014;7:117–120. doi: 10.3892/etm.2013.1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ke D, Fang J, Fan L, Chen Z, Chen L. Regulatory T cells contribute to rosuvastatin-induced cardioprotection against ischemia-reperfusion injury. Coron Artery Dis. 2013;24:334–341. doi: 10.1097/MCA.0b013e3283608c12. [DOI] [PubMed] [Google Scholar]
- 31.Shen Ma, Liu DW, Pan J, Yu J, Shi L, Deng W, Zhu L, Yang L, Liu FJ, et al. Statin function as an anti-inflammation therapy for depression in patients with coronary artery disease by downregulating interleukin-1beta. J Cardiovasc Pharmacol. 2016;67:129–135. doi: 10.1097/FJC.0000000000000323. [DOI] [PubMed] [Google Scholar]
- 32.Greque GV, Serrano CV, Jr, Strunz CM, Soeiro A, Santos M, Pivateli F, Jacob JL, Pesaro AE, Nicolau JC, Kalil-Filho R. Preprocedural statin therapy, inflammation, and myocardial injury in low-risk stable coronary artery disease patients submitted to coronary stent implantation. Catheter Cardiovasc Interv. 2016;87:222–229. doi: 10.1002/ccd.24937. [DOI] [PubMed] [Google Scholar]
- 33.Bonsu KO, Reidpath DD, Kadirvelu A. Effects of statin treatment on inflammation and cardiac function in heart failure: An adjusted indirect comparison meta-analysis of randomized trials. Cardiovasc Ther. 2015;33:338–346. doi: 10.1111/1755-5922.12150. [DOI] [PubMed] [Google Scholar]
- 34.Fei L, Zhang J, Niu H, Yuan C, Ma X. Effects of rosuvastatin and MiR-126 on myocardial injury induced by acute myocardial infarction in rats: Role of vascular endothelial growth factor A (VEGF-A) Med Sci Monit. 2016;22:2324–2334. doi: 10.12659/MSM.896983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim YH, Park SM, Kim M, Kim SH, Lim SY, Lim SY, Ahn JC, Song WH, Shim WJ. Cardioprotective effects of rosuvastatin and carvedilol on delayed cardiotoxicity of doxorubicin in rats. Toxicol Mech Methods. 2012;22:488–498. doi: 10.3109/15376516.2012.678406. [DOI] [PubMed] [Google Scholar]
- 36.Xu L, Zang P, Feng B, Qian Q. Atorvastatin inhibits the expression of RAGE induced by advanced glycation end products on aortas in healthy Sprague-Dawley rats. Diabetol Metab Syndr. 2014;6:102. doi: 10.1186/1758-5996-6-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin D, Wu Y, Zhao L, Guo J, Zhang K, Chen Z. Atorvastatin reduces serum HMGB1 levels in patients with hyperlipidemia. Exp Ther Med. 2012;4:1124–1126. doi: 10.3892/etm.2012.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang H, Wang H, Wang Y, Addorisio M, Li J, Postiglione MJ, Chavan SS, Al-Abed Y, Antoine DJ, Andersson U, et al. The haptoglobin beta subunit sequesters HMGB1 toxicity in sterile and infectious inflammation. J Intern Med. 2017;282:76–93. doi: 10.1111/joim.12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao XJ, Qu YY, Liu XW, Zhu M, Ma CY, Jiao YL, Cui B, Chen ZJ, Zhao YR. Immune complexes induce TNF-alpha and BAFF production from U937 cells by HMGB1 and RAGE. Eur Rev Med Pharmacol Sci. 2017;21:1810–1819. [PubMed] [Google Scholar]
- 40.Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger CJ, Arnold B, Nawroth P, Andersson U, Harris RA, Harris HE. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61:1–9. doi: 10.1111/j.0300-9475.2005.01534.x. [DOI] [PubMed] [Google Scholar]
- 41.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luan ZG, Zhang H, Yang PT, Ma XC, Zhang C, Guo RX. HMGB1 activates nuclear factor-kappaB signaling by RAGE and increases the production of TNF-alpha in human umbilical vein endothelial cells. Immunobiology. 2010;215:956–962. doi: 10.1016/j.imbio.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 43.Dumitriu IE, Baruah P, Bianchi ME, Manfredi AA, Rovere-Querini P. Requirement of HMGB1 and RAGE for the maturation of human plasmacytoid dendritic cells. Eur J Immunol. 2005;35:2184–2190. doi: 10.1002/eji.200526066. [DOI] [PubMed] [Google Scholar]
- 44.Saidi H, Bras M, Formaglio P, Melki MT, Charbit B, Herbeuval JP, Gougeon ML. HMGB1 is involved in IFN-alpha production and TRAIL expression by HIV-1-exposed plasmacytoid dendritic cells: Impact of the crosstalk with NK cells. PLoS Pathog. 2016;12:e1005407. doi: 10.1371/journal.ppat.1005407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sirois CM, Jin T, Miller AL, Bertheloot D, Nakamura H, Horvath GL, Mian A, Jiang J, Schrum J, Bossaller L, et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J Exp Med. 2013;210:2447–2463. doi: 10.1084/jem.20120201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang LF, Yao YM, Zhang LT, Dong N, Yu Y, Sheng ZY. The effect of high-mobility group box 1 protein on activity of regulatory T cells after thermal injury in rats. Shock. 2009;31:322–329. doi: 10.1097/SHK.0b013e3181834070. [DOI] [PubMed] [Google Scholar]
- 47.Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP, Arnold B, Bianchi ME, Manfredi AA, Rovere-Querini P. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol. 2005;174:7506–7515. doi: 10.4049/jimmunol.174.12.7506. [DOI] [PubMed] [Google Scholar]
- 48.Liu J, Wang H, Li J. Inflammation and inflammatory cells in myocardial infarction and reperfusion injury: A double-edged sword. Clin Med Insights Cardiol. 2016;10:79–84. doi: 10.4137/CMC.S33164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu B, Su Z, Lin R, Dai R, Chen C, Wu H. Short-time pretreatment of rosuvastatin attenuates myocardial ischemia and reperfusion injury by inhibiting high mobility group box 1 protein expression. Int J Cardiol. 2013;168:4946–4948. doi: 10.1016/j.ijcard.2013.07.074. [DOI] [PubMed] [Google Scholar]
- 50.Hu X, Xu C, Zhou X, Cui B, Lu Z, Jiang H. PI3K/Akt signaling pathway involved in cardioprotection of preconditioning with high mobility group box 1 protein during myocardial ischemia and reperfusion. Int J Cardiol. 2011;150:222–223. doi: 10.1016/j.ijcard.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 51.Ding HS, Yang J, Gong FL, Yang J, Ding JW, Li S, Jiang YR. High mobility group [corrected] box 1 mediates neutrophil recruitment in myocardial ischemia-reperfusion injury through toll like receptor 4-related pathway. Gene. 2012;509:149–153. doi: 10.1016/j.gene.2012.07.072. [DOI] [PubMed] [Google Scholar]
- 52.Wang JQ, Wu GR, Wang Z, Dai XP, Li XR. Long-term clinical outcomes of statin use for chronic heart failure: A meta-analysis of 15 prospective studies. Heart Lung Circ. 2014;23:105–113. doi: 10.1016/j.hlc.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 53.Henninger C, Huelsenbeck S, Wenzel P, Brand M, Huelsenbeck J, Schad A, Fritz G. Chronic heart damage following doxorubicin treatment is alleviated by lovastatin. Pharmacol Res. 2015;91:47–56. doi: 10.1016/j.phrs.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 54.Mansouri E, Jangaran A, Ashtari A. Protective effect of pravastatin on doxorubicin-induced hepatotoxicity. Bratisl Lek Listy. 2017;118:273–277. doi: 10.4149/BLL_2017_054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.