

Abstract
Clear cell renal cell carcinoma (ccRCC) is the most frequent type of renal cell carcinoma (RCC). The present study aimed to examine prognostic markers and construct a prognostic prediction system for ccRCC. The mRNA sequencing data of ccRCC was downloaded from The Cancer Genome Atlas (TCGA) database, and the GSE40435 dataset was obtained from the Gene Expression Omnibus database. Using the Limma package, the differentially expressed genes (DEGs) in the TCGA dataset and GSE40435 dataset were obtained, respectively, and the overlapped DEGs were selected. Subsequently, Cox regression analysis was applied for screening prognosis-associated genes. Following visualization of the co-expression network using Cytoscape software, the network modules were examined using the GraphWeb tool. Functional annotation for genes in the network was performed using the clusterProfiler package. Finally, a prognostic prediction system was constructed through Bayes discriminant analysis and confirmed with the GSE29609 validation dataset. The results revealed a total of 263 overlapped DEGs and 161 prognosis-associated genes. Following construction of the co-expression network, 16 functional terms and three pathways were obtained for genes in the network. In addition, red, yellow (Involving chemokine ligand 10 (CXCL10), CD27 molecule (CD27) and runt-related transcription factor 3 (RUNX3)], green (Involving angiopoietin-like 4 (ANGPTL4), stannio-calcin 2 (STC2), and sperm associated antigen 4 (SPAG4)], and cyan modules were extracted from the co-expression network. Additionally, the prognostic prediction system involving 44 signature genes, including ANGPTL4, STC2, CXCL10, SPAG4, CD27, matrix metalloproteinase (MMP9) and RUNX3, was identified and confirmed. In conclusion, the 44-gene prognostic prediction system involving ANGPTL4, STC2, CXCL10, SPAG4, CD27, MMP9 and RUNX3 may be utilized for predicting the prognosis of patients with ccRCC.
Keywords: clear cell renal cell carcinoma, co-expression network, module analysis, signature genes, prognosis
Introduction
Renal cell carcinoma (RCC), which is derived from the lining of the proximal convoluted tubule, represents 90-95% of cases of kidney cancer (1). The early symptoms of RCC are usually undetected, which leads to advanced disease stages in patients newly diagnosed with RCC (2). With the progression of RCC, tumor cells may metastasize to other organs, including the liver, lymph nodes, lungs, brain, adrenal glands and bones (3). RCC has a relatively higher incidence in men than women, particularly in those >65 years old (4). Clear cell RCC (ccRCC), characterized by the clear cytoplasm in cells, is the most frequent type of RCC (5). Therefore, examining the pathogenesis of ccRCC is necessary and of significance.
There are several studies reporting the molecular mechanisms of ccRCC. For example, metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is upregulated in patients with ccRCC, which may be utilized to predict overall survival (OS) and serve as a promising therapeutic target for the disease (6,7). Hypoxia-inducible factor-1α, which is involved in tumoral adaptation to hypoxic conditions, can be a critical prognostic factor in metastatic ccRCC (8). The overexpression of cannabinoid receptor 2 is important in the cell cycle, cellular proliferation and migration of RCC cells, and predicts poor outcomes for patients with RCC (9). Previous studies have demonstrated that X-linked inhibitor of apoptosis protein is a potential prognostic marker in RCC, and that its downregulation may weaken immune resistance (10,11). Carbonic anhydrase 9 belongs to the carbonic anhydrase family, and its low expression is correlated with the poor prognosis of patients with ccRCC (12,13). However, these studies are insufficient and the prognostic mechanisms of ccRCC remain to be fully elucidated. In addition, the majority of the aforementioned results obtained from these microarray data were not validated by other datasets.
Bioinformatics analysis has been increasingly applied for revealing the genetic changes in the high-throughput data of tumors (14,15). In the present study, comprehensive bioinformatics analyses for gene expression data were performed to construct a prognostic prediction system for ccRCC with specific signature genes. Additionally, these predicted genes were validated by another dataset. The aim of the present study was to provide additional, and reliable, prognostic markers for patients with ccRCC, and shed light on the molecular mechanisms of disease progression.
Materials and methods
Data source and data preprocessing
From The Cancer Genome Atlas (TCGA; https://cancergenome.nih.gov/) database, the mRNA sequencing data of ccRCC, which was sequenced on the Illumina HiSeq 2000 RNA Sequencing platform, were downloaded with relevant clinical information on December 18th, 2016. There were a total of 606 samples, and 605 of these had information on sample source (normal or tumor tissues), including 533 ccRCC and 72 normal samples. The expression profile dataset GSE40435 (GPL10558 platform) in the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) database was also obtained, which included 101 ccRCC and 101 normal samples. According to the annotation platform, probes in the raw data of GSE40435 were converted into gene symbols. If multiple probes matched one gene, the average expression value of the gene was obtained. Subsequently, the expression data underwent logarithmic transformation using the R package Limma (http://www.bioconductor.org/packages/release/bioc/html/limma.html) (16), to reach an approximately normal distribution from the skewed distribution. Thereafter, the median normalization method (17) was utilized to normalize the data.
Analysis of differentially expressed genes (DEGs)
The TCGA data was combined with the 19,004 protein coding gene annotation information in the HUGO Gene Nomenclature Committee (HGNC) database (18), and mRNAs in the TCGA dataset were identified. The differential expression analysis of genes or mRNAs was then performed between ccRCC and normal samples in the TCGA dataset and GSE40435 dataset separately, using the R package Limma (16). Subsequently, the false discovery rate (FDR) values were calculated by the R package multtest (http://www.bioconductor.org/packages/release/bioc/html/multtest.html) (19). An FDR <0.05 and |log2fold change (FC)|>0.585 were the thresholds for selection of the DEGs. In addition, the overlapped DEGs in the TCGA and GSE40435 datasets were identified for the following analyses.
Screening of prognosis-associated genes
From the TCGA dataset, 596 samples (525 ccRCC samples and 71 normal samples) that matched with survival information (survival time and survival status) were identified. Subsequently, the 525 ccRCC samples were used for screening prognosis-associated genes. Cox regression analysis in the survival package (20) was applied for selecting prognosis-associated genes, and the log-rank test (21) was then utilized to calculate significant p-values. The top six genes were selected according to −logRank (P-value), following which Kaplan-Meier (KM) survival curves (22) were produced for the six genes.
Co-expression network analysis for significant prognostic genes and functional annotation
The expression values were extracted from TCGA database for the prognosis-associated genes, following which the correlation coefficients between the expression values of two genes were calculated using the COR function (23) in R. P<0.05 and a correlation coefficient |r|≥0.6 were used for selecting gene co-expression interactions. Subsequently, the co-expression network was visualized using Cytoscape software 3.4.0 (http://www.cyto-scape.org/) (24).
Using Fisher’s exact test method in the R package clusterProfiler (http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html) (25), the significant Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were enriched for the gene nodes in the co-expression network. In addition, the transcription factors (TFs) significantly associated with the nodes in the co-expression network were searched using The Database for Annotation, Visualization and Integrated Discovery (DAVID; https://david.ncifcrf.gov/) (26). Additionally, sub modules in the co-expression network were examined using the GraphWeb tool (http://biit.cs.ut.ee/graphweb/) (27).
Construction of the prognostic prediction system
The 525 ccRCC samples with survival information in the TCGA dataset were considered as the training dataset for the prognostic prediction system. First, the above samples were classified into two groups based on their survival status: Alive and deceased groups. Subsequently, combining survival status with survival times, the samples were further divided into good prognosis (alive and survival time ≥15 months) and bad prognosis (succumbed to mortality and survival time <15 months) groups. As the median OS time was ~15 months for all the ccRCC samples, the cut-off value for grouping was set as 15 months. The prior probability based on the Bayesian approach was then determined. The nodes of the co-expression network were ranked (−logRank P-values from largest to smallest). Using the discriminant Bayes function in the e1071 package of R (https://cran.r-project.org/web/packages/e1071/index.html) (28). Bayes discriminant analysis was performed for the network nodes through adding genes one by one and removing the genes that influenced prediction accuracy.
The prognostic score was defined as the discriminant coefficient of each sample when the prediction accuracy was the highest, and the gene set was considered as signature genes. The prediction system under the highest prediction accuracy was defined as the prognostic prediction system.
Validation of the prognostic prediction system
To examine the predictive effect of the constructed prediction system, KM survival analysis (22) was performed for the TCGA dataset to assess the association between the classification results of the prognostic prediction system and the real survival time and survival status. In addition, GSE29609 in the GEO data-base was obtained as the validation dataset, which included 39 ccRCC samples (32 samples with survival information). The expression values of the aforementioned signature genes were extracted from the GSE29609 dataset, and the prognostic scores of the 32 samples were obtained based on the prognostic prediction system. Subsequently, the 32 samples were divided into good prognosis and bad prognosis groups according to the aforementioned criteria. In addition, the correlations between the classification results of the prognostic prediction system and the actual survival time and survival status were detected by KM survival analysis (22).
Results
DEG analysis
Based on the HGNC database, the expression values of 18,531 protein-coding mRNAs were obtained from the TCGA dataset. A total of 12,669 mRNAs remained following removal of the mRNAs with low-abundance expression. There were numerous mRNAs expressed at low levels prior to filtering, but the peak of expression density was markedly elevated following filtering (Fig. 1).
Figure 1.
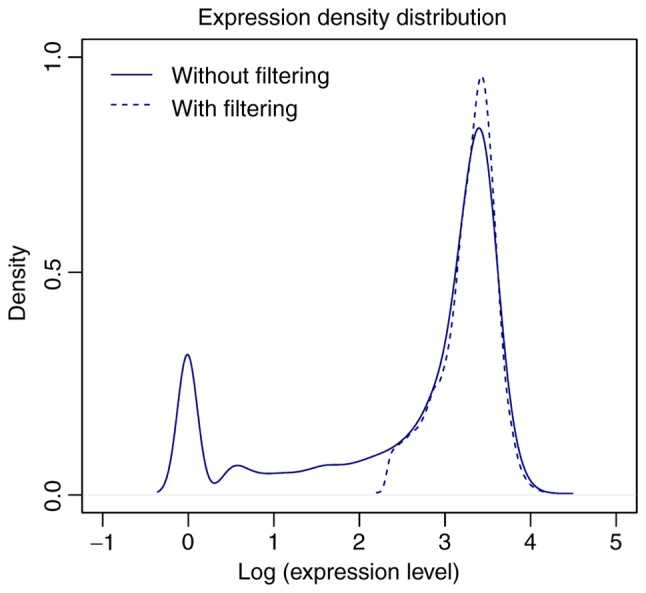
Expression density distributions of the mRNAs prior to (solid line) and following (dotted line) filtering.
Through differential expression analysis, a total of 621 and 2,764 DEGs were identified in the TCGA dataset and GSE40435 dataset, respectively. Among them, 263 overlapping DEGs in both the TCGA dataset and GSE40435 dataset were selected.
According to the logFC values, the top 50 DEGs (top 25 upregulated and top 25 downregulated) in the TCGA dataset (Fig. 2A) and GSE40435 dataset (Fig. 2B), respectively, were selected and subjected to bidirectional hierarchical clustering analysis. The heatmap showed that it was possible to divide the samples into two groups by the above DEGs.
Figure 2.

Bidirectional hierarchical clustering heatmaps for the top 50 genes differentially expressed between clear cell renal cell carcinoma and normal samples in (A) The Cancer Genome Atlas dataset and (B) GSE40435 dataset. The blue and yellow colors in the sample strip represent normal samples and tumor samples, respectively.
Screening of prognosis-associated genes
A total of 161 prognosis-associated genes were identified using Cox regression analysis from the TCGA dataset. According to −logRank (P-value), the top six genes [transcription factor AP-2a (TFAP2A); family with sequence similarity 167, member A (FAM167A); interleukin 20 receptor b (IL20RB); leucine-rich repeat LGI family, member 4 (LGI4); sodium channel, nonvoltage-gated 1b (SCNN1B); and solute carrier family 17, member 2 (SLC17A2)] were selected. The KM survival curves were generated (Fig. 3). It was shown that low expression of these genes was linked to high survival time, indicating that these six genes may be used as predictors for prognosis.
Figure 3.

Kaplan-Meier survival curves for the top six prognosis-associated genes of (A) TFAP2A, (B) FAM167A, (C) IL20RB, (D) LGI4, (E) SCNN1B, and (F) SLC17A2. Red and black lines represent samples with high expression and low expression, respectively. HR, hazard ratio; TFAP2A, transcription factor AP-2α; FAM167A, family with sequence similarity 167, member A; IL20RB, interleukin 20 receptor β; LGI4, leucine-rich repeat LGI family, member 4; SCNN1B, sodium channel, nonvoltage-gated 1β; SLC17A2, solute carrier family 17, member 2.
Co-expression network analysis and functional annotation
Following the selection of gene co-expression interactions for the prognosis-associated genes, the co-expression network (involving 141 nodes and 1,937 edges) was constructed (Fig. 4).
Figure 4.

Co-expression network of the prognosis-associated genes. The circles indicate red, yellow, green and cyan modules. The annotations represent the most significantly enriched terms for each module. Red and grey lines represent negative and positive correlation coefficients, respectively. Inverted and regular triangles represent downregulated and upregulated genes, respectively.
In addition, four significant network modules were identified, including a red module, a yellow module, involving chemokine ligand 10 (CXCL10), CD27 molecule (CD27), and runt-related transcription factor 3 (RUNX3), a green module, involving angiopoietin-like 4 (ANGPTL4), stanniocalcin 2 (STC2), and sperm associated antigen 4 (SPAG4), and a cyan module. Nervous system development, immune system process, regulation of secretion, and NAD metabolic process were the most significantly enriched terms for genes in the red, yellow, green and cyan modules, respectively (Fig. 4).
By performing functional and pathway enrichment analysis, a total of 16 GO terms, including immune response, three KEGG pathways, including cytokine-cytokine receptor interaction, and 11 TFs, including nuclear factor-kB, were obtained for genes in the network nodes (Fig. 5A and B).
Figure 5.

Functional and pathway enrichment analysis. (A) Functional terms and pathways enriched for the network nodes. (B) Transcription factors targeting the network nodes.
Construction and validation of prognostic prediction system
The 525 ccRCC samples in the TCGA dataset were divided into good prognosis (202 samples) and bad prognosis (323 samples) groups. Through a series of processes (Fig. 6A), the prognostic prediction system containing 44 signature genes, including ANGPTL4, STC2, CXCL10, SPAG4, CD27, matrix metallo-peptidase 9 (MMP9), and RUNX3, were constructed (Table I). The 44-gene prediction system had the highest prognostic accuracy for the patients with ccRCC.
Figure 6.

Construction and validation of prognostic prediction system. (A) Construction processes of the prognostic prediction system based on the TCGA dataset. (B) Prognostic scores of the samples in the good and bad prognosis groups. Kaplan-Meier survival curves for validating the prognostic prediction system based on the (C) TCGA dataset and (D) GSE29609 dataset. Blue and green lines represent good and bad prognosis, respectively. TCGA, The Cancer Genome Atlas.
Table I.
Information of the 44 signature genes in The Cancer Genome Atlas dataset.
| Gene | logFC | P-value | FDR |
|---|---|---|---|
| C10orf99 | −2.344 | 6.88×10−64 | 3.23×10−61 |
| ADAMDEC1 | −1.896 | 1.81×10−55 | 6.03×10−53 |
| HS3ST2 | −1.641 | 2.44×10−48 | 5.72×10−46 |
| IL20RB | −1.514 | 3.55×10−47 | 7.49×10−45 |
| SLC17A2 | −1.182 | 7.10×10−28 | 5.23×10−26 |
| LAG3 | −0.863 | 6.59×10−22 | 3.17×10−20 |
| SPAG4 | −0.856 | 1.76×10−29 | 1.43×10−27 |
| CD27 | −0.820 | 2.44×10−22 | 1.24×10−20 |
| SH2D1A | −0.804 | 1.55×10−18 | 5.34×10−17 |
| STC2 | −0.802 | 7.73×10−32 | 7.47×10−30 |
| MMP9 | −0.797 | 5.24×10−22 | 2.54×10−20 |
| ANGPTL4 | −0.786 | 5.94×10−37 | 7.60×10−35 |
| OSCAR | −0.753 | 2.63×10−17 | 7.89×10−16 |
| HHLA2 | −0.742 | 1.70×10−24 | 9.79×10−23 |
| RASD2 | −0.734 | 6.34×10−20 | 2.43×10−18 |
| NKG7 | −0.728 | 2.15×10−21 | 9.80×10−20 |
| INHBB | −0.723 | 1.73×10−24 | 9.92×10−23 |
| CD96 | −0.718 | 1.58×10−18 | 5.42×10−17 |
| NOD2 | −0.687 | 2.47×10−14 | 5.39×10−13 |
| P2RX7 | −0.674 | 1.82×10−16 | 5.02×10−15 |
| PGF | −0.655 | 7.44×10−22 | 3.54×10−20 |
| RASAL3 | −0.627 | 5.19×10−16 | 1.36×10−14 |
| ST8SIA4 | −0.627 | 2.64×10−19 | 9.66×10−18 |
| CDCA7 | −0.624 | 1.16×10−11 | 1.96×10−10 |
| CHSY3 | −0.624 | 3.61×10−13 | 7.12×10−12 |
| CXCL10 | −0.622 | 4.34×10−17 | 1.27×10−15 |
| CD6 | −0.622 | 1.38×10−14 | 3.09×10−13 |
| RUNX3 | −0.615 | 1.35×10−16 | 3.79×10−15 |
| SLC1A3 | −0.609 | 6.02×10−16 | 1.56×10−14 |
| CXCL9 | −0.609 | 2.13×10−18 | 7.18×10−17 |
| TRIM9 | −0.605 | 5.80×10−15 | 1.36×10−13 |
| SEMA5B | −0.599 | 3.69×10−20 | 1.47×10−18 |
| PDGFRA | 0.605 | 9.58×10−21 | 4.11×10−19 |
| ADH1B | 0.667 | 8.24×10−26 | 5.35×10−24 |
| G6PC | 0.693 | 3.07×10−21 | 1.37×10−19 |
| KRT7 | 0.718 | 3.93×10−29 | 3.09×10−27 |
| TMEM30B | 0.754 | 1.11×10−29 | 9.13×10−28 |
| FAM167A | 0.911 | 8.99×10−37 | 1.13×10−34 |
| TFAP2A | 0.948 | 4.13×10−36 | 5.13×10−34 |
| SLC13A3 | 1.009 | 9.21×10−49 | 2.29×10−46 |
| TMEM45B | 1.036 | 1.43×10−47 | 3.12×10−45 |
| C1orf116 | 1.062 | 5.48×10−47 | 1.14×10−44 |
| SCNN1B | 1.404 | 2.16×10−85 | 2.74×10−82 |
| ATP6V1B1 | 1.456 | 1.36×10−92 | 1.91×10−89 |
FC, fold change; FDR, false discovery rate.
The prognostic scores of the samples varied between −1.5 and 1.5 (good prognosis group between −1.5 and 0; bad prognosis group between 0 and 1.5; Fig. 6B). The discriminant scoring system of the prognostic prediction system was as follows:
Prognostic score = (Bayes discriminant) = [0-1.5, bad; −1.5-0, good], where represents the prognostic score, and i means gene.
To assess the effect of the prediction system, KM survival analysis was performed in the TCGA dataset first. The result showed that the survival ratio of the good prognosis group was significantly higher than that of the bad prognosis group (P=7.008×10−15; Fig. 6C).
In addition, in the GSE29609 validation dataset, the survival ratio of the good prognosis group was also significantly higher that of the bad prognosis group (P=8.46×10−8; Fig. 6D). These results suggested that the prognostic prediction system was able to accurately and practically classify ccRCC samples according to their prognosis.
Discussion
Previously, several studies have utilized microarray analysis to identify prognostic genes for ccRCC. By genome-wide expression analyses of the expression profiles of patients with primary ccRCC with different disease-free survival rates, platelet and endothelial cell adhesion molecule 1, endothelin receptor type B and tetraspanin 7 have been considered as prognostic markers potentially involved in tumor metastases (29). Based on array-comparative genomic hybridization, the genetic clustering of ccRCC was considered as a potential prognostic indicator in patients with RCC that is closely associated with DNA methylation alteration (30). However, these studies are insufficient for prognostic marker identification in ccRCC. In the present study, there were a total of 263 DEGs overlapped in the TCGA dataset and GSE40435 dataset, and 161 of these were associated with prognosis. Enrichment analysis showed that they were correlated with three KEGG pathways, including the cytokine-cytokine receptor interaction pathway. In addition, four significant network modules (red, yellow, green, and cyan modules) were identified from the co-expression network. Notably, ANGPTL4, STC2 and SPAG4 were present in the green module; whereas CXCL10, CD27 and RUNX3 were present in the yellow module. Through a series of bioinformatics methods, a prognostic prediction system was established comprising 44 signature genes, including ANGPTL4, STC2, CXCL10, SPAG4, CD27, MMP9, and RUNX3, and its prediction accuracy was confirmed.
ANGPTL4, which is a member of the angiopoietin/ANGPTL family, has a high expression and can be used as a diagnostic marker in primary ccRCC (31,32). It is suggested that an increased serum level of ANGPTL4 may function as a promising diagnostic and prognostic marker for patients with RCC (33). The overexpression of STC2 is involved in the metastasis of RCC and may be an indicator for the shorter OS of patients with RCC (34,35). In addition, multilevel whole-genome analysis has revealed that STC2 is one of the genes hypomethylated in copy number gains in ccRCC (36). SPAG4 is upregulated in human RCC and has influences on the growth and invasion capability of tumor cells (37,38). The mRNA expression of SPAG4 is negatively correlated with tumor stage, grade and size, suggesting that SPAG4 can act as a marker for the diagnosis and prognosis of RCC (39). SPAG4 contributes to the survival of cancer cells via suppressing hypoxia-induced tetraploid formation, and thus SPAG4 can independently predict poor prognosis in RCC (40). In the present study, ANGPTL4, STC2 and SPAG4 were all involved in the green module. Therefore, ANGPTL4, STC2 and SPAG4 may function in ccRCC through their co-expression, making them the prognostic factors for ccRCC.
CXCL10 inhibits tumor growth in RCC via restraining angiogenesis and decreasing the expression levels of vascular endothelial growth factor, platelet derived growth factor, fibroblast growth factor, and MMP9 (41). In addition, the interferon-inducible CXCR3 ligands score based on expression levels of CXCL9, CXCL10 and CXCL11, is suggested to be linked with different risk subgroups of recurrence and mortality in patients with ccRCC (42). CD27+ lymphocyte infiltration and the overexpression of CD70 are correlated with poor prognosis in ccRCC, and an elevated serum level of CD27 may be used for anti-CD70 therapy by predicting CD70-expressing ccRCC (43,44). RUNX3 is closely associated with RCC progression, and its high expression can significantly suppress the migration, invasion and angiogenesis in RCC (45-47). RUNX3 inhibits RCC migration and invasion through mediating the microRNA-6780a-5p/ E-cadherin/epithelial-mesenchymal transition signaling pathway, therefore, RUNX3 serves as a potential prognostic factor of RCC (48). In terms of the association of this gene with ccRCC, RUNX3 is decreased in ccRCC tissues, and it functions as an inhibitor of ccRCC cell growth and metastasis via regulating cyclins and tissue inhibitors of matrix metalloproteinase 1 (TIMP1) (49). In the present study, CXCL10, CD27 and RUNX3 were all involved in the yellow module, indicating that these co-expressed genes may also be associated with the prognosis of patients with ccRCC.
MMPs and their inhibitors (TIMPs) have an important function in the maintenance of extracellular matrix homeostasis. In RCC, the mRNA or protein expression of MMP2, MMP3, MMP9, TIMP1 and TIMP2 are relevant to the clinicopathological parameters (50). MMP9 correlates with high metastasis and poor survival rates in RCC, indicating that MMP9 may be utilized for predicting the disease-free survival rates of patients with RCC (51,52). The Notch ligand d-like 4 (DLL4) is tied up with tumor invasion and metastasis. It has been found that DLL4 facilitates RCC cell migration and invasion via upregulating MMP2 and MMP9 (53). It is known that, in the majority of ccRCC cases, inaction of the von Hippel-Lindau (VHL) tumor suppressor gene is an important hallmark, and the protein isoform of VHL coordinately regulates the metastasis-associated genes CXCR4/CXCL12 and MMP2/MMP9 (54). MMP9 has been selected as one of the 10 important genes in the protein-protein interaction network that associates with the progression of ccRCC (55). In the present study, this gene was one of the 44 signature genes predicting ccRCC prognosis, suggesting it may be used as a prognostic factor for patients with ccRCC.
Previously, a study reported that cytokine-cytokine receptor interaction was the most significant pathway for DEGs in RCC tissue (56). In the present study, this pathway was also significantly enriched for DEGs identified in ccRCC samples, suggesting that these crucial DEGs may function through the regulation of this pathway.
The predictive accuracy of the 44-signature gene-prognostic prediction system was confirmed by the validation dataset (GSE29609), indicating this system may be applied for the prognosis of patients with ccRCC. Although comprehensive bioinformatics analysis was performed, and hundreds of samples were used in the present study, a limitation remained that the validation dataset had a relatively small sample size, and thus the results require experimental validation, particularly the co-expression of genes identified in the same module.
In conclusion, the 44-gene prognostic prediction system, involving ANGPTL4, STC2, CXCL10, SPAG4, CD27, MMP9, and RUNX3, may be important in predicting the prognosis of patients with ccRCC. However, these key genes and the 44-gene prognostic prediction system require further validation by experimental investigations.
Funding
No funding was received.
Availability of data and materials
The datasets used during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
YoW and YaW were involved in the design of this study and performed the statistical analysis. YaW collected important background information. FL drafted the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Not applicable.
References
- 1.Cohen HT, Mcgovern FJ. Renalcell carcinoma. N Engl Med. 2005;353:2477–2490. doi: 10.1056/NEJMra043172. [DOI] [PubMed] [Google Scholar]
- 2.Jonasch E, Gao J, Rathmell WK. Renal cell carcinoma. BMJ. 2014;349:g4797. doi: 10.1136/bmj.g4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ljungberg B, Cowan NC, Hanbury DC, Hora M, Kuczyk MA, Merseburger AS, Patard JJ, Mulders PF, Sinescu IC, European Association of Urology Guideline Group EAU guidelines on renal cell carcinoma: the 2010 update. Eur Urol. 2010;58:398–406. doi: 10.1016/j.eururo.2010.06.032. [DOI] [PubMed] [Google Scholar]
- 4.Znaor A, Lortet-Tieulent J, Laversanne M, Jemal A, Bray F. International variations and trends in renal cell carcinoma incidence and mortality. Eur Urol. 2015;67:519–530. doi: 10.1016/j.eururo.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Young JR, Margolis D, Sauk S, Pantuck AJ, Sayre J, Raman SS. Clear cell renal cell carcinoma: Discrimination from other renal cell carcinoma subtypes and oncocytoma at multi-phasic multidetector CT. Radiology. 2013;267:444–453. doi: 10.1148/radiol.13112617. [DOI] [PubMed] [Google Scholar]
- 6.Zhang HM, Yang FQ, Chen SJ, Che J, Zheng JH. Upregulation of long non-coding RNA MALAT1 correlates with tumor progression and poor prognosis in clear cell renal cell carcinoma. Tumour Biol. 2015;36:2947–2955. doi: 10.1007/s13277-014-2925-6. [DOI] [PubMed] [Google Scholar]
- 7.Hirata H, Hinoda Y, Shahryari V, Deng G, Nakajima K, Tabatabai ZL, Ishii N, Dahiya R. Long noncoding RNA MALAT1 promotes aggressive renal cell carcinoma through Ezh2 and interacts with miR-205. Cancer Res. 2015;75:1322–1331. doi: 10.1158/0008-5472.CAN-14-2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klatte T, Seligson DB, Riggs SB, Leppert JT, Berkman MK, Kleid MD, Yu H, Kabbinavar FF, Pantuck AJ, Belldegrun AS. Hypoxia-inducible factor 1α in clear cell renal cell carcinoma. Clin Cancer Res. 2007;13:7388–7393. doi: 10.1158/1078-0432.CCR-07-0411. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Xu Y, Zhu L, Zou Y, Kong W, Dong B, Huang J, Chen Y, Xue W, Huang Y, Zhang J. Cannabinoid receptor 2 as a novel target for promotion of renal cell carcinoma prognosis and progression. J Cancer Res Clin Oncol. 2018;144:39–52. doi: 10.1007/s00432-017-2527-y. [DOI] [PubMed] [Google Scholar]
- 10.Mizutani Y, Nakanishi H, Li YN, Matsubara H, Yamamoto K, Sato N, Shiraishi T, Nakamura T, Mikami K, Okihara K, et al. Overexpression of XIAP expression in renal cell carcinoma predicts a worse prognosis. Int J Oncol. 2007;30:919–925. [PubMed] [Google Scholar]
- 11.Yamada T, Horinaka M, Shinnoh M, Yoshioka T, Miki T, Sakai T. A novel HDAC inhibitor OBP-801 and a PI3K inhibitor LY294002 synergistically induce apoptosis via the suppression of survivin and XIAP in renal cell carcinoma. Int J Oncol. 2013;43:1080–1086. doi: 10.3892/ijo.2013.2042. [DOI] [PubMed] [Google Scholar]
- 12.Tostain J, Li G, Gentilperret A, Gigante M. Carbonic anhydrase 9 in clear cell renal cell carcinoma: A marker for diagnosis, prognosis and treatment. Eur J Cancer. 2010;46:3141–3148. doi: 10.1016/j.ejca.2010.07.020. [DOI] [PubMed] [Google Scholar]
- 13.Choueiri TK, Regan MM, Rosenberg JE, Oh WK, Clement J, Amato AM, McDermott D, Cho DC, Atkins MB, Signoretti S. Carbonic anhydrase IX and pathological features as predictors of outcome in patients with metastatic clear-cell renal cell carcinoma receiving vascular endothelial growth factor-targeted therapy. Bju Int. 2010;106:772–778. doi: 10.1111/j.1464-410X.2010.09218.x. [DOI] [PubMed] [Google Scholar]
- 14.Yang W, Huang P, Zhao M, Lau YL. International Conference on Biomedical Engineering and Informatics. Sanya; Hainan, China: 2008. Biomarker identification for early tumor detection aided by bioinformatics gene expression analysis; pp. 469–473. [Google Scholar]
- 15.Fonseca AL, da Silva VL, da Fonsêca MM, Meira ITJ, da Silva TE, Kroll JE, Ribeiro-dos-Santos AM, Freitas CR, Furtado R, de Souza JE, et al. Bioinformatics analysis of the human surfaceome reveals new targets for a variety of tumor types. Int J Genomics. 20162016:1–7. doi: 10.1155/2016/8346198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smyth GK. Limma: Linear Models for Microarray Data. Springer; New York: 2005. [Google Scholar]
- 17.Wang Y, Zeigler MM, Lam GK, Hunter MG, Eubank TD, Khramtsov VV, Tridandapani S, Sen CK, Marsh CB. The role of the NADPH oxidase complex, p38 MAPK, and Akt in regulating human monocyte/macrophage survival. Am J Respir Cell Mol Biol. 2007;36:68–77. doi: 10.1165/rcmb.2006-0165OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruford EA, Lush MJ, Wright MW, Sneddon TP, Sue P, Ewan B. The HGNC Database in 2008: A resource for the human genome. Nucleic Acids Res. 2008;36:445–448. doi: 10.1093/nar/gkm881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pollard KS, Dudoit S, van der Laan MJ. Multiple testing procedures: The multtest package and applications to genomics. In: Gentleman R, Carey VJ, Huber W, Irizarry RA, Du ST, editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer; New York: 2005. pp. 465–465. [Google Scholar]
- 20.Therneau TM. Survival analysis. R package survival, version 2. 2016:39–5. [Google Scholar]
- 21.Kleinbaum DG, Klein M. Statistics for Biology and Health. Springer; New York: 2005. Kaplan-meier survival curves and the log-rank test; pp. 45–82. [Google Scholar]
- 22.Porcher R. CORR Insights(®): Kaplan-meier survival analysis overestimates the risk of revision arthroplasty: A meta-analysis. Clin Orthop Relat Res. 2015;473:3443–3445. doi: 10.1007/s11999-015-4291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nowicka-Zagrajek J, Weron R. COR: MATLAB function to compute the correlation coefficients. Hsc software. 2008 [Google Scholar]
- 24.Kohl M, Wiese S, Warscheid B. Cytoscape: Software for visualization and analysis of biological networks. Methods Mol Biol. 2011;696:291–303. doi: 10.1007/978-1-60761-987-1_18. [DOI] [PubMed] [Google Scholar]
- 25.Yu G, Wang LG, Han Y, He QY. Cluster profiler: An R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, Lempicki RA. DAVID bioinformatics resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:169–175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reimand J, Tooming L, Peterson H, Adler P, Vilo J. GraphWeb: Mining heterogeneous biological networks for gene modules with functional significance. Nucleic Acids Res. 2008;36:452–459. doi: 10.1093/nar/gkn230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dimitriadou E, Hornik K, Leisch F, Meyer D, Weingesse A. The e1071 package. Ethnos J Anthropol. 2006;23:55–56. [Google Scholar]
- 29.Wuttig D, Zastrow S, Füssel S, Toma MI, Meinhardt M, Kalman K, Junker K, Sanjmyatav J, Boll K, Hackermüller J, et al. CD31, EDNRB and TSPAN7 are promising prognostic markers in clear-cell renal cell carcinoma revealed by genomewide expression analyses of primary tumors and metastases. Int J Cancer. 2012;131:E693–E704. doi: 10.1002/ijc.27419. [DOI] [PubMed] [Google Scholar]
- 30.Arai E, Ushijima S, Tsuda H, Fujimoto H, Hosoda F, Shibata T, Kondo T, Imoto I, Inazawa J, Hirohashi S, Kanai Y. Genetic clustering of clear cell renal cell carcinoma based on array-comparative genomic hybridization: Its association with DNA methylation alteration and patient outcome. Clin Cancer Res. 2008;14:5531–5539. doi: 10.1158/1078-0432.CCR-08-0443. [DOI] [PubMed] [Google Scholar]
- 31.Verine J, Lehmann-Che J, Soliman H, Feugeas JP, Vidal JS, Mongiat-Artus P, Belhadj S, Philippe J, Lesage M, Wittmer E, et al. Determination of angptl4 mRNA as a diagnostic marker of primary and metastatic clear cell renal-cell carcinoma. PLoS One. 2010;5:e10421. doi: 10.1371/journal.pone.0010421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galaup A, Cazes A, Le Jan S, Philippe J, Connault E, Le Coz E, Mekid H, Mir LM, Opolon P, Corvol P, et al. Angiopoietin-like 4 prevents metastasis through inhibition of vascular permeability and tumor cell motility and invasiveness. Proc Natl Acad Sci USA. 2006;103:18721–18726. doi: 10.1073/pnas.0609025103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dong D, Jia L, Zhou Y, Ren L, Li J, Zhang J. Serum level of ANGPTL4 as a potential biomarker in renal cell carcinoma. Urol Oncol. 2017;35:279–285. doi: 10.1016/j.urolonc.2016.12.017. [DOI] [PubMed] [Google Scholar]
- 34.Xin M, Gu L, Li H, Gao Y, Li X, Shen D, Gong H, Li S, Niu S, Zhang Y, et al. Hypoxia-induced overexpression of stannio-calcin-1 is associated with the metastasis of early stage clear cell renal cell carcinoma. J Transl Med. 2015;13:56. doi: 10.1186/s12967-015-0421-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyer HA, Tölle A, Jung M, Fritzsche FR, Haendler B, Kristiansen I, Gaspert A, Johannsen M, Jung K, Kristiansen G. Identification of stanniocalcin 2 as prognostic marker in renal cell carcinoma. Eur Urol. 2009;55:669–678. doi: 10.1016/j.eururo.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 36.Girgis AH, Iakovlev VV, Beheshti B, Bayani J, Squire JA, Bui A, Mankaruos M, Youssef Y, Khalil B, Khella H, et al. Multilevel whole-genome analysis reveals candidate biomarkers in clear cell renal cell carcinoma. Cancer Res. 2012;72:5273–5284. doi: 10.1158/0008-5472.CAN-12-0656. [DOI] [PubMed] [Google Scholar]
- 37.Knaup KX, Monti J, Hackenbeck T, Jobst-Schwan T, Klanke B, Schietke RE, Wacker I, Behrens J, Amann K, Eckardt KU, et al. Hypoxia regulates the sperm associated antigen 4 (SPAG4) via HIF, which is expressed in renal clear cell carcinoma and promotes migration and invasion in vitro. Mol Carcinog. 2014;53:970–978. doi: 10.1002/mc.22065. [DOI] [PubMed] [Google Scholar]
- 38.Kennedy C, Sebire K, de Kretser DM, O’Bryan MK. Human sperm associated antigen 4 (SPAG4) is a potential cancer marker. Cell Tissue Res. 2004;315:279–283. doi: 10.1007/s00441-003-0821-2. [DOI] [PubMed] [Google Scholar]
- 39.Shiraishi T, Terada N, Zeng Y, Mooney S, Takahashi S, Takaha N, Miki T, Getzenberg R, Kulkarni P. 433 sperm associated antigen 4 is a novel biomarker for renal cell carcinoma. J Urol. 2012;187:e177-e178. doi: 10.1016/j.juro.2012.02.500. [DOI] [Google Scholar]
- 40.Shoji K, Murayama T, Mimura I, Wada T, Kume H, Goto A, Ohse T, Tanaka T, Inagi R, van der Hoorn FA, et al. Sperm-associated antigen 4, a novel hypoxia-inducible factor 1 target, regulates cytokinesis, and its expression correlates with the prognosis of renal cell carcinoma. Am J Pathol. 2013;182:2191–2203. doi: 10.1016/j.ajpath.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 41.Jin HA, Heo JH, Kang YH, Kim KH, Han KS, Hong SJ. Abstract B18: CXCL10 suppresses tumor angiogenesis and impedes expression of critical angiogenic factors in renal cell carcinoma. Cancer Res. 2016;76:B18. doi: 10.1158/1538-7445.TME16-B18. [DOI] [Google Scholar]
- 42.Liu W, Liu Y, Qiang F, Zhou L, Chang Y, Xu L, Zhang W, Xu Ji. Elevated expression of IFN-inducible CXCR3 ligands predicts poor prognosis in patients with non-metastatic clear-cell renal cell carcinoma. Oncotarget. 2016;7:13976. doi: 10.18632/oncotarget.7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruf M, Moch H, Schraml P. Interaction of tumor cells with infiltrating lymphocytes via CD70 and CD27 in clear cell renal cell carcinoma. Oncoimmunology. 2015;4:e1049805. doi: 10.1080/2162402X.2015.1049805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruf M, Mittmann C, Nowicka AM, Hartmann A, Hermanns T, Poyet C, van den Broek M, Sulser T, Moch H, Schraml P. pVHL/HIF-regulated CD70 expression is associated with infiltration of CD27+ lymphocytes and increased serum levels of soluble CD27 in clear cell renal cell carcinoma. Clin Cancer Res. 2015;21:889–898. doi: 10.1158/1078-0432.CCR-14-1425. [DOI] [PubMed] [Google Scholar]
- 45.Chen F, Bai J, Li W, Mei P, Liu H, Li L, Pan Z, Wu Y, Zheng J. RUNX3 suppresses migration, invasion and angiogenesis of human renal cell carcinoma. PLoS One. 2013;8:e56241. doi: 10.1371/journal.pone.0056241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang G, Qin W, Zheng J, Wei M, Zhou X, Wang H, Wen W. Expressions of EZH2 and RUNX3 in renal cell carcinoma and their clinical significance. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2013;29:82–84. In Chinese. [PubMed] [Google Scholar]
- 47.Parmar KM, Singla M, Mandal AK, Bhattacharya S, Singh SK. The expression of RUNX3 gene in renal cell cancer and its clinical relevance with serum vascular endothelial growth factor. Int J Mol Immunooncol. 2017;2:73. doi: 10.18203/issn.2456-3994.IntJMolImmunoOncol20172646. [DOI] [Google Scholar]
- 48.Chen F, Liu X, Cheng Q, Zhu S, Bai J, Zheng J. RUNX3 regulates renal cell carcinoma metastasis via targeting miR-6780a5p/E-cadherin/EMT signaling axis. Oncotarget. 2016;8:101042–101056. doi: 10.18632/oncotarget.13205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao He, Wang XL, Zhang H, Guo P, Huang C, Liu C, Yao X, Chen F, Lou YW, et al. RUNX3 mediates suppression of tumor growth and metastasis of human CCRCC by regulating cyclin related proteins and TIMP-1. PLoS One. 2012;7:e32961. doi: 10.1371/journal.pone.0032961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhuvarahamurthy V, Kristiansen GO, Johannsen M, Loening SA, Schnorr D, Jung K, Staack A. In situ gene expression and localization of metalloproteinases MMP1, MMP2, MMP3, MMP9, and their inhibitors TIMP1 and TIMP2 in human renal cell carcinoma. Oncol Rep. 2006;15:1379–1384. [PubMed] [Google Scholar]
- 51.Cho NH, Shim HS, Rha SY, Kang SH, Hong SH, Choi YD, Hong SJ, Cho SH. Increased expression of matrix metallo-proteinase 9 correlates with poor prognostic variables in renal cell carcinoma. Eur Urol. 2003;44:560–566. doi: 10.1016/S0302-2838(03)00362-2. [DOI] [PubMed] [Google Scholar]
- 52.Sato A, Nagase H, Obinata D, Fujiwara K, Fukuda N, Soma M, Yamaguchi K, Kawata N, Takahashi S. Inhibition of MMP-9 using a pyrrole-imidazole polyamide reduces cell invasion in renal cell carcinoma. Int J Oncol. 2013;43:1441–1446. doi: 10.3892/ijo.2013.2073. [DOI] [PubMed] [Google Scholar]
- 53.Huang QB, Ma X, Li HZ, Ai Q, Liu SW, Zhang Y, Gao Y, Fan Y, Ni D, Wang BJ, Zhang X. Endothelial Delta-like 4 (DLL4) promotes renal cell carcinoma hematogenous metastasis. Oncotarget. 2014;5:3066–3075. doi: 10.18632/oncotarget.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Struckmann K, Mertz K, Steu S, Storz M, Staller P, Krek W, Schraml P, Moch H. pVHL co-ordinately regulates CXCR4/CXCL12 and MMP2/MMP9 expression in human clear-cell renal cell carcinoma. J Pathol. 2008;214:464–471. doi: 10.1002/path.2310. [DOI] [PubMed] [Google Scholar]
- 55.Yuan L, Zeng G, Chen L, Wang G, Wang X, Cao X, Lu M, Liu X, Qian G, Xiao Y, Wang X. Identification of key genes and pathways in human clear cell renal cell carcinoma (ccRCC) by co-expression analysis. Int J Biol Sci. 2018;14:266–279. doi: 10.7150/ijbs.23574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feng JY, Diao XW, Fan MQ, Wang PX, Xiao Y, Zhong X, Wu RH, Huang CB. Screening of feature genes of the renal cell carcinoma with DNA microarray. Eur Rev Med Pharmacol Sci. 2013;17:2994–3001. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used during the current study are available from the corresponding author on reasonable request.