
Figure 1.
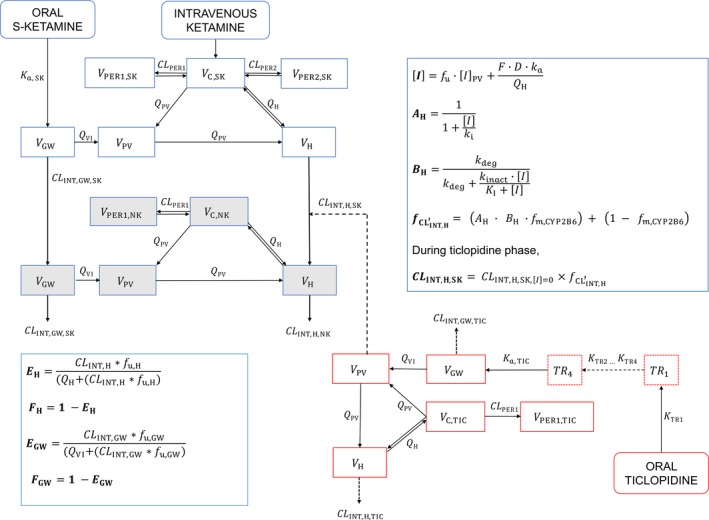
A diagrammatic representation of the final semiphysiologically based pharmacokinetic model that includes S‐ketamine model, norketamine model, ticlopidine model, and a dynamic drug‐drug interaction model. A H, reversible component of CYP2B6 inhibition; B H, the non‐reversible component of CYP2B6 inhibition; C, central compartment; CLINT, intrinsic clearance; CYP, cytochrome P450; D, amount of inhibitor (ticlopidine) administered; F, bioavailability of ticlopidine into the first depot compartment; , inhibition parameter signifying hepatic CYP2B6 degradation; f m,CYP2B6, fraction of S‐ketamine metabolized by CYP2B6; f u, fraction unbound of ticlopidine in the blood; GW, gut wall; H, hepatic; [I], ticlopidine concentration at the enzyme site; [I]PV, ticlopidine concentration at the portal vein; k a, absorption rate constant; k deg, the physiological degradation rate of CYP2B6 at inhibitor concentration of [I] = 0; K I, degradation rate of CYP2B6 at inhibitor concentration of [I]; k i, the dissociation constant of ticlopidine for CYP2B6; k inact, the maximal inhibition rate achieved at inhibitor concentration [I] = ∞; NK, norketamine; PER1, first peripheral compartment; PER2, second peripheral compartment; PV, portal vein; Q, blood flow; SK, S‐ketamine; TIC, ticlopidine; V, volume; VI, gut wall villous mucosa.