

Here, Aubrey et al. researched how mutant TRP53 drives tumor development. Their findings demonstrate a strong functional synergy between deregulated expression of c-MYC and the hot spot mutant TRP53 proteins that distinguishes them from the less frequently occurring TRP53 mutants and that mutant TRP53 drives tumorigenesis primarily through dominant-negative effects.
Keywords: TRP53, dominant-negative effect, tumorigenesis, TRP53 target genes
Abstract
Mutations in Trp53, prevalent in human cancer, are reported to drive tumorigenesis through dominant-negative effects (DNEs) over wild-type TRP53 function as well as neomorphic gain-of-function (GOF) activity. We show that five TRP53 mutants do not accelerate lymphomagenesis on a TRP53-deficient background but strongly synergize with c-MYC overexpression in a manner that distinguishes the hot spot Trp53 mutations. RNA sequencing revealed that the mutant TRP53 DNE does not globally repress wild-type TRP53 function but disproportionately impacts a subset of wild-type TRP53 target genes. Accordingly, TRP53 mutant proteins impair pathways for DNA repair, proliferation, and metabolism in premalignant cells. This reveals that, in our studies of lymphomagenesis, mutant TRP53 drives tumorigenesis primarily through the DNE, which modulates wild-type TRP53 function in a manner advantageous for neoplastic transformation.
TRP53 gene mutations are found in many human cancers and are typically associated with poor patient outcomes (Vogelstein et al. 2000; Vousden and Lane 2007). Over 2000 different TRP53 mutations have been reported in human cancer (Bouaoun et al. 2016); however, they occur at a disproportionately high rate at six specific amino acid “hot spot” residues (R175, G245, R248, R249, R273, and R282) and result in a limited number of specific single amino acid substitutions. The mechanism underlying the strong selection for hot spot mutant TRP53 proteins in human cancer remains unknown (Baugh et al. 2018). Hot spot mutant TRP53 proteins have lost wild-type TRP53 transcriptional function but can still bind and inhibit the function of the wild-type TRP53 protein expressed from the nonmutated allele (dominant-negative effect [DNE]). They are also thought to exhibit neomorphic gain-of-function (GOF) effects involving interactions with diverse transcriptional regulators (Farmer et al. 1992; Freed-Pastor and Prives 2012). Through these mechanisms, mutant TRP53 protein overexpression has been reported to influence cancer development and response to therapy (Freed-Pastor and Prives 2012; Muller and Vousden 2014).
The DNE is thought to rely on the prolonged half-life of mutant TRP53 (Freed-Pastor and Prives 2012) and the formation of mixed tetramers with the wild-type TRP53 protein (Milner et al. 1991; Farmer et al. 1992; Sturzbecher et al. 1992; Jeffrey et al. 1995) that impair normal transcriptional control of wild-type TRP53 target genes (Willis et al. 2004). Mutant TRP53 DNE and GOF are supported by findings in the Li-Fraumeni cancer predisposition syndrome (Li and Fraumeni 1969), which typically results from the inheritance of a germline TRP53 mutation (Malkin et al. 1990; Srivastava et al. 1990), where patients with a hot spot TRP53 mutation, such as R248Q, develop cancer at a younger age compared with those with a TRP53-null mutation (Bougeard et al. 2008). Mutant TRP53 GOF is also supported by the observation that mice with homozygous germline Trp53 gene missense mutations (murine: R172H and R270H) develop a different spectrum of tumors with increased metastasis compared with mice lacking TRP53 (Trp53−/−) (Lang et al. 2004; Olive et al. 2004).
Notably, TRP53 mutations frequently co-occur with deregulated c-MYC proto-oncogene expression across a range of human cancer types (Ulz et al. 2016), including Burkitt lymphoma (Gaidano et al. 1991; Giulino-Roth et al. 2012; Love et al. 2012). These combined genetic alterations are associated with highly aggressive disease and poor patient outcomes. Accordingly, 20%–30% of lymphomas arising in Eμ-Myc mice, which overexpress the c-Myc oncogene under control of the IgH gene enhancer (Adams et al. 1985), acquire Trp53 mutations (Eischen et al. 1999; Schmitt et al. 1999). Altogether, these observations suggest an important functional interplay between mutant TRP53 proteins and deregulated c-MYC expression during tumorigenesis.
We conducted a systematic evaluation of five different mutant TRP53 proteins across three contrasting tumor development models, including a model of c-MYC overexpression, to interrogate the relative importance of the DNE and GOF effect of mutant TRP53 during cancer development. This has highlighted the importance of the DNE during the early stages of MYC-driven lymphoma development and uncovered previously unrecognized features of the DNE.
Results
To investigate the relative contributions from the loss-of-function effect, DNE, and GOF effect of mutant TRP53 during tumor development (Fig. 1A), we assessed the impact of five different TRP53 mutations in three different tumor-prone settings: Trp53−/−, Trp53+/−, and Eµ-Myc/Trp53+/+. Each Trp53 mutation that we studied had been observed to arise spontaneously in Eµ-Myc lymphomas, including two hot spot Trp53 mutations (R246Q and R270H), two less common mutations (V170M and I192S), and one rare mutation (insG280) (Fig. 1B; Kelly et al. 2014). The full-length mouse Trp53 cDNA sequences were cloned into a constitutive retroviral expression vector (pMIG) (Supplemental Fig. S1A). Expression of each mutant TRP53 protein and the ability to form multimeric complexes were confirmed (Supplemental Fig. S1B,C). The impact of all five TRP53 mutants on tumor development was evaluated using hematopoietic stem/progenitor cell (HSPC) reconstitution models (Fig. 1A). The Trp53−/− setting was anticipated to reveal only GOF effects because the wild-type TRP53 protein is absent. The Trp53+/− condition was anticipated to be sensitive to both the DNE and GOF effect. The Eμ-Myc;Trp53+/+ setting provides an assessment of the interplay between mutant TRP53, both DNE and GOF, with c-MYC overexpression.
Figure 1.

Mutant TRP53 proteins do not accelerate lymphoma development in the Trp53−/− and Trp53+/− genetic backgrounds. (A) HSPC reconstitution model to examine the impact of mutant TRP53 protein expression on tumor development. (B) The mutant TRP53 proteins studied, with the corresponding human amino acid position indicated. (*) Hot spot mutation. (C–F) Kaplan-Meier survival curves for mice reconstituted with Trp53−/− (C,D) or Trp53+/− (E,F) HSPCs transduced with empty vector (pMIG) or expression vectors for each of the five mutant TRP53 proteins that were tested. (n) Number of mice. C and E represent composite survival curves of all TRP53 mutants (N = 5) combined. P-values were determined by log rank (Mantel-Cox) test. (G) Tumor phenotype summary for mutant TRP53 transduced Trp53+/− HSPC reconstitution experiments (from E and F). The tumor spectra for the individual TRP53 mutants are shown in Supplemental Figure S1G.
Transduction of Trp53−/− HSPCs with mutant TRP53 expression vectors and transplantation into lethally irradiated wild-type C57BL/6-Ly5.1 mice did not result in accelerated lymphoma development (Fig. 1C,D). Tumors from these mice rarely expressed the mutant TRP53 protein (7.5%; three out of 40) (Supplemental Fig. S1D), arguing against a strong selective advantage. Mice reconstituted with Trp53+/− HSPCs that had been transduced with mutant TRP53 proteins also displayed no acceleration in tumor development (Fig. 1E,F). However, expression of hot spot (R270H, R246Q, and, to a lesser extent, G280) mutant TRP53 proteins altered tumor spectrum with the emergence of myeloid neoplasms (38%) and lineage marker-negative tumors (14%) that were not observed in control mice (Fig. 1G; Supplemental Fig. S1F,G). Notably, 57% (16 out of 28) of these tumors expressed the mutant TRP53 protein (Supplemental Fig. S1E), demonstrating a selective advantage conferred by the mutant TRP53 proteins.
In striking contrast, mutant TRP53 markedly accelerated lymphoma development in the setting of c-MYC overexpression (i.e., mice reconstituted with mutant TRP53 transduced Eμ-Myc;Trp53+/+ HSPCs) (Fig. 2A). Remarkably, the two hot spot mutant TRP53 proteins had the most potent impact (Fig. 2A, red box). This suggests a distinctive functional interaction between the hot spot mutant TRP53 proteins and c-MYC expression; however, differences in immunogenicity underlying the observed differences between the mutant TRP53 proteins cannot be excluded. Notably, mutant TRP53 did not accelerate lymphoma development to the same extent as complete deletion of wild-type Trp53 using CRISPR/Cas9 technology (Fig. 2A, dotted line). This demonstrates that exogenous overexpression of a mutant TRP53 protein cannot recapitulate complete loss of TRP53 function and implies that, despite mutant TRP53 overexpression, the endogenous wild-type TRP53 protein retains the capacity for tumor suppression, consistent with findings from a lung adenocarcinoma model (Turrell et al. 2017). The immunophenotype of the mutant TRP53-accelerated MYC lymphomas was similar to that of control lymphomas (Supplemental Fig. S2A), and, while they displayed biological heterogeneity, all expressed readily detectable mutant TRP53 protein in their nuclei (Fig. 2B; Supplemental Fig. S2B).
Figure 2.
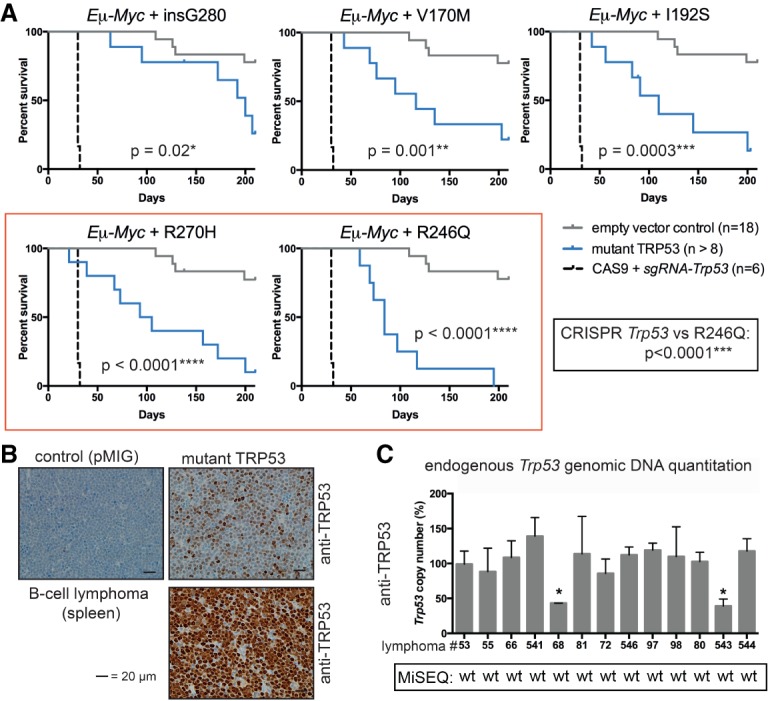
Overexpression of mutant TRP53 proteins accelerates lymphoma development in an Eμ-Myc;Trp53+/+ background and relieves selective pressure for mutation of endogenous Trp53 genes. (A) Kaplan-Meier survival curves for mice reconstituted with Eμ-Myc;Trp53+/+ HSPCs comparing empty vector control (pMIG), CRISPR/Cas9 Trp53 knockout, and each mutant TRP53 protein (V170M, I192S, G280, R246Q, and R270H). P-values were determined by log rank (Mantel-Cox) test. (B) Selected TRP53 protein immunohistochemistry in lymphomas from the Eμ-Myc hematopoietic reconstitution experiments (mice #88 and #541 plus control mouse #53). (C) Endogenous Trp53 allele copy number in lymphomas from the Eμ-Myc hematopoietic reconstitution experiments as determined by genomic DNA quantitative PCR (pMIG/control: #53; V170M: #55, #66, and #541; G280: #68 and #81; I192S: #72 and #546; R246Q: #97 and #98; R270H: #80, #543, and #544). Primary cells from Trp53−/− and Trp53+/− mice were used as controls. Data from MiSeq analysis throughout the coding region of the DNA-binding domain (exons 4–10) are indicated. (wt) Wild-type sequence. Data represent mean ± SEM. (*) P < 0.05, comparing lymphoma sample with wild-type control as determined by paired t-test.
Given that all mutant TRP53-accelerated lymphomas were derived from Eμ-Myc;Trp53+/+ HSPCs and that a high rate (20%–30%) of spontaneous Trp53 mutation occurs in this model (Eischen et al. 1999; Schmitt et al. 1999; Michalak et al. 2009), we assessed the genetic and functional status of the endogenous Trp53 alleles in these lymphomas. Most mutant TRP53-accelerated lymphomas retained both copies of the endogenous wild-type Trp53 gene (10 out of 12; 83%) (Fig. 2C) and, as such, showed a significant response to etoposide treatment (Supplemental Fig. S2C). Only two lymphomas (17%) had lost one allele of endogenous wild-type Trp53 and, during the derivation of cell lines, underwent selection for functional inactivation of the remaining wild-type Trp53 allele (Supplemental Fig. S2D), precluding them from further investigations. Importantly, no missense mutations of the endogenous Trp53 alleles were observed in any of the lymphomas that arose with retroviral expression of mutant TRP53. Based on these findings, we conclude that the exogenous mutant TRP53 protein had a powerful effect in reducing the selective pressure for spontaneous mutations in the endogenous wild-type Trp53 gene. We hypothesized that this may be due to either suppression of endogenous wild-type TRP53 function by the DNE or GOF effect of mutant TRP53 that may obviate selection for endogenous missense Trp53 mutation.
Mutant TRP53 proteins are considered to drive tumorigenesis by deregulating gene expression through their DNE and GOF effect (Freed-Pastor and Prives 2012). In order to examine the transcriptional effects of the mutant TRP53 proteins in a controlled setting, we expressed each of the five mutant TRP53 proteins in Eμ-Myc;Trp53+/+ lymphoma cell lines (Fig. 3A). The Trp53 wild-type status was confirmed by functional studies and sequencing (data not shown). Notably, in these lymphoma lines, but not in Eµ-Myc;Trp53−/− lymphoma cell lines, R246Q and R270H hot spot TRP53 mutant proteins were found to be expressed at lower levels compared with the other mutant TRP53 proteins examined (Fig. 3B) despite equivalent construct expression (Fig. 3C) and ability to reduce TRP53-dependent induction of apoptosis (Fig. 3D). Therefore, interestingly, the tumor-initiating potency was inversely correlated with mutant TRP53 protein level in cells that also expressed wild-type TRP53. To test whether the presence of wild-type TRP53 negatively impacts the expression of the hot spot mutant TRP53 proteins, we used CRISPR/Cas9 technology to inactivate the endogenous wild-type Trp53 gene in Trp53+/+;Eμ-Myc lymphoma cell lines prior to introducing mutant TRP53 protein expression. This resulted in substantial accumulation of the R246Q and R270H mutant TRP53 proteins (Supplemental Fig. S3A). The finding that loss of endogenous wild-type Trp53 permits higher-level expression of the hot spot TRP53 mutant proteins was corroborated in studies using primary mouse embryonic fibroblasts (Supplemental Fig. S3B). Strikingly, even in the absence of endogenous wild-type TRP53, MDM2 inhibition by nutlin-3a (Vassilev et al. 2004) resulted in a further increase of these TRP53 mutant proteins in Eµ-Myc lymphoma lines (Supplemental Fig. S3A). This confirms that MDM2 is a critical negative regulator of hot spot mutant TRP53 protein levels (Terzian et al. 2008) and that it can drive mutant TRP53 protein degradation even in the absence of wild-type TRP53.
Figure 3.
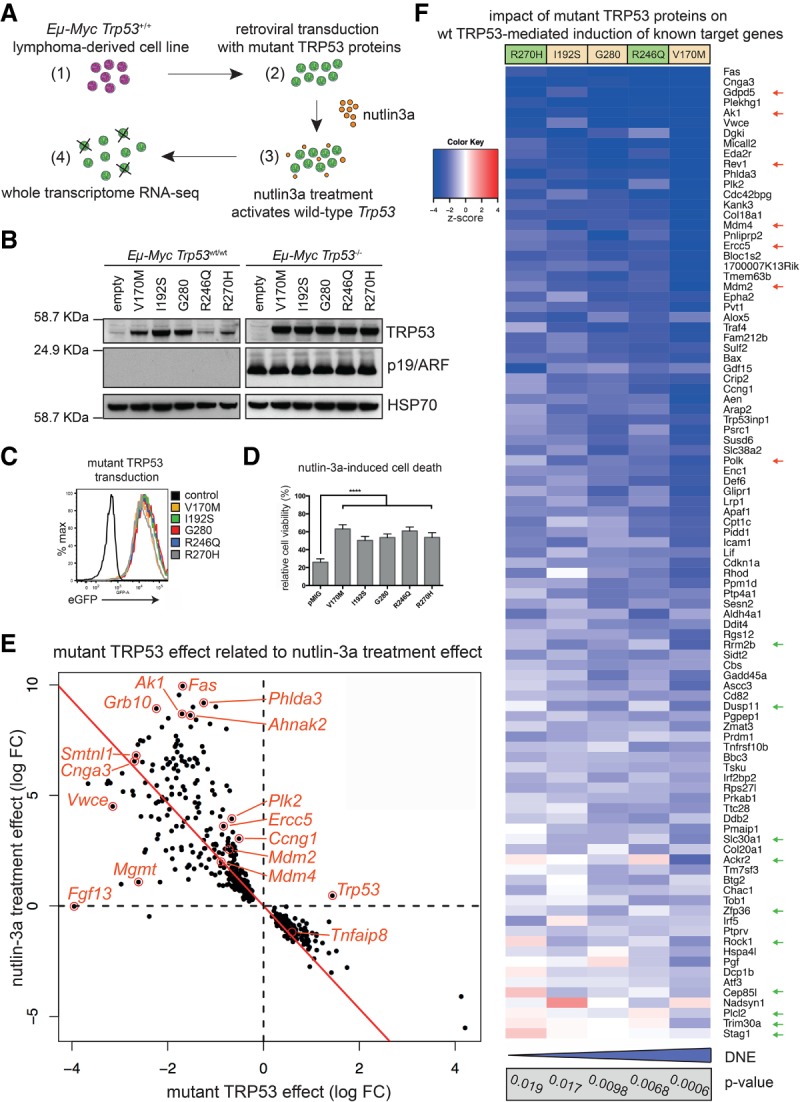
Mutant TRP53 proteins exert a target gene-selective DNE. (A) Experimental approach to examine the DNE exerted by the mutant TRP53 proteins in Eμ-Myc lymphoma-derived cell lines. (B) Analysis of mutant TRP53 transduced Eμ-Myc;Trp53+/+ and Eμ-Myc;Trp53−/− lymphoma-derived cell lines by Western blot with HSP70 as a loading control. (C) Uniform eGFP expression in the mutant TRP53 transduced cell lines shown in B, assessed by flow cytometry. (D) Mutant TRP53 proteins inhibit nutlin-3a-induced apoptosis. n = 5 different cell lines with two to four independent experiments for each cell line. Data represent mean ± SEM. Paired two-tailed t-test was performed comparing pMIG (empty vector control) with each mutant TRP53 individually. For each mutant, P < 0.0001 (****). (E) Differentially expressed genes in mutant TRP53 transduced Eμ-Myc lymphoma lines after treatment with nutlin-3a. The scatter plot shows log fold changes following nutlin-3a treatment versus mutant TRP53 effect log fold changes. The plot displays the 455 genes that were differentially expressed in the nutlin-3a-treated mutant TRP53 transduced samples (false discovery rate [FDR] < 0.1) versus the nutlin-3a treatment effect in pMIG (control) samples. The red line shows the least squares line with zero intercept. The mutant TRP53 effect shows a strong inverse correlation to the treatment effect. (F) Heat map depicting the impact of each mutant TRP53 protein on the induction of known wild-type TRP53 target genes after nutlin-3a treatment, color-coded by z-score (2: P < 0.05; 3: P < 0.003; 4: P < 0.0001). The P-value for individual mutant TRP53 proteins under the full TRP53 target gene set test is indicated (gray box) with the relative overall strength of the DNE indicated (blue bar). Gene expression distinguishing hot spot TRP53 mutations (green arrows) and relatively strongly repressed genes of interest are indicated (red arrows).
We next proceeded to globally characterize the transcriptional changes associated with mutant TRP53 expression after activation of the endogenous wild-type TRP53 protein with the MDM2 antagonist nutlin-3a using next-generation mRNA sequencing (RNA-seq). The RNA-seq data are illustrated according to treatment, cell line, and Trp53 mutation (Supplemental Fig. S3C). Treatment of control Eµ-Myc lymphoma cell lines with nutlin-3a altered the expression of >5000 genes (false discovery rate [FDR] < 0.05) (Supplemental Table S1). Uniform and modest (approximately fourfold) overexpression of each mutant Trp53 transcript was confirmed (Supplemental Fig. S3D, top panel). Differentially expressed genes were identified between mutant TRP53 transduced and control Eμ-Myc lymphoma cell lines following nutlin-3a treatment (n = 455 genes with FDR < 0.1) (Fig. 3E; Supplemental Table S2). Differentially expressed genes included some previously reported mutant TRP53 GOF targets (Dck, Wdr13, E2F1, and Hmgcs1) (Freed-Pastor and Prives 2012; Kollareddy et al. 2015). However, the predominant effect resulted from opposition of the nutlin-3a treatment effect observed in the control cell lines (Fig. 3E), consistent with the DNE of mutant TRP53 being the primary underlying mechanism for differential gene expression in this setting. Gene set signature analysis was performed using a list of known wild-type TRP53 target genes (n = 253 genes total; n = 185 assigned for analysis; n = 95 induced after nutlin-3a) (Supplemental Table S3). Nutlin-3a treatment caused significant induction of this gene set in the control cell lines (P = 3.94 × 10−8), and, overall, this gene set was significantly repressed in all mutant TRP53 transduced lines (P = 3 × 10−4), confirming the presence of an overall DNE. Surprisingly, however, 39% (37 out of 95 genes) of the wild-type TRP53-induced target genes (Supplemental Table S4, yellow) were not significantly repressed (P > 0.05) by mutant TRP53 (Supplemental Table S4, gray). For example, the key proapoptotic TRP53 target genes Puma/Bbc3 and Noxa/Pmaip1 (Oda et al. 2000; Jeffers et al. 2003; Villunger et al. 2003) showed comparable baseline expression and nutlin-3a-driven induction between mutant TRP53-expressing and control cell lines (Supplemental Fig. S3D). This is consistent with the ability of the mutant TRP53 proteins to accelerate lymphoma development because these targets are not critical mediators of wild-type TRP53 tumor suppressor function (Brady et al. 2011; Li et al. 2012; Valente et al. 2013; Janic et al. 2018). Furthermore, mutant TRP53 did not significantly repress induction of other known wild-type TRP53 target genes (e.g., Rock1, Btg2, Dusp11, Stag1, and Zmat3), whereas Mdm4, Fas, Ercc5, Mgmt, Rev1, Bax, Plk2, and Ak1 were significantly repressed (Supplemental Table S4, gray). Many strongly repressed genes function in DNA damage repair (e.g., Ercc5, Polk, Mgmt, and Rev1), which is critical for TRP53-dependent tumor suppression (Janic et al. 2018), negative feedback to TRP53 signaling (Fig. 3F, red arrows), or metabolic pathways (Supplemental Table S5). Quantitative RT–PCR (qRT–PCR) showed comparable expression of Rock1 in the presence or absence of mutant TRP53 following nutlin-3a treatment, while other targets, including Mdm2 and Ercc5, were strongly repressed by mutant TRP53 (Supplemental Fig. S3E).
Variation in the nature of the DNE was revealed when comparing differentially expressed genes for the distinct mutant TRP53 proteins individually (Fig. 3F; Supplemental Table S4). Individual P-values were determined for each mutant TRP53 protein in the wild-type TRP53 gene set test to indicate their respective DNE strength (Fig. 3F, gray box). The hot spot mutant TRP53 proteins could be distinguished from the less frequent TRP53 mutants by permitting higher-level expression of certain TRP53 target genes following nutlin-3a treatment, including Rock1, Ackr2, and Trim30a (Fig. 3F, green arrows). This suggests that preserved expression of select wild-type TRP53 target genes may be advantageous for neoplastic transformation and/or sustained tumor growth. Various effector functions of the wild-type TRP53 protein are required for homeostasis and adaption to cellular stress (Basak et al. 2008; Maddocks et al. 2013) such that an ideal mutant TRP53 dominant-negative protein would be predicted to preserve certain beneficial wild-type TRP53 functions while repressing its tumor suppressor function.
To functionally evaluate the consequences of the target gene-selective DNE exerted by mutant TRP53 during early tumor development, we analyzed preleukemic Eμ-Myc;Trp53+/+ B lymphoid cells expressing the two hot spot TRP53 mutants R246Q and R270H or a control vector. For this, mice were analyzed 4 wk after reconstitution, prior to the development of lymphoma (Supplemental Fig. S4A). All reconstituted mice displayed similar retroviral transduction efficiency in their leukocytes (Supplemental Fig. S4B). Mice reconstituted with mutant TRP53 transduced Eμ-Myc;Trp53+/+ HSPCs exhibited an expansion of pro-B/pre-B cells in their bone marrow (Fig. 4A), the tumor-initiating cells (i.e., cancer stem cells) in the Eμ-Myc lymphoma model (Langdon et al. 1986). Analysis of B lymphoid cells ex vivo showed that, consistent with the relative lack of repression of Puma and Noxa, mutant TRP53 expression did not affect spontaneous cell death (Supplemental Fig. S4C,D) or killing by several TRP53-dependent (nutlin-3a and γ-irradiation) or TRP53-independent (docetaxel) agents (Fig. 4B). In contrast, preleukemic B cells expressing hot spot mutant TRP53 showed an increase in mitochondrial number and function (Fig. 4C), consistent with a previous report (Wang et al. 2013); increased numbers of cells in the S/G2M phases of the cell cycle (Fig. 4D); and an increase in nuclear γH2AX foci (Fig. 4E,F). The latter indicates increased DNA damage, and this was supported by comet assays (Supplemental Fig. S4E). Of note, the preleukemic B lymphoid cells did not show high-level hot spot mutant TRP53 protein expression, although efficient vector transduction was confirmed by eGFP positivity (Supplemental Fig. S4F–H).
Figure 4.

The hot spot mutant TRP53 proteins R246Q and R270H selectively deregulate metabolic, cell proliferation, and DNA repair pathways in preleukemic cells. Mice were reconstituted with empty vector (pMIG) control or mutant TRP53 (R246Q and R270H) transduced Eμ-Myc;Tp53+/+ HSPCs, and their preleukemic cells were analyzed at 4 wk. (A) Bone marrow-derived pre-B/pro-B cells calculated from flow cytometry and total femur cell counts. Mutant TRP53 R246Q (n = 6) and R270H (n = 6) are compared with pMIG control (n = 5). P-values were determined by unpaired t-test. (B) Cell death after administration of 5 µM nutlin-3a or 0.5 µg/mL docetaxel or exposure to 2.5 Gy of γ-irradiation. Data represent mean ± SEM. Mutant TRP53 R246Q (n = 3) and R270H (n = 3) are compared with pMIG control (n = 3). P-values were determined by paired Student's t-test. (C) Mitochondria number and activity assessed in bone marrow-derived preleukemic pre-B/pro-B cells. Representative histogram for R246Q mutant TRP53 protein transduced preleukemic B lymphoid cells as compared with empty vector (pMIG) control transduced cells. Median fluorescence intensity summary data represent mean ± SEM. pMIG: n = 3; mutant TRP53: N = 2 mutations; n = 8 replicates. P-value was determined by unpaired t-test. (D) Cell cycle analysis of preleukemic B lymphoid cells using DAPI staining and the Watson pragmatic model for analysis. Data represent mean ± SEM. Mutant TRP53 R246Q (n = 6) and R270H (n = 6) are compared with pMIG control (n = 5). P-values were determined by unpaired t-test. (E) Representative images from confocal microscope. Images shown are deconvoluted maximum projection images using Fiji software. (Blue) DAPI for nucleus; (red) γH2AX foci. (F) Quantitation of γH2AX focus number per cell nucleus and γH2AX focus area per cell nucleus. Data represent mean ± SEM. Control: n = 4; mutant TRP53 (R246Q and R270H): n = 14. P-value was determined by unpaired t-test. (*) P < 0.05; (**) P < 0.01. (G,H) RNA-seq analysis of untreated (directly ex vivo) preleukemic Eµ-Myc B lymphoid cells expressing mutant TRP53 proteins (data for all five TRP53 mutants combined) compared with empty vector (pMIG) transduced control cells. (G) Log2 fold changes with differentially expressed genes highlighted (FDR < 0.05). (H) Barcode enrichment plot depicting down-regulation of the TRP53 gene set test (P-value by FRY test). (I) Model: Mutant TRP53 exerts a selective DNE that modulates wild-type TRP53 function.
Finally, RNA-seq was performed on preleukemic Eμ-Myc;Trp53+/+ B lymphoid cells expressing each of the five mutant TRP53 proteins or a pMIG control vector (Supplemental Table S6). Consistent with the DNE, this confirmed that mutant TRP53 proteins primarily repressed gene expression (Fig. 4G), and TRP53 gene set testing revealed repression of wild-type TRP53 target genes (Fig. 4H). Comparison of the DNE for each mutant TRP53 protein showed variation in strength and selectivity similar to that observed in Eµ-Myc lymphoma-derived cell lines (Supplemental Fig. S5A,B).
Discussion
This study systematically evaluated the biological effects of five different Trp53 mutations in three tumor settings: Trp53−/−, Trp53+/−, and Eµ-Myc/Trp53+/+. While there are limitations to the latter model (specifically, the Trp53mutant/+/+ genotype does not exactly mimic the Trp53 heterozygous state; i.e., one wild-type allele and one mutant Trp53 allele), it is impossible to use the Eμ-Myc;Trp53+/− background in these experiments due to rapid lymphoma development in these animals such that they cannot even be bred, thus also precluding the generation of Eμ-Myc;Trp53−/− mice. Given that TRP53 protein levels are primarily regulated at the post-translational level, we considered the Eµ-Myc;Trp53mutant/+/+ model to represent a meaningful and tractable system to study the impact of mutant TRP53 during the early stages of lymphoma development when wild-type TRP53 is still expressed and functional alongside mutant TRP53 in the cells undergoing neoplastic transformation. In our scenario, the ratio of wild-type TRP53 emanating from two wild-type Trp53 alleles to retrovirally expressed mutant TRP53 likely recapitulates the heterozygous state in which endogenous wild-type and mutant Trp53 alleles are at a 1:1 ratio.
The findings from our investigations highlighted the importance of the DNE of mutant TRP53 in driving lymphoma development, with the proposed GOF effect of mutant TRP53 playing only a minor role in our experimental systems. Interestingly, we report for the first time a marked synergy between MYC overexpression and mutant TRP53 proteins that functionally distinguishes the hot spot mutant TRP53 proteins from other TRP53 mutants. This has important implications for human cancer where MYC overexpression and mutant TRP53 frequently co-occur and are associated with dismal patient outcomes.
Our findings reveal that the DNE of mutant TRP53 is a primary process driving neoplastic transformation, such as in c-MYC-driven lymphoma development when wild-type Trp53 remains intact. This reflects the heterozygous Trp53 state during the early stages of tumorigenesis in humans before loss of heterozygosity (LOH) at the Trp53 locus (Freed-Pastor and Prives 2012) and in established mutant TRP53-driven cancers that have retained a copy of wild-type TRP53. Importantly, tumors arising in Li-Fraumeni patients that have undergone LOH at the TRP53 locus have usually been preceded by a long period of latency during which the normal wild-type TRP53 allele remains intact, and the impact of the DNE may therefore be of great clinical importance. The DNE of all TRP53 mutants demonstrated a remarkable wild-type TRP53 target gene selectivity by which certain processes were preferentially deregulated, particularly DNA damage repair, cell proliferation, metabolism, and negative feedback to wild-type TRP53 signaling.
Notably, the selectivity of the DNE permitted expression of a number of TRP53 target genes that are likely advantageous to tumor development, such as Rock1. In accordance with this hypothesis, expression of Rock1, which is not affected by the R270H and R246Q TRP53 mutant proteins (Fig. 3F), has been reported to be critical for neoplastic transformation (Kumper et al. 2016) and has been associated previously with mutant TRP53 (Zhang et al. 2013). This target gene preference of the DNE may explain the previous observation that heterozygous Trp53 mutation, unlike complete loss of TRP53, cannot rescue the embryonic lethality caused by Mdm2 gene knockout in mice (Terzian et al. 2008).
Remarkably, the hot spot R270H mutant TRP53 displayed the weakest DNE, permitting unrestrained expression of the largest number of wild-type TRP53 target genes, yet was among the most potent TRP53 mutants in accelerating lymphoma development. This challenges dogma and indicates that the target gene-selective and, consequently, effector process-selective nature of the DNE of mutant TRP53 modulates wild-type TRP53 function—rather than globally repressing its function—in a manner that may be advantageous for tumorigenesis and cancer growth (Fig. 4I). Many potential mechanisms could account for the observed target gene selectivity: These may include features of the mutant TRP53 and wild-type TRP53 protein–protein interaction, differences in DNA binding relating to sequence differences or chromatin structure, interactions with other transcription factors, and/or the gene-specific kinetics for activation of gene expression.
In conclusion, the DNE is the primary mediator of mutant TRP53-driven tumorigenesis. Early during neoplastic transformation, the DNE can preferentially deregulate certain pathways (metabolism, cell cycle, and DNA damage repair), while other pathways remain relatively unaffected (apoptosis), and that these effects occur prior to mutant TRP53 protein stabilization.
Materials and methods
Mice
All experiments with mice followed the guidelines of the Melbourne Directorate Animal Ethics Committee, according to the Walter and Eliza Hall Institute of Medical Research Ethics Committee. C57BL/6-Ly5.1 (wild type) were obtained from the Walter and Eliza Hall Institute's breeding facility (Kew, Victoria, Australia). The Eμ-Myc transgenic mice (Adams et al. 1985) and the Trp53−/− mice have been described (Jacks et al. 1994), and these strains were backcrossed to C57BL/6-Ly5.2 mice for 30 or 20 generations prior to use in the present experiments.
Fetal liver-derived HSPC reconstitutions
For hematopoietic reconstitutions, embryonic day 13.5 (E13.5) fetal liver cells (a rich source of HSPCs) were obtained from Trp53+/−, Trp53−/−, or Eμ-Myc;Trp53+/+ mice on a C57BL/6J-Ly5.2 background and cultured as described previously (Aubrey et al. 2015). Cells were infected with the constitutive expression mutant TRP53 retrovirus or an empty control vector (viral constructs are described in the Supplemental Material). Transduced cells were collected, washed in PBS, and injected intravenously into lethally irradiated (2 × 5.5 Gy) C57BL/6J-Ly5.1 mice. All transplanted mice were maintained on prophylactic antibiotics with neomycin (Sigma, N1876) for 4 wk following γ-irradiation and injection of HSPCs.
RNA isolation, cDNA synthesis, and qRT–PCR analysis
Samples for RNA collection were obtained in Trizol reagent (Invitrogen) for all quantitative real-time PCR experiments, and total RNA was purified according to the manufacturer's instruction. Total isolated RNA was reverse-transcribed using the SuperScript III first strand synthesis kit (Invitrogen) using random hexamer primers. cDNA synthesis was carried out according to the manufacturer's instructions. qRT–PCR reactions were performed in triplicate using the TaqMan gene expression assays (Applied Biosystems) for mRNA. Thermal cycling and data collection were performed using the ABI Prism 7900 real-time PCR system (Applied Biosystems) using the SDS software package (Applied Biosystems). Gene expression levels were standardized to the expression of the housekeeping gene Hmbs.
qPCR assay for Trp53 genomic DNA copy number and Trp53 exon sequencing
Genomic DNA was purified from samples using the DNeasy DNA extraction kit (Qiagen, 69506). For wild-type Trp53 copy number analysis, two genomic loci were amplified and quantitated using real-time PCR. Primer pairs were designed to exon 4 and exon 10 of the mouse genomic DNA Trp53 sequence, and primers were designed to target exonic and intronic regions of the DNA locus. The Rosa locus was used as a control to normalize values obtained for exon 4 and exon 10 of the Trp53 locus. Primer details are in the Supplemental Material. qPCR amplification was carried out directly from genomic DNA samples. For exon sequence analysis, indexing PCR primers spanning exons 4–10 were designed. Primer sequences will be provided on request. PCR fragments were sequenced using the MiSeq platform (Illumina).
Single-guide RNA (sgRNA) lentiviral constructs
The inducible lentiviral platform for CRISPR/Cas9 gene modification using doxycycline-inducible sgRNA expression and stable constitutive expression of CAS9 and the sgRNA sequences targeting exon 4 and exon 5 of the mouse Trp53 gene have been described previously (Aubrey et al. 2015).
Virus production and transduction of cell lines
Lentiviral and retroviral particles were generated, and target cells were infected by spin inoculation as described previously (Aubrey et al. 2015).
Flow cytometric analysis and cell sorting
Assessment of fluorescent protein expression in cell lines as well as immunophenotyping of Eμ-Myc lymphomas (staining with surface marker-specific monoclonal antibodies that had been conjugated to fluorochromes) were performed using an LSR IIW, LSR IIC, or FACSCalibur flow cytometer (Becton Dickinson) as described previously (Kelly et al. 2014; Aubrey et al. 2015).
Drug treatments and functional assays
To perform cell death assays, cells were plated at 5 × 104 cells per well of a 96-well plate and treated with drugs in tissue culture medium. Nutlin-3a stock solution in DMSO [Cayman chemical, 18585; synonym: (-)-nutlin-3], docetaxel solution (Sandoz), and 20 mg/mL Etoposide solution (Pfizer, 503289) were diluted in tissue culture medium to the concentrations indicated. At set time points, as indicated in each figure, the viability of cultured cells was determined by exclusion of propidium iodide (PI) as assessed by flow cytometry using an LSR-IIC flow cytometer (Becton Dickinson). PI-negative cells were considered viable, and data are expressed as a percentage of the viability of a matched untreated (vehicle-treated) control cell population.
Western blot analysis
Total protein extracts were prepared by lysis in RIPA buffer (50 mM Tris-HCL, 150 mM NaCl, 1% NP-40, 0.5% DOC, 0.1% SDS) supplemented with protease inhibitors (complete protease inhibitor cocktail; Roche), and Western blot analysis (using 10–25 µg of protein) was performed as described previously (Aubrey et al. 2015). The antibodies used included HSP70 (clone N-6; a gift from Dr. Robert Anderson, Peter MacCallum Cancer Centre), p19/ARF (Rockland, 5.C3.1), and TRP53 (Novocastra/Leica, CM5).
Blue native polyacrylamide gel electrophoresis (BN-PAGE)
The method for BN-PAGE was reviewed recently (Dewson 2015).
DNA damage analysis
The single-cell gel electrophoresis (comet assay) was used to measure DNA damage and was performed according to the manufacturer's instructions (The Trevigen Comet assay, 4250-050-K). To assess nuclear staining for γH2AX, preleukemic B lymphoid cells were stained with Alexa fluor 647 (AF647)-conjugated antibodies against phospho-histone H2A.X (Ser139; “γ-H2AX”; EMD Millipore, 05-636-AF647). Images were obtained using a DeltaVision microscope (GE LifeScience). Foci were measured in an automated fashion using a custom-written macro in Fiji (Schindelin et al. 2012) with the method detailed in the Supplemental Material.
Metabolic assay, cell cycle analysis, and RNA-seq on preleukemic cells
Preleukemic B lymphoid cells were harvested from the femora of mice at 4 wk after hematopoietic reconstitution. For the metabolic and cell cycle analyses, the cells were plated at 5 × 104 cells per well in a 96-well format with OP9 supportive cell layer and medium supplemented with IL-7 for 72 h. Mitochondrial activity in preleukemic B lymphoid cells was assessed using Mitotracker Orange CMTMRos (Invitrogen, M7510). Cells were stained at a final working concentration of 200 nM for 30 min at 37°C, and fluorescence was assessed by flow cytometry. For cell cycle analysis, whole bone marrow from femora of preleukemic mice was analyzed. Cells were first fixed and permeabilized using the BD Fixation and Permeabilization Solutions (554722) and then stained with antibodies against CD19 and B220 followed by counterstaining with DAPI. DAPI staining was analyzed using an LSR IIW flow cytometer, and cell cycle analysis in B lymphoid cells (B220+CD19+) was performed using FloJo software by the Watson Pragmatic model. For the RNA-seq analysis on preleukemic cells, B220+L5.2+GFP+ cells were isolated from the bone marrow by FACS, and RNA was extracted directly using the Trizol method. The mRNA libraries were prepared and the sequencing was performed as described below.
Library preparation for RNA-seq analysis
The experiments were performed in a 24-well format with 5 × 105 cells per well. After 12 h of treatment with 5 µM nutlin-3a in the presence of 25 µM broad-spectrum caspase inhibitor QVD-OPH (MP-Biomedicals, OPH-109) to prevent cell death, cells were collected in 1 mL of Trizol, and purification of mRNA was performed using the RNeasy (Qiagen) kit. The mRNA libraries were prepared using the Agilent Technologies SureSelect strand-specific RNA library preparation. Multiplexed sequencing was performed on an Illumina HiSeq 2000 at the Australian Genome Research Facility (AGRF) to produce between 7 million and 11 million 100-base-pair single-end reads per sample.
RNA-seq analysis
Detailed methods for RNA-seq analysis are in the Supplemental Material.
TRP53 immunohistochemistry
Immunohistochemistry was performed on paraffin-embedded tissue sections; this was provided as a service by the Walter and Eliza Hall Institute Histology Service. TRP53 staining (anti-TRP53 rabbit polyclonal; 1:500; Novocastra, CM5) was performed using the automated system Omnis (Dako). All immunohistochemistry images were obtained using the Eclipse 90i microscope (Nikon).
Statistical analysis
Graphpad Prism software was used for generating Kaplan-Meier animal survival curves and statistical analysis comparing animal survival curves by log rank test. Graphpad software was also used in the analysis of cell death assays. Paired and unpaired two-tailed t-tests were performed to compare samples where indicated, and a two-way ANOVA was performed to compare transcription induction of TRP53 target genes using qRT–PCR. Stata (version 12.1) software was used to calculate linear regression and adjusted r-squared.
Supplementary Material
Acknowledgments
We thank all members of the Molecular Genetics of Cancer Division at The Walter and Eliza Hall Institute (WEHI) for support and advice; S. Haupt, Y. Haupt, A. Kallies, S. Nutt, T. Johanson, P. Hodgkin, S. Heinzel, M. Pang, P. Colman, T. Burgess, M.J. Herold, K. Rogers, C.E. Teh, D.H.D. Gray, and S. Wilcox for insightful discussion, advice, and reagents; G. Siciliano, H. Johnson, C. Gatt, and their team for animal husbandry; S. Monard and the team at the WEHI Flow Cytometry Unit; K. Rogers and team for support from the WEHI Dynamic Imaging Facility; C. Tsui, E. Tsui, and the team from the WEHI Histology Service; J. McManus and J. Corbin for hematology support; J. Zhang for providing cytokines; and D. Quilici and T. Nikolaou for irradiation service. This work was supported by Leukaemia Foundation National Research Program Clinical PhD Scholarship (to B.J.A.), National Health and Medical Research Council (NHMRC) Research Fellowship 1081421 (to A.K.V.), NHMRC project grant 1086291 (to G.L.K.), NHMRC program grant 1016701 (to A.S.), NHMRC Senior Principal Research Fellowship 1020363 (to A.S.), Leukemia and Lymphoma Society of America Specialized Center of Research (SCOR) grant 7001-13 (to A.S.), NHMRC Principal Research Fellowship 1058892 (to G.K.S.), NHMRC program grant 1054618 (to G.K.S.), Cancer Council Victoria grant-in-aids 1086157 and 1147328 (to G.L.K.), Victorian Cancer Agency Fellowship 17028 (to G.L.K), a Leukemia Foundation Australia grant (to G.L.K. and A.S.), and bequests from the Anthony Redstone Estate and Craig Perkins Cancer Research Foundation. This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government National Health and Medical Research Council Independent Research Institutes Infrastructure Support Scheme.
Author contributions: B.J.A., G.L.K., and A.S. wrote the manuscript and conceptualized and planned the study. A.K.V. performed data analysis. Y.C. and G.K.S performed bioinformatics analysis and contributed to writing the manuscript. L.W. assisted with confocal microscopy and performed automated image analysis. A.J.K., G.D., and L.A.O. provided valuable advice and important reagents. B.J.A., A.J., C.C., E.C.L., S.T.D., J.P.B., A.S., and G.L.K performed experiments and analyzed data.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.314286.118.
References
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. 1985. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318: 533–538. [DOI] [PubMed] [Google Scholar]
- Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O'Connor L, Milla L, Wilcox S, Tai L, Strasser A, Herold MJ. 2015. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep 10: 1422–1432. [DOI] [PubMed] [Google Scholar]
- Basak S, Jacobs SB, Krieg AJ, Pathak N, Zeng Q, Kaldis P, Giaccia AJ, Attardi LD. 2008. The metastasis-associated gene Prl-3 is a p53 target involved in cell-cycle regulation. Mol Cell 30: 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh EH, Ke H, Levine AJ, Bonneau RA, Chan CS. 2018. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ 25: 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, Olivier M. 2016. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat 37: 865–876. [DOI] [PubMed] [Google Scholar]
- Bougeard G, Sesboue R, Baert-Desurmont S, Vasseur S, Martin C, Tinat J, Brugieres L, Chompret A, de Paillerets BB, Stoppa-Lyonnet D, et al. 2008. Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J Med Genet 45: 535–538. [DOI] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. 2011. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 145: 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewson G. 2015. Blue native PAGE and antibody gel shift to assess Bak and Bax conformation change and oligomerization. Cold Spring Harbor Protoc 2015: 485–489. [DOI] [PubMed] [Google Scholar]
- Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. 1999. Disruption of the ARF–Mdm2–p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev 13: 2658–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer G, Bargonetti J, Zhu H, Friedman P, Prywes R, Prives C. 1992. Wild-type p53 activates transcription in vitro. Nature 358: 83–86. [DOI] [PubMed] [Google Scholar]
- Freed-Pastor WA, Prives C. 2012. Mutant p53: one name, many proteins. Genes Dev 26: 1268–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidano G, Ballerini P, Gong JZ, Inghirami G, Neri A, Newcomb EW, Magrath IT, Knowles DM, Dalla-Favera R. 1991. p53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc Natl Acad Sci 88: 5413–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulino-Roth L, Wang K, MacDonald TY, Mathew S, Tam Y, Cronin MT, Palmer G, Lucena-Silva N, Pedrosa F, Pedrosa M, et al. 2012. Targeted genomic sequencing of pediatric Burkitt lymphoma identifies recurrent alterations in antiapoptotic and chromatin-remodeling genes. Blood 120: 5181–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. 1994. Tumor spectrum analysis in p53-mutant mice. Curr Biol 4: 1–7. [DOI] [PubMed] [Google Scholar]
- Janic A, Valente LJ, Wakefield MJ, Di Stefano L, Milla L, Wilcox S, Yang H, Tai L, Vandenberg CJ, Kueh AJ, et al. 2018. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat Med 24: 947–953. [DOI] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al. 2003. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4: 321–328. [DOI] [PubMed] [Google Scholar]
- Jeffrey PD, Gorina S, Pavletich NP. 1995. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 267: 1498–1502. [DOI] [PubMed] [Google Scholar]
- Kelly GL, Grabow S, Glaser SP, Fitzsimmons L, Aubrey BJ, Okamoto T, Valente LJ, Robati M, Tai L, Fairlie WD, et al. 2014. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev 28: 58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, Carrero ZI, Ramakrishnan G, Watabe K, Haupt Y, et al. 2015. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat Commun 6: 7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumper S, Mardakheh FK, McCarthy A, Yeo M, Stamp GW, Paul A, Worboys J, Sadok A, Jorgensen C, Guichard S, et al. 2016. Rho-associated kinase (ROCK) function is essential for cell cycle progression, senescence and tumorigenesis. Elife 5: e12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. 2004. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119: 861–872. [DOI] [PubMed] [Google Scholar]
- Langdon WY, Harris AW, Cory S, Adams JM. 1986. The c-myc oncogene perturbs B lymphocyte development in Eµ-myc transgenic mice. Cell 47: 11–18. [DOI] [PubMed] [Google Scholar]
- Li FP, Fraumeni JF Jr. 1969. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med 71: 747–752. [DOI] [PubMed] [Google Scholar]
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. 2012. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149: 1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love C, Sun Z, Jima D, Li G, Zhang J, Miles R, Richards KL, Dunphy CH, Choi WW, Srivastava G, et al. 2012. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet 44: 1321–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH. 2013. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493: 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin D, Li FP, Strong LC, Fraumeni JFJ, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, et al. 1990. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250: 1233–1238. [DOI] [PubMed] [Google Scholar]
- Michalak EM, Jansen ES, Happo L, Cragg MS, Tai L, Smyth GK, Strasser A, Adams JM, Scott CL. 2009. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ 16: 684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner J, Medcalf EA, Cook AC. 1991. Tumor suppressor p53: analysis of wild-type and mutant p53 complexes. Mol Cell Biol 11: 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PA, Vousden KH. 2014. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25: 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. 2000. Noxa, a BH3-only member of the bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288: 1053–1058. [DOI] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. 2004. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119: 847–860. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW. 1999. INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev 13: 2670–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Zou ZQ, Pirollo K, Plattner W, Chang EH. 1990. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 348: 747–749. [DOI] [PubMed] [Google Scholar]
- Sturzbecher HW, Brain R, Addison C, Rudge K, Remm M, Grimaldi M, Keenan E, Jenkins JR. 1992. A C-terminal α-helix plus basic region motif is the major structural determinant of p53 tetramerization. Oncogene 7: 1513–1523. [PubMed] [Google Scholar]
- Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, Van Pelt CS, Lozano G. 2008. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev 22: 1337–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrell FK, Kerr EM, Gao M, Thorpe H, Doherty GJ, Cridge J, Shorthouse D, Speed A, Samarajiwa S, Hall BA, et al. 2017. Lung tumors with distinct p53 mutations respond similarly to p53 targeted therapy but exhibit genotype-specific statin sensitivity. Genes Dev 31: 1339–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulz P, Heitzer E, Speicher MR. 2016. Co-occurrence of MYC amplification and TP53 mutations in human cancer. Nat Genet 48: 104–106. [DOI] [PubMed] [Google Scholar]
- Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A, Strasser A. 2013. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep 3: 1339–1345. [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Kein C, et al. 2004. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303: 844–848. [DOI] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. 2003. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302: 1036–1038. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. 2000. Surfing the p53 network. Nature 408: 307–310. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lane DP. 2007. p53 in health and disease. Nat Rev Mol Cell Biol 8: 275–283. [DOI] [PubMed] [Google Scholar]
- Wang PY, Ma W, Park JY, Celi FS, Arena R, Choi JW, Ali QA, Tripodi DJ, Zhuang J, Lago CU, et al. 2013. Increased oxidative metabolism in the Li-Fraumeni syndrome. N Engl J Med 368: 1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis A, Jung EJ, Wakefield T, Chen X. 2004. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 23: 2330–2338. [DOI] [PubMed] [Google Scholar]
- Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, Lin M, Yu H, Liu L, Levine AJ, et al. 2013. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun 4: 2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.