

Abstract
Pseudaminic acid is an amino deoxy sialic acid whose glycosides are essential components of many pathogenic Gram-negative bacterial cell walls including those from Pseudomonas aeruginosa, Vibrio cholerae, Campylobacter jejuni, Campylobacter coli, Vibrio vulnificus, and Pseudoalteromonas distincta. The study of pseudaminic acid glycosides is however hampered by poor availability from nature, the paucity of good synthetic methods, and limited to no understanding of the factors controlling stereoselectivity. Conformational analysis of the side chains of various stereoisomeric sialic acids suggested that the side chain of pseudaminic acid would take up the most electron-withdrawing trans,gauche-conformation, as opposed to the gauche,gauche conformation of N-acetyl neuraminic acid and the gauche,trans-conformtion of 7-epi N-acetyl neuraminic acid, leading to the prediction of high equatorial selectivity. This prediction is borne out by the synthesis of a suitably protected pseudaminic acid donor from N-acetyl neuraminic acid in 20 steps and 5% overall yield, and by the exquisite equatorial selectivity it displays in coupling reactions with typical glycosyl acceptors. The selectivity of the glycosylation reactions is further buttressed by the development and implementation of conditions for the regioselective release of the two amines from the corresponding azides, such as required for the preparation of the lipopolysaccharides. These findings open the way to the synthesis and study of pseudaminic acid-based bacterial lipopolysaccharides and, importantly in the broader context of glycosylation reactions in general, underline the significant role played by side chain conformation in the control of reactivity and selectivity.
Graphical Abstract
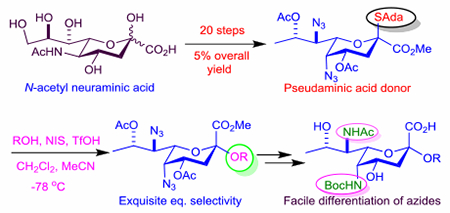
Introduction
In spite of the many advances in chemical and enzymatic glycosidation, whether automated or classical, in recent years,1–16 the stereocontrolled synthesis of many classes of glycosidic bond continues to be a significant challenge that retards the preparation of saccharides, oligosaccharides and their conjugates for biomedical research. The problem arises because of the location of typical glycosylation reactions at the interface between SN1 and SN2 reactions where minor changes in structure and conditions cause major shifts in mechanism,17–19 is compounded by the iterative nature of oligosaccharide synthesis, and is particularly significant in the preparation of the microbial glycans with their great structural diversity and complexity.20–24 Indeed, notwithstanding the several spectacular syntheses published in recent years,25–35 the surface of the microbial glycan problem has barely been scratched leaving many challenges for the ingenuity of the organic chemist.
The bacterial sialic acids legionaminic (Leg) 1 and pseudaminic acid (Pse) 2 and their glycosides, congeners of the ubiquitous N-acetyl neuraminic acid (NeuAc) 3 glycosides, are a case in point (Figure 1).36 Leg and Pse are found in a diverse range of bacterial capsular and lipopolysaccharides in the form of both axial and equatorial glycosides,23,24 and offer broad opportunities for the development of antibacterial therapeutics and/or vaccines.37–39 NeuAc on the other hand is found exclusively in the form of its equatorial glycosides,40–44 for whose preparation a number of effective chemical methods are now available.45–51 Leg differs from NeuAc, with whom it shares the D-glycero-D-galacto configuration only by the absence of a C-O bond at the 9-position and by replacement of a C-O by a C-N bond at the 7-position, whereas Pse with the L-glycero-L-manno configuration differs from NeuAc in configuration at both the 5- and 7-positions in addition to the deoxygenation at C9 and the N for O substitution at C7 (Figure 1). Other members of the class include acinetaminic acid 4 and fusaminic acid 5 (Figure 1), and the 4- and 8-epimers of legionaminic acid (not shown).23,24,52–55
Figure 1.
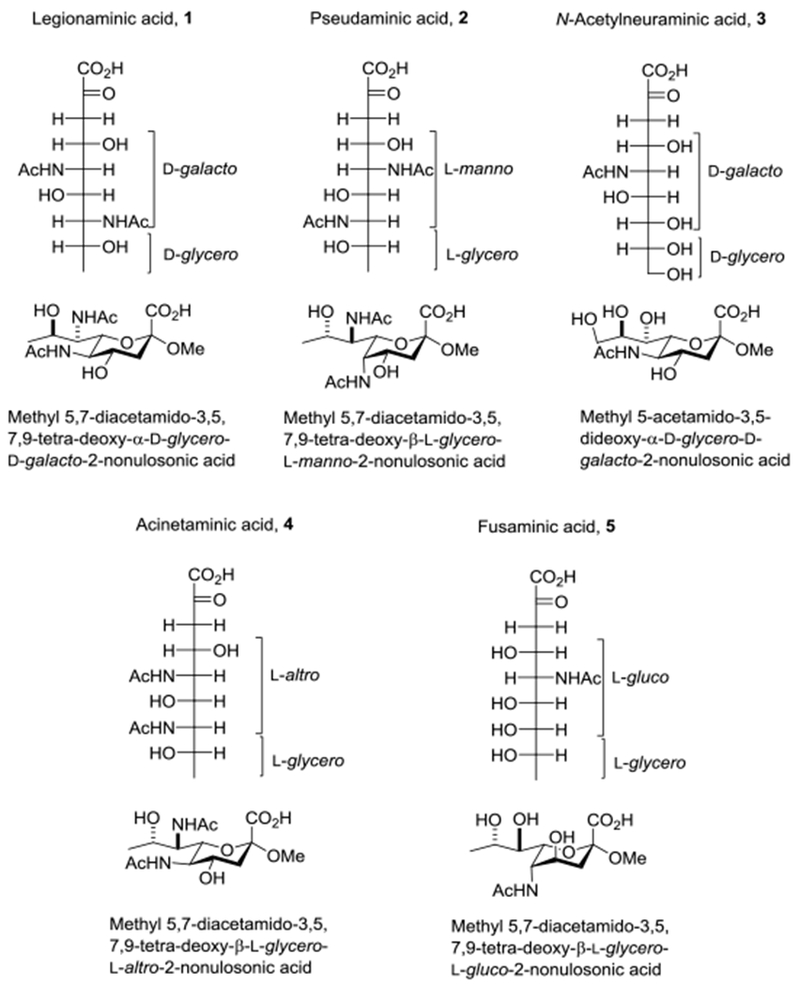
Fischer Projections of Legionaminic, Pseudaminic, N-Acetyl Neuraminic, N-Acetylacinetaminic and Fusaminic Acid, and Structures and Formal Names of their Equatorial Methyl Pyranosides.
Investigations of the glycosylation reactions of Leg and Pse are necessarily preceded by the synthesis of suitable donors as neither substance is presently available from nature in sufficient quantities. Tsvetkov and coworkers described syntheses of both Leg and Pse and their stereoisomers by homologation of hexose sugars,56,57 while Ito and coworkers adopted an analogous approach to a Pse donor 6 that was found to be axially selective in a single glycosylation reaction conducted with a primary acceptor in acetonitrile at 0 °C.58 Seeberger and co-workers reported the synthesis of the Leg donor 7 from D-threonine and found it to be axially selective in a single glycosylation with a primary acceptor at −78 °C in dichloromethane.38 Subsequently, Li and coworkers adopted an analogous approach to the Pse donors 8-10 from L-threonine, each of which showed modest to excellent axial selectivity in dichloromethane at −78 °C (Figure 2).59 Payne, Kiefel and coworkers reported the synthesis of N,N-diacetyl Pse from NeuAc but did not describe its conversion into a glycosyl donor and use in glycosylation reactions.60,61 Biosynthetic and chemoenzymatic approaches to legionaminic acid and its equatorial glycosides have been described.62,63
Figure 2.
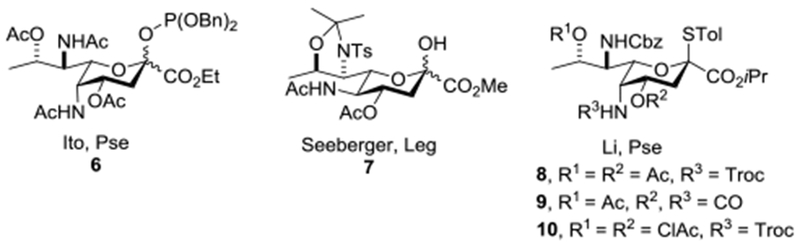
Axially Selective Leg and Pse Donors
Our laboratory has a long-standing interest in the stereoselective synthesis of the equatorial sialic acid glycosides,46–48,64,65 and, following the seminal work of Bols,66 in the role played by side chain conformation in anomeric reactivity and selectivity.50,67–71 We viewed Leg and especially Pse as proving grounds for our hypotheses on the manner in which side chain conformation influences the reactivity and selectivity of glycosyl donors and, with Pse in mind first investigated the influence of configuration at the 7-position with the NeuAc and 7-epi-NeuAc donors 11 and 12, respectively (Figure 3).68 We found, consistent with the earlier NMR studies of Zbiral,72,73 that the 7-epi-NeuAc donor 12 showed a change in predominant side chain conformation from the gg-conformer74 observed in 11 and in NeuAc derivatives in general,72,75–79 to the gt-conformation so as to avoid unfavorable dipolar and steric interactions with the C5-N5 bond (Figure 3). We also found, consistent with the work of Bols and ourselves using rigid bicyclic systems,66,67 that the change in predominant side chain conformation from gg to gt was accompanied by a considerable loss of reactivity. Subsequently, working with the NeuAc donor 13 and its C5 epimer 14, we found that inversion of configuration at C5 was accompanied by a change of side chain conformation from the gg to the gt conformer, again to minimize dipolar and steric interaction with the C5-N5 bond (Figure 3).50 We found that the anticipated80–83 increase in reactivity on replacement of an equatorial C-N bond at the C5 position in 13 by an axial one in 14 was offset by the change in side chain conformation such that 13 and 14 displayed similar selectivity in glycosylation reactions. Extrapolating from these results we predicted that inversion of configuration at C7 of the 5-epi-donor 14 would again result in unfavorable dipolar and steric interactions in the hypothetical 5,7-bis-epi system 15, minimization of which would result in a change in side conformation from the gt to the most-disarming66,67 tg conformer. This analysis of side chain conformation is consistent with the 3J coupling constant of 10.5 Hz for the H6,H7 spin system in a glycoside of 6 reported by Ito and coworkers,58 and in both epimers of Pse itself by Tsvetkov and coworkers57 and by Payne and coworkers,61 albeit no predictions of reactivity were offered by those workers.
Figure 3.
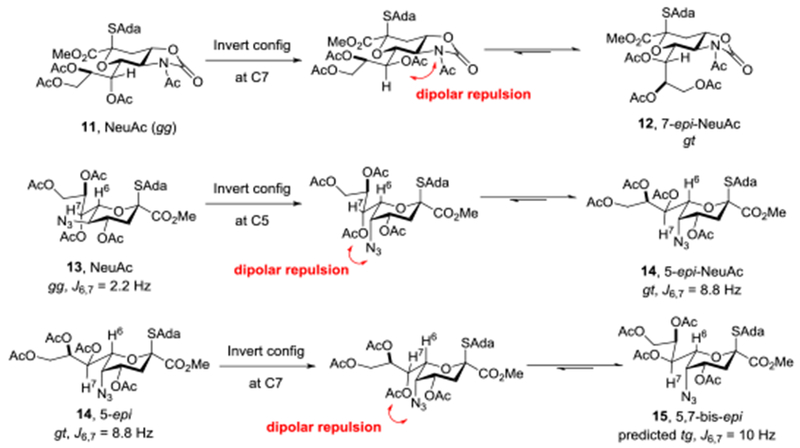
Influence of Configuration at C5 and C7 on the Side Chain Conformation of NeuAc Donors.
On the basis of this analysis of the influence of configuration at the C5 and C7 on side chain configuration, drawing on experience from our synthesis of the 5-epi-NeuAc donor 1450 and of moderately equatorially selective Leg donor 16,69 we designed the Pse donor 17 in the expectation that its side chain would adopt the tg-conformation very predominantly and consequently afford excellent equatorial selectivity in its glycosylation reactions (Figure 4). We report here on the synthesis of 17 from NeuAc, the analysis of its side chain conformation, its highly equatorially selective glycosylation reactions, and on the selective functionalization of the two azido groups in the coupled products such as will be required for the synthesis of many natural Pse oligosaccharides.
Figure 4.
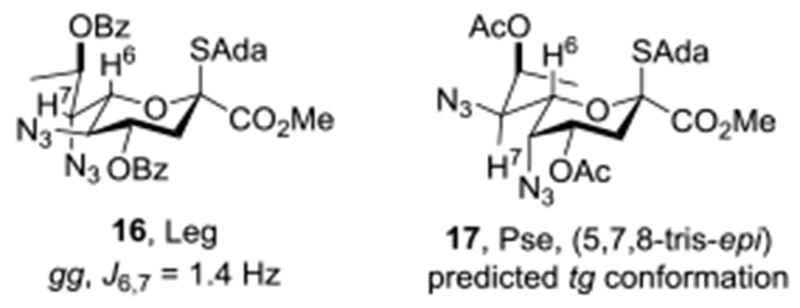
Equatorially Selective Leg and Pse Donors
Results and Discussion
Donor Synthesis
Our strategy for the synthesis of 17 sought to make use of the readily available NeuAc as starting material taking advantage of the lessons learnt in the previous synthesis of 16.69 However, in addition to the operations of deoxygenation at the 9-position and replacement of the C-O by a C-N bond at the 7-position executed in the synthesis of 16, the preparation of 17 necessitates inversion of configuration at positions 5, 7, and 8. Analogously to Kiefel, Payne and coworkers in their synthesis of N,N-diacetyl Pse from NeuAc (Scheme 1), we anticipated that displacement of suitably activated alcohols from the 5- and 7-positions by the azide anion would afford the two C-N bonds both with inversion of configuration. This approach in turn requires initial replacement of the equatorial acetamide at C5 by a hydroxyl group with retention of configuration, for which we anticipated using the Zbiral oxidative deamination84–86 as in our earlier synthesis of a 3-deoxy-D-glycero-D-galacto-nonulosonic acid (KDN) donor, our synthesis of the 5-epi-NeuAc donor 14,50 and the Kiefel-Payne synthesis of N,N-diacetyl-Pse.60,61 However, distinct from the Kiefel-Payne synthesis which employed acetic acid as nucleophile in the deamination step thereby necessitating extra steps to isolate the 5-position for inversion (Scheme 1), we planned to employ levulinic acid as nucleophile as in our synthesis of 14 to facilitate selective manipulation of the ensuing ester at the 5-position. Tactically, in view of the generally modest yields of the Zbiral reaction which rarely exceed 55–60%, we initially elected to conduct the oxidative deamination of the 5-position at a late stage of the synthesis. We also selected the 1-adamantanyl thioglycoside for the glycosyl donor in view of its ready activation at the low temperatures envisaged for the eventual glycosylation reactions.47
Scheme 1.
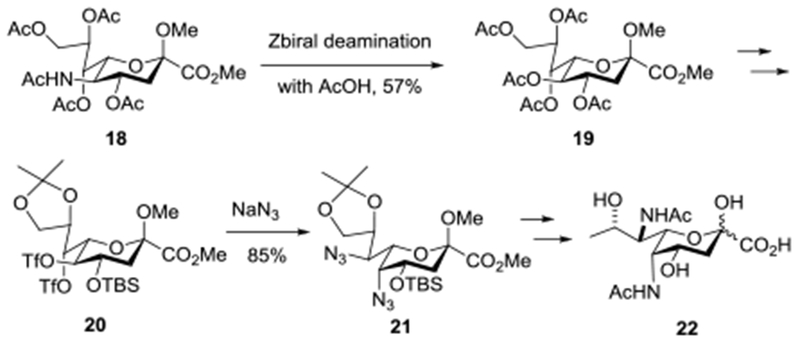
Key Steps in the Kiefel-Payne Synthesis of N,N-diacetyl-Pse 22.
Thus, NeuAc was converted by a sequence of three well-established literature steps to the known thioglycoside 23 on a 20 g scale in 70% overall yield.47,87 Global Zemplen deacetylation was followed by installation of an 8,9-O-acetonide in the usual manner to give 24 in 93% yield. Regioselective monoacetylation followed the established pattern88 and afforded the 4-O-acetate 25, which on silylation gave 26 cleanly. Removal of the acetonide was followed by selective sulfonylation of the primary hydroxyl group with 2,4,6-tri-isopropylbenzenesulfonyl chloride to give 27. Oxidation with the Dess-Martin periodinane89 was followed by Luche reduction90 giving the ketone 28 and the inverted alcohol 29, respectively. Displacement of the sulfonate group from 29 with sodium iodide afforded 30 which, on hydrogenolysis in a mixture of ethyl acetate and triethylamine, gave the 9-deoxy derivative 31. It is noteworthy that these conditions, with the incorporation of triethylamine, enabled selective hydrogenolysis of the C-I bond without detriment to the thioglycoside moiety, something that we had previously been unable to accomplish cleanly in our synthesis of the Leg donor 16.69 Reaction with acetic anhydride and DMAP then gave 32, the substrate for the deamination step. Adopting thermal conditions for the oxidative deamination similar to the ones employed in the Kiefel-Payne synthesis (Scheme 1), treatment of 32 with nitrosyl tetrafluoroborate and pyridine gave the corresponding N-nitrosoacetamide, that on warming to 50 °C in levulinic acid resulted in the formation of the oxidative deamination product 33 as a mixture of diastereomers. Heating was required in this step as the intermediate N-nitroso amide did not undergo reaction with levulinic acid at lower temperatures under standard conditions. To facilitate purification the crude reaction mixture containing 33 was treated with hydrazine hydrate, acetic acid and pyridine resulting in removal of the levulinate ester and the isolation of 34 in 38% overall yield from 32 in the form of an approximately 1:1 mixture of stereoisomers (Scheme 2). Based on our previous studies of substituent effects in the Zbiral oxidative deamination reaction,86 the poor stereoselectivity in the final deamination reaction presumably arises from the absence of a C-O bond at the 9-position, which reduces the need for stereodirecting participation by the ring oxygen and supports a more carbenium ion-like intermediate at the 5-position. Whatever the reason, the modest yield and unacceptably poor selectivity at this late stage of the synthesis caused us to abandon this route and adopt a sequence in which the deamination reaction was conducted before deoxygenation at the 9-position.
Scheme 2.
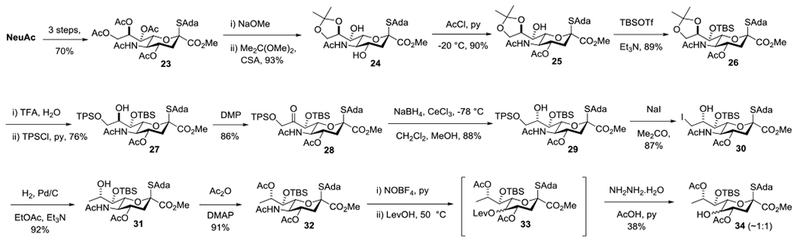
First Approach with Late Stage Oxidative Deamination
Accordingly, intermediate 25 was converted to the 7-O-naphthylmethyl ether 35 by treatment with sodium hydride and 2-naphthylmethyl bromide in the usual manner91 on a multigram scale in 82% yield. Reverting to conditions for the Zbiral reaction previously developed in our laboratories,92 reaction of 35 with nitrosyl tetrafluoroborate and pyridine gave a crude N-nitrosoacetamide derivative that was treated with preformed sodium trifluoroethoxide and 18-crown-6 in dichloromethane at −10 °C, with addition of levulinic acid after minutes followed by warming to 0 °C before quenching. In this manner, working with >5 g of 35, we isolated 21% of the alkene 36, which is the typical byproduct for this class of reaction,86 and 55% of the desired levulinate 37 in the form of a single equatorial diastereomer. Removal of the acetonide with aqueous trifluoroacetic acid followed by installation of the triisopropylbenzenesulfonyl group on the primary alcohol with triisopropylbenzenesulfonyl chloride, pyridine and dibutyltin oxide93,94 gave 38 in 83% yield. Application of the Lattrell-Dax protocol95–97 then gave the 8-epi-isomer 39 in 72% yield. It is noteworthy in this sequence that a secondary triflate is displaced in preference to a primary arenesulfonate ester, testifying to the power of the trifloxy group as a nucleofuge in substitution reactions.98 Subsequently, the triisopropylbenzenesulfonyl group was displaced with sodium iodide in hot acetone to give an 81% yield of 40, which on hydrogenolysis over palladium on carbon in ethyl acetate and triethylamine afforded 91% of the 9-deoxy compound 41. Standard acetylation then gave the triester 42 in excellent yield, from which the naphthylmethyl ether and the levulinate ester were removed sequentially with DDQ and hydrazine hydrate giving 43 and 44, respectively, in good yields. Finally, triflation of the diol 44 followed by reaction with sodium azide in DMF gave the desired Pse donor 17 in 70% yield (Scheme 3). Overall, we describe a practical synthesis of donor 17 that, with the possible exception of the Zbiral deamination employs well-established, simple and scalable reactions and affords the product on the scale of multiple hundreds of milligrams, suitable for the investigation of the glycosylation reaction.
Scheme 3.
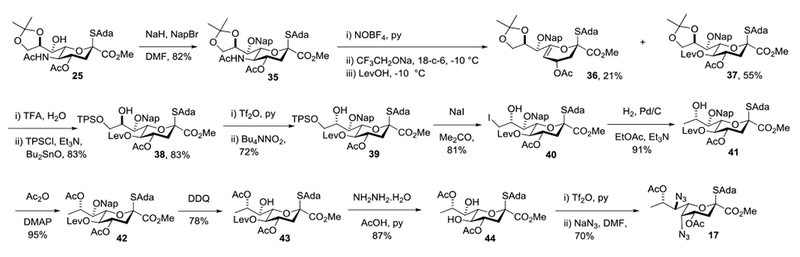
Synthesis of Pse Donor 17.
Glycosylation
Turning to glycosylation we employed the widely used combination of N-iodosuccinimide and triflic acid for activation of the thioglycoside and conducted all reactions in a 2:1 mixture of acetonitrile and dichloromethane at −78 °C for ease of comparison with our earlier work. In the event activation of 17 in the presence of benzyl alcohol as acceptor under these conditions afforded the equatorial glycoside 45 as a single anomer in 89% isolated yield (Scheme 4). Similarly, use of methyl 2,3,4-tri-O-benzyl-β-D-galactoside and methyl 2,4,6-tri-O-benzyl-β-D-galactoside as acceptor alcohols afforded the equatorial glycosides 46 and 47, as single anomers in 83 and 77% isolated yield. Finally, use of the highly sterically hindered methyl 2,3,6-tri-O-benzyl-β-D-galactoside as acceptor gave 53% of the equatorial glycoside 48, without competing formation of the axial isomer (Scheme 4). The anomeric configuration of 45-48 was determined by measurement of the 3JC1-H3axial heteronuclear coupling constant which in each case fell in the range 6.8–7.5 Hz that is diagnostic of the equatorial glycoside, as opposed to the ~0 Hz expected for the opposite anomer.68,99–102 We selected galactopyranosyl 3- and 6-alcohols as acceptors for this study as they afford what are by far the most common types of linkage in the realm of sialic acid glycosides. The highly hindered galactopyranosyl 4-alcohol leading to the glycoside 48 was selected as a model study for the very demanding Pse-β-(2→4)-6-deoxy-N-acetylgalactosaminide linkage found in the repeating unit from the Pseudomonas aeruginosa O10 lipopolysaccharide.103
Scheme 4.
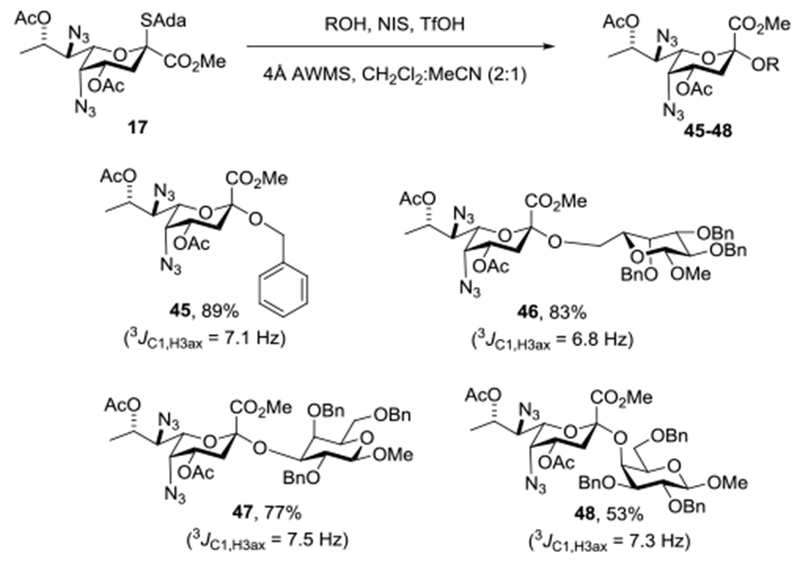
Stereoselective Synthesis of Equatorial Pse Glycosides
The results presented in Scheme 4 validate our design hypothesis (Figure 3) particularly in so far the selectivities observed, with the exception of the benzyl glycoside, are superior to those seen with the Leg donor 16 under the same conditions and with the same alcohols.69 We conclude that donor side chain conformation is an important control element in the preparation of glycosidic bonds, whether the donor is a simple monocyclic pyranosyl system as presented here or the more familiar 4,6-O-benzylidene-protected and related donors from the synthesis of β-mannosides and related systems. The contrast between the excellent equatorial selectivity observed here with Pse donor 17 and the mostly unselective or axially selective donors Pse donors 8-10 reported by Li and coworkers59 is striking, especially as the conditions employed are similar (solvent, temperature, and triflate-based activating systems). Inspection of the spectral data for 8–10 reveals each of them to display a 10.0 Hz 3JH6,H7 coupling constant and so the tg conformation, indicating that the difference in selectivity must arise from the difference in amine protecting groups, perhaps for steric or hydrogen bonding/association reasons,19,104–106 or from the use of the more bulky isopropyl ester that is less easily accommodated in the axial position. The Ito study with donor 6 was performed under different conditions (acetonitrile, 0 °C) that do not permit meaningful comparison with the present work.
Deprotection
Reaction of the benzyl glycoside 45 with excess thioacetic acid in pyridine107 for 40 h at room temperature afforded 73% of the bisacetamide 49 (Scheme 5). Heating of this bisamide to 60 °C with aqueous barium hydroxide in 1,4-dioxane followed after workup by hydrogenolysis also in aqueous 1,4-dioxane over palladium charcoal afforded N,N-diacetyl Pse 22, whose spectral data were consistent with those provided by Tsvetkov and by Kiefel and Payne.57,61
Scheme 5.
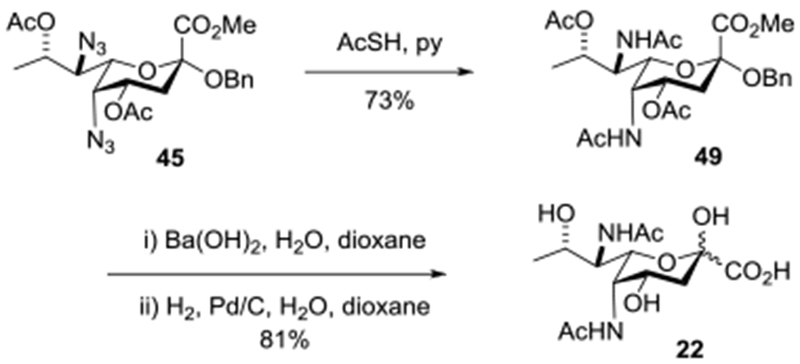
Synthesis of N,N-Diacetyl Pse
Many Pse glycosides,23 including the axially-linked Pseudomonas aeruginosa 1244 pilin glycoside prepared by Li and coworkers59 and the Pse-β-(2→4)-6-deoxy-N-acetylgalactosaminide linkage found in the repeating unit from the Pseudomonas aeruginosa O10 lipopolysaccharide,103 are characterized by the presence of two different amides at positions 5 and 7. This requires either the synthesis of donors with differentially protected amines at the 5- and 7-positions, with all the associated complexity, as in 8-10, or the regioselective unmasking of one of two identically protected amines. We anticipated that the differing steric environments of the two azides in glycosides 45-48 would permit the latter option and were encouraged by the work of Kiefel and coworkers who showed in a model system, albeit one with the incorrect configuration at the 8-position and so a different steric environment, that the side chain azide was more reactive than the axial azide in the pyranose ring toward Staudinger reaction with triphenylphosphine.60 In the event, heating of 47 with thioacetic acid and lutidine in chloroform at reflux for 10 h yielded 68% of a single monoamide 50 arising from conversion of the side chain azide (Scheme 6). Subsequent treatment with 1,3-propanedithiol and triethylamine in wet pyridine108 at room temperature followed by installation of a Boc group afforded the amido carbamate 51 in 72% yield. Finally, heating with aqueous barium hydroxide in dioxane followed by hydrogenolysis afforded the Pse glycoside 52 with the two amines differentially protected, one in the form of a tert-butylcarbamate suitable for selective cleavage and further elaboration (Scheme 6).
Scheme 6.
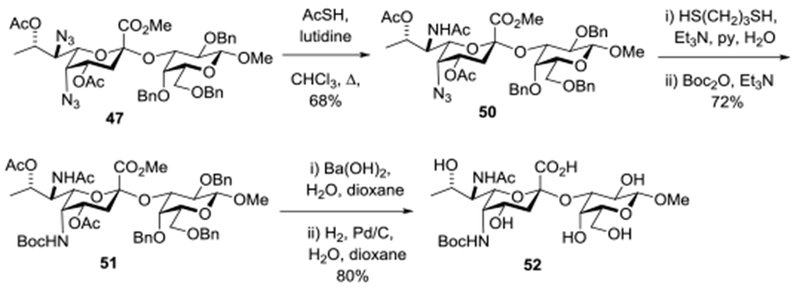
Regioselective Azide Cleavage.
Finally, the sequence of azide cleavage reactions was reversed providing first the 5-azido-7-N-Boc derivative 53, then the 5-N-acetyl-7-N-Boc derivative 54, and ultimately the Pse glycoside 55 in which the amino group at the 7-position is poised for use in further steps following cleavage of the Boc group (Scheme 7).
Scheme 7.
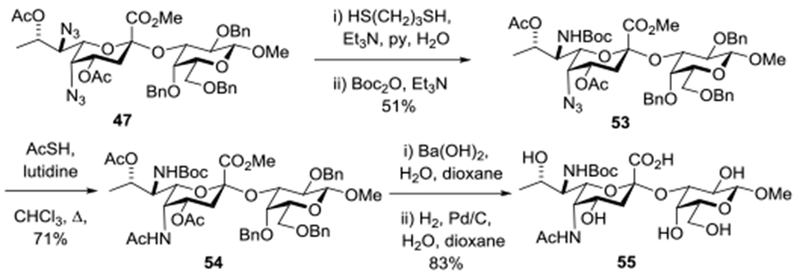
Alternative Regioselective Azide Cleavage.
Conclusion.
Readily available N-acetylneuraminic acid is shown to be a suitable starting material for the synthesis of a pseudaminic acid donor, in which both amines are protected in the form of azides. The synthesis employs operationally simple chemistry and proceeds in 20 steps and 5% overall yield on such as scale as to afford multi-hundred milligram quantities for the study of glycosylation reactions. The thioglycoside serves as an effective donor for coupling to a range of primary and hindered secondary alcohols and affords the corresponding equatorial glycosides with exquisite selectivity. Conformational analysis of the side chain reveals this selectivity to be a function of the trans,gauche conformation of the side chain, with its maximal electron-withdrawing capacity, which is a function of the 5,7-bis-epi-configuration when compared to the prototypical N-acetylneuraminic acid and the gauche,gauche conformation of its side chain. This study underlines the role played by side chain conformation in the reactivity and selectivity of glycosyl donors and further enables the mechanism-based development of stereoselective glycosylation reactions. The development of two different modes of regioselective deprotection sequence enables conversion of the azide-protected glycosides to differently substituted amine functionality in the final pseudaminic acid glycosides, suitable for further elaboration to bacterial lipopolysaccharides.
Supplementary Material
Acknowledgements.
We thank the NIH (GM62160) for support of this work and acknowledge the NSF (MRI-084043) for funds in support of the purchase of the 600 MHz NMR spectrometer in the Lumigen Instrument Center at Wayne State University.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs8b09654.
Full experimental details and copies of the 1H and 13C NMR spectra of all new compounds (PDF)
References
- (1).Peng P; Schmidt RR Acid–Base Catalysis in Glycosidations: A Nature Derived Alternative to the Generally Employed Methodology. Acc. Chem. Res 2017, 50, 1171–1183. [DOI] [PubMed] [Google Scholar]
- (2).Williams R; Galan MC Recent Advances in Organocatalytic Glycosylations. Eur. J. Org. Chem 2017, 6247–6264. [Google Scholar]
- (3).Yu B Gold(I)-Catalyzed Glycosylation with Glycosyl o-Alkynylbenzoates as Donors. Acc. Chem. Res 2018, 51, 507–516. [DOI] [PubMed] [Google Scholar]
- (4).Panza M; Pistorio SG; Stine KJ; Demchenko AV Automated Chemical Oligosaccharide Synthesis: Novel Approach to Traditional Challenges. Chem. Rev 2018, 108, 8105–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sasaki K; Tohda K Recent Topics in β-Selective Mannosylation. Tetrahedron Lett 2018, 59, 496–503. [Google Scholar]
- (6).Seeberger PH The Logic of Automated Glycan Assembly. Acc. Chem. Res 2015, 48, 1450–1463. [DOI] [PubMed] [Google Scholar]
- (7).Bennett CS; Galan MC Methods for 2-Deoxyglycoside Synthesis. Chem. Rev 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wen L; Edmunds G; Gibbons C; Zhang J; Gadi MR; Zhu H; Fang J; Liu X; Kong Y; Wang PG Toward Automated Enzymatic Synthesis of Oligosaccharides. Chem. Rev, 2018, 108, 8151–8187. [DOI] [PubMed] [Google Scholar]
- (9).Kulkarni SS; Wang C-C; Sabbavarapu NM; Podilapu AR; Liao P-H; Hung S-C “One-Pot” Protection, Glycosylation, and Protection–Glycosylation Strategies of Carbohydrates. Chem. Rev 2018, 108. [DOI] [PubMed] [Google Scholar]
- (10).Codée JDC; Christina AE; Walvoort MTC; Overkleeft HS; van der Marel GA Uronic Acids in Oligosaccharide and Glycoconjugate Synthesis. Top. Curr. Chem 2011, 301, 253–290. [DOI] [PubMed] [Google Scholar]
- (11).Spell ML; Deveaux K; Bresnahan CG; Ragains JR O-Glycosylation Enabled by Remote Activation. Synlett 2017, 28, 751–761. [Google Scholar]
- (12).Ragains JR In Selective Glycosylations: Synthetic Methods and Catalysis; Bennett CS, Ed.; Wiley-VCH: Weinheim, 2017, p 211–230. [Google Scholar]
- (13).Mao R-Z; Xiong D-C; Guo F; Li Q; Duan J; Ye X-S Light-driven Highly Efficient Glycosylation Reactions. Org. Chem. Front 2016, 3, 737–743. [Google Scholar]
- (14).Nokami I,T; Mong KKT Chemical Glycosylation by Single Electron Transfer. Israel J. Chem 2015, 55, 297–305. [Google Scholar]
- (15).Yu B; Wang L-X In Organic Chemistry-Breakthroughs and Perspectives; Ding K, Dai L-X, Eds.; Wiley-VCH: Weinheim, 2012, p 181–219. [Google Scholar]
- (16).Guinchard X; Picard S; Crich D In Modern Tools for the Synthesis of Complex Bioactive Molecules; Cossy J, Arseniyadis S, Eds.; Wiley: Hoboken, 2012, p 395–432. [Google Scholar]
- (17).Crich D Mechanism of a Chemical Glycosylation. Acc. Chem. Res 2010, 43, 1144–1153. [DOI] [PubMed] [Google Scholar]
- (18).Crich D Methodology Development and Physical Organic Chemistry: A Powerful Combination for the Advancement of Glycochemistry. J. Org. Chem 2011, 76, 9193–9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Adero PO; Amarasekara H; Wen P; Bohé L; Crich D The Experimental Evidence in Support of Glycosylation Mechanisms at the SN1-SN2 Interface. Chem. Rev 2018, 118, 8242–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Elshahawi SI; Shaaban KA; Kharel MK; Thorson JS A Comprehensive Review of Glycosylated Bacterial Natural Products. Chem. Soc. Rev 2015, 44, 7591–7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Microbial Glycobiology; Moran AP; Holst O; Brennan PJ; von Itzstein M, Eds.; Elsevier: Amsterdam, 2009. [Google Scholar]
- (22).Kosma P Occurrence, Synthesis and Biosynthesis of Bacterial Heptoses. Curr. Org. Chem 2008, 12, 1021–1039. [Google Scholar]
- (23).Zunk M; Kiefel MJ The Occurrence and Biological Significance of the α-Keto-Sugars Pseudaminic Acid and Legionaminic Acid within Pathogenic Bacteria. RSC Adv 2014, 4, 3413–3421. [Google Scholar]
- (24).Knirel YA; Shashkov AS; Tsvetkov YE; Jansson P-E; Zaehringer U 5,7-Diamino-3,5,7,9-tetradeoxy-non-2-ulosonic Acids in Bacterial Glyco-Polymers: Chemistry and Biochemistry. Adv. Carbohydr. Chem. Biochem 2003, 58, 371–417. [DOI] [PubMed] [Google Scholar]
- (25).Fraser-Reid B; Lu J; Jayaprakash KN; Lopez JC Synthesis of a 28-mer Oligosaccharide Core of Mycobacterial Lipoarabinomannan (LAM) Requires Only Two n-Pentenyl Ortho-Ester Progenitors. Tetrahedron: Asymmetry 2006, 17, 2449–2463. [Google Scholar]
- (26).Joe M; Bai Y; Nacario RC; Lowary TL Synthesis of the Docosanasaccharide Arabinan Domain of Mycobacterial Arabinogalactan and a Proposed Octadecasaccharide Biosynthetic Precursor. J. Am. Chem. Soc 2007, 129, 9885–9901. [DOI] [PubMed] [Google Scholar]
- (27).Wu y; Xiong D-C; Chen S-C; Wang Y-S; Ye X-S Total synthesis of Mycobacterial Arabinogalactan Containing 92 Monosaccharide Units. Nat. Commun 2017, 8, 14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zheng RB; Jegouzo SAF; Joe M; Bai Y; Tran H-A; Shen K; Saupe J; Xia L; Ahmed MF; Liu Y-H; Patil PS; Tripathi A; Hung S-C; Taylor ME; Lowary TL; Drickamer K Insights into Interactions of Mycobacteria with the Host Innate Immune System from a Novel Array of Synthetic Mycobacterial Glycans. ACS Chem. Biol 2017, 12, 2990–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kosma P Recent Advances in Kdo-Glycoside Formation. Carbohydr. Chem 2017, 42, 116–164. [Google Scholar]
- (30).Trattnig N; Farcet J-B; Gritsch P; Christler A; Pantophlet R; Kosma P Synthesis of a Pentasaccharide Fragment Related to the Inner Core Region of Rhizobial and Agrobacterial Lipopolysaccharides. J. Org. Chem 2017, 82, 12346–12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hollaus R; Ittig S; Hofinger A; Haegman M; Beyaert R; Kosma P; Zamyatina A Chemical Synthesis of Burkholderia Lipid A Modified with Glycosyl Phosphodiester-Linked 4-Amino-4-deoxy-β-L-arabinose and Its Immunomodulatory Potential. Chem. Eur. J 2015, 21, 4102–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Li W; Silipo A; Andersen Gersby LB; Newman M-A; Molinaro A; Yu B Synthesis of Bradyrhizose Oligosaccharides Relevant to the Bradyrhizobium O-Antigen. Angew. Chem. Int. Ed 2017, 56, 2092–2096. [DOI] [PubMed] [Google Scholar]
- (33).Kenfack MT; Mazur M; Nualnoi T; Shaffer TL; Ngassimou A; Blériot Y; Marrot J; Marchetti R; Sintiprungrat K; Chantratita N; Silipo A; Molinaro A; AuCoin DP; Burtnick MN; Brett PJ; Gauthier C Deciphering Minimal Antigenic Epitopes Associated with Burkholderia pseudomallei and Burkholderia mallei Lipopolysaccharide O-Antigens. Nat. Commun 2017, 8:115, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Eradi P; Ghosh S; Andreana PR Total Synthesis of Zwitterionic Tetrasaccharide Repeating Unit from Bacteroides fragilis ATCC 25285/NCTC 9343 Capsular Polysaccharide PS A1 with Alternating Charges on Adjacent Monosaccharides. Org. Lett 2018, 20, 4526–4530. [DOI] [PubMed] [Google Scholar]
- (35).Ishiwata A; Ito Y In Glycochemical Synthesis: Strategies and Applications; Hung S-C, Zulueta MML, Eds.; Wiley: Hoboken, 2016, p 361–406. [Google Scholar]
- (36).Note that under the Rosanoff convention the equatorial glycosides of Leg and NeuAc are defined as the α-anomers and their axial glycosides as the β-anomers. In contrast, the equatorial glycosides of Pse are the β-anomers while the axial Pse glycosides have the α-anomeric configuration.; McNaught AD Nomenclature of Carbohydrates. Carbohydr. Res 1997, 297, 1–92 [DOI] [PubMed] [Google Scholar]
- (37).Ud-Din AIMS; Roujeinikova A Flagellin Glycosylation with Pseudaminic Acid in Campylobacter and Helicobacter: Prospects for Development of Novel Therapeutics. Cell. Mol. Life Sci 2018, 75, 1163–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Matthies S; Stallforth P; Seeberger PH Total Synthesis of Legionaminic Acid as Basis for Serological Studies. J. Am. Chem. Soc 2015, 137, 2848–2851. [DOI] [PubMed] [Google Scholar]
- (39).Ménard R; Schoenhofen IC; Tao L; Aubry A; Bouchard P; Reid CW; Lachance P; Twine SM; Fulton KM; Cui Q; Hogues H; Purisima EO; Sulea T; Logan SM Small-Molecule Inhibitors of the Pseudaminic Acid Biosynthetic Pathway: Targeting Motility as a Key Bacterial Virulence Factor. Antimicrob. Agent. Chemother 2014, 58, 7430–7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Schauer R Chemistry, Metabolism, and Biological Functions of Sialic Acids. Adv. Carbohydr. Chem. Biochem 1982, 40, 131–234. [DOI] [PubMed] [Google Scholar]
- (41).Schauer R Sialic Acids: Fascinating Sugars in Higher Animals and Man Zoology 2004, 107, 49–64. [DOI] [PubMed] [Google Scholar]
- (42).Miyagi T; Yamaguchi K In Comprehensive Glycoscience; Kamerling JP, Ed.; Elsevier: Amsterdam, 2007; Vol. 3, p 297–323. [Google Scholar]
- (43).Angata T; Varki A Chemical Diversity in the Sialic Acids and Related α-Keto Acids: An Evolutionary Perspective Chem. Rev 2002, 102, 439–469. [DOI] [PubMed] [Google Scholar]
- (44).Chen X; Varki AP Advances in the Biology and Chemistry of Sialic Acids. ACS Chem. Biol 2010, 5, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Tanaka H; Nishiura Y; Takahashi T Stereoselective Synthesis of Oligo-α-(2,8)-Sialic Acids. J. Am. Chem. Soc 2006, 128, 7124–7125. [DOI] [PubMed] [Google Scholar]
- (46).Crich D; Li W O-Sialylation with N-Acetyl-5-N,4-O-Carbonyl Protected Thiosialoside Donors in Dichloromethane; Facile and Selective Cleavage of the Oxazolidinone Ring. J. Org. Chem 2007, 72, 2387–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Crich D; Li W α-Selective Sialylations at −78 °C in Nitrile Solvents with a 1-Adamantanyl Thiosialoside. J. Org. Chem 2007, 72, 7794–7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Mandhapati AR; Rajender S; Shaw J; Crich D The Isothiocyanato Moiety. An Ideal Protecting Group for Stereoselective Sialic Acid Glycoside Synthesis and Subsequent Diversification. Angew. Chem. Int. Ed 2015, 54, 1275–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Navuluri C; Crich D In Glycochemical Synthesis: Strategies and Applications; Hung S-C, Zulueta MML, Eds.; Wiley: New York, 2016, p 131–154. [Google Scholar]
- (50).Dhakal B; Buda S; Crich D Stereoselective Synthesis of 5-Epi-α-Sialosides Related to the Pseudaminic Acid Glycosides. Reassessment of the Stereoselectivity of the 5-Azido-5-deacetamidosialyl Thioglycosides and Use of Triflate as Nucleophile in the Zbiral Deamination of Sialic Acids. J. Org. Chem 2016, 81, 10617–10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lih Y-H; Wu C-Y In Selective Glycosylations: Synthetic Methods and Catalysts; Bennett CS, Ed.; Weinheim: 2017, p 353–370. [Google Scholar]
- (52).Kenyon JJ; Marzaioli AM; De Castro C; Hall RM 5,7-Di-N-acetyl-acinetaminic Acid: A Novel Non-2-ulosonic Acid Found in the Capsule of an Acinetobacter baumannii Isolate. Glycobiology 2015, 25, 644–654. [DOI] [PubMed] [Google Scholar]
- (53).Vinogradov E; St. Michael F; Cox AD The Structure of the LPS O-Chain of Fusobacterium nucleatum strain 25586 Containing Two Novel Monosaccharides, 2-Acetamido-2,6-dideoxy-L-altrose and a 5-Acetimidoylamino-3,5,9-trideoxy-gluconon-2-ulosonic Acid. Carbohydr. Res 2017, 440-441, 10–15. [DOI] [PubMed] [Google Scholar]
- (54).Knirel YA; Sheelev SD; Perepelov AV Higher Aldulosonic Acids: Components of Bacterial Glycans. Mendeleev, Commun 2011, 21, 173–182. [Google Scholar]
- (55).Kenyon JJ; Notaro A; Hsu LY; De Castro C; Hall RM 5,7-Di-N-acetyl-8-epiacinetaminic Acid: A New Non-2-ulosonic Acid Found in the K73 Capsule Produced by an Acinetobacter baumannii Isolate from Singapore. Sci. Rep 2017, 7, 11357 (1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Tsvetkov YE; Shashkov AS; Knirel YA; Zähringer U Synthesis and Identification in Bacterial Lipopolysaccharides of 5,7-Diacetamido-3,5,7,9-tetradeoxy-D-glycero-D-galacto and -D-Glycero-D-talo-non-2-ulosonic Acids. Carbohydr. Res 2001, 331, 233–237. [DOI] [PubMed] [Google Scholar]
- (57).Tsvetkov YE; Shashkov AS; Knirel YA; Zähringer U Synthesis and NMR Spectroscopy of Nine Stereoisomeric 5,7-Diacetamido-3,5,7,9-tetradeoxynon-2-ulosonic Acids. Carbohydr. Res 2001, 335, 221–243. [DOI] [PubMed] [Google Scholar]
- (58).Lee YJ; Kubota A; Ishiwata A; Ito Y Synthesis of Pseudaminic Acid, A Unique Nonulopyranoside Derived from Pathogenic Bacteria Through 6-Deoxy-AltdiNAc. Tetrahedron Lett 2011, 52, 418–421. [Google Scholar]
- (59).Liu H; Zhang Y; Wei R; Andolina G; Li X Total Synthesis of Pseudomonas aeruginosa 1244 Pilin Glycan via de Novo Synthesis of Pseudaminic Acid. J. Am. Chem. Soc 2017, 139, 13420–13428. [DOI] [PubMed] [Google Scholar]
- (60).Zunk M; Williams J; Carter J; Kiefel MJ A New Approach Towards the Synthesis of Pseudaminic Acid Analogues. Org. Biomol. Chem 2014, 12, 2918–2925. [DOI] [PubMed] [Google Scholar]
- (61).Williams JT; Corcilius L; Kiefel MJ; Payne RJ Total Synthesis of Native 5,7-Diacetylpseudaminic Acid from N‑Acetylneuraminic Acid. J. Org. Chem 2016, 81, 2607–2611. [DOI] [PubMed] [Google Scholar]
- (62).Hassan MI; Lundgren BR; Chaumun M; Whitfield DM; Clark B; Schoenhofen IC; Boddy CN Total Biosynthesis of Legionaminic Acid, A Bacterial Sialic Acid Analog. Angew. Chem. Int. Ed 2016, 55, 12018–12021. [DOI] [PubMed] [Google Scholar]
- (63).Santra A; Xiao A; Yu H; li W; Li Y; Ngo N; McArthur JB; Chen X A Diazido Mannose Analogue as Chemoenzymatic Synthon for Synthesizing Di-N-acetyllegionaminic Acid-Containing Glycosides. Angew. Chem. Int. Ed 2018, 57, 2929–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Crich D; Li W Efficient Glycosidation of a Phenyl Thiosialoside Donor with Diphenyl Sulfoxide and Triflic Anhydride in Dichloromethane. Org. Lett 2006, 8, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Crich D; Navuluri C Efficient, Highly Stereoselective Synthesis of α-Keto-deoxy-D-glycero-D-galacto-nonulosonic Acid (KDN) Glycosides by Means of the 4,5-O-Carbonate Protecting Group. Angew. Chem. Int. Ed 2010, 49, 3049–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Jensen HH; Nordstrøm LU; Bols M The Disarming Effect of the 4,6-Acetal Group on Glycoside Reactivity: Torsional or Electronic. J. Am. Chem. Soc 2004, 126, 9205–9213. [DOI] [PubMed] [Google Scholar]
- (67).Moumé-Pymbock M; Furukawa T; Mondal S; Crich D Probing the Influence of a 4,6-O-Acetal on the Reactivity of Galactopyranosyl Donors: Verification of the Disarming Influence of the trans-gauche Conformation of C5-C6 Bonds. J. Am. Chem. Soc 2013, 135, 14249–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Kancharla PK; Crich D Influence of Side Chain Conformation and Configuration on Glycosyl Donor Reactivity and Selectivity as Illustrated by Sialic Acid Donors Epimeric at the 7-Position. J. Am. Chem. Soc 2013, 135, 18999–19007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Popik O; Dhakal B; Crich D Stereoselective Synthesis of the Equatorial Glycosides of Legionaminic Acid. J. Org. Chem 2017, 82, 6142–6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Dharuman S; Crich D Determination of the Influence of Side Chain Conformation on Glycosylation Selectivity Using Conformationally Restricted Donors. Chem. Eur. J 2016, 22, 4535–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Dharuman S; Amarasekara H; Crich D Interplay of Protecting Groups and Side Chain Conformation in Glycopyranosides. Modulation of the Influence of Remote Substituents on Glycosylation? J. Org. Chem 2018, 83, 10334–10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Christian R; Schulz G; Brandstetter HH; Zbiral E On the Side-Chain Conformation of N-Acetylneuraminic Acid and its Epimers at C-7, C-8, and C-7,8. Carbohydr. Res 1987, 162, 1–11. [DOI] [PubMed] [Google Scholar]
- (73).Bandgar BP; Zbiral E Strukturelle Abwandlungen an N-Acetylneuraminsaure, 25. Mitt. [I] : Synthese von Methyl-2-α-glycosiden von 4-epi-, 7-epi-, 8-epi- und 7,8-Bis-epi-N-acetylneuraminsaure. Monat für Chemie 1991, 122, 1075–1088. [Google Scholar]
- (74).The conformation about the exocyclic C6-C7 bond is discussed in terms of three staggered conformers: gauche,gauche (gg), gauche,trans (gt) and trans,gauche (tg) where the first term describes the relationship of O7 to O6, and the second the relationship of O7 to C5.; Bock K; Duus JOA Conformational Study of Hydroxymethyl Groups in Carbohydrates Investigated by 1H NMR Spectroscopy, J. Carbohydr. Chem 1994, 13, 513–543. [Google Scholar]
- (75).Brown EB; Brey WS Jr.; Weltner W Jr. NMR of N-Acetyl Neuraminic Acid. Biochim. Biophys. Acta 1975, 399, 124–130. [DOI] [PubMed] [Google Scholar]
- (76).Flippen JL Crystal Structure of β-D-N-Acetylneuraminic Acid Dihydrate (Sialic Acid). Acta Cryst 1973, B29, 1881–1886. [Google Scholar]
- (77).Veluraja K; Rao VSR Theoretical Studies on the Conformation of β-D-N-Acetyl Neuraminic Acid (Sialic Acid). Biochim. Biophys. Acta 1980, 630, 442–446. [DOI] [PubMed] [Google Scholar]
- (78).Rao VSR; Qasba PK; Balaji PV; Chandrasekaran R Conformation of Carbohydrates; Harwood Academic Publishers: Amsterdam, 1998. [Google Scholar]
- (79).Sabesan S; Bock K; Lemieux RU The Conformational Properties of the Gangliosides GMz and GM1 Based on 1H and 13C Nuclear Magnetic Resonance Studies. Can. J. Chem 1984, 62, 1034–1045. [Google Scholar]
- (80).Miljkovic M; Yeagley D; Deslongchamps P; Dory YL Experimental and Theoretical Evidence of Through-Space Electrostatic Stabilization of the Incipient Oxocarbenium Ion by an Axially Oriented Electronegative Substituent During Glycopyranoside Acetolysis. J. Org. Chem 1997, 62, 7597–7604. [Google Scholar]
- (81).Bülow A; Meyer T; Olszewski TK; Bols M The C-4 Configuration as a Probe for the Study of Glycosidation Reactions. Eur. J. Org. Chem 2004, 323–329. [Google Scholar]
- (82).Jensen HH; Bols M Stereoelectronic Substituent Effects. Acc. Chem. Res 2006, 39, 259–265. [DOI] [PubMed] [Google Scholar]
- (83).Smith DM; Woerpel KA Electrostatic Interactions in Cations and their Importance in Biology and Chemistry. Org. Biomol. Chem 2006, 4, 1195–1201. [DOI] [PubMed] [Google Scholar]
- (84).Schreiner E; Zbiral E A Convenient Approach to 3-Deoxy-D-glycero-D-galacto-nonulosonic Acid (KDN), 5-Azido-5-deoxy-KDN and 5-Deoxy-KDN, and their 4-Methylumbelliferyl 2α-Glycosides Liebigs Ann. Chem 1990, 581–586. [Google Scholar]
- (85).Shirai R; Nakamura M; Hara S; Takayanagi H; Ogura H Thermal Rearrangement of N-Acetyl-N-nitrosoneuraminic Acid Derivative: Synthesis of 3-Deoxy-D-Nonulosonic Acid (KDN). Tetrahedron Lett 1988, 29, 4449–4452. [Google Scholar]
- (86).Buda S; Crich D Oxidative Deamination of N-Acetyl Neuraminic Acid: Substituent Effects and Mechanism. J. Am. Chem. Soc 2016, 138, 1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Marra A; Sinaÿ P Stereoselective Synthesis of 2-Thioglycosides of N-Acetylneuraminic Acid. Carbohydr. Res 1989, 187, 35–42. [Google Scholar]
- (88).Furuhata K Chemistry of N-Acetylneuraminic Acid (Neu5Ac). Trends Glycosci. Glycobiol 2004, 16, 143–169. [Google Scholar]
- (89).Dess PB; Martin JC Readily Accessible 12-I-5 Oxidant for the Conversion of Primary and Secondary Alcohols to Aldehydes and Ketones. J. Org. Chem 1983, 48, 4155–4156. [Google Scholar]
- (90).Luche J-L; Rodriguez-Hahn L; Crabbe P Reduction of Natural Enones in the Presence of Cerium Trichloride. J. Chem. Soc., Chem. Commun 1978, 601–602. [Google Scholar]
- (91).Xia J; Abbas SA; Locke RD; Piskorz CF; Alderfer JL; Matta KL Use of 1,2-Dichloro 4,5-dicyanoquinone (DDQ) for Cleavage of the 2-Naphthylmethyl (NAP) Group. Tetrahedron Lett 2000, 41, 169–173. [Google Scholar]
- (92).Navuluri C; Crich D Chemical Diversification of Sialic Acid Glycosides by Stereospecific, Chemoselective Deamination. Angew. Chem. Int. Ed 2013, 52, 11549–11552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Martinelli MJ; Nayyar NK; Moher ED; Dhokte UP; Pawlak JM; Vaidyanathan R Dibutyltin Oxide Catalyzed Selective Sulfonylation of α-Chelatable Primary Alcohols. Org. Lett 1999, 1, 447–450. [Google Scholar]
- (94).Martinelli MJ, Vaidyanathan R, Pawlak JM, Nayyar NK, Dhotke UP, Doecke CW, Zollars LMH, Moher ED, Khau VV, Kosmrlj B, Catalytic Regioselective Sulfonylation of α-Chelatable Alcohols: Scope and Mechanistic Insight. J. Am. Chem. Soc 2002, 124, 3578–3585. [DOI] [PubMed] [Google Scholar]
- (95).Lattrell R; Lohaus G Attempted Total Synthesis of Cephalosporin Derivatives. II. Substitution Reactions with trans-3-(Sulfonyloxy)-2-azetidinones. Synthesis of cis-3-(Acylamino)-4-(alkylthio)-2-azetidinones. Liebigs Ann. Chem 1974, 901–920. [Google Scholar]
- (96).Albert R; Dax K; Link RW; Stuetz AE Carbohydrate Triflates: Reaction with Nitrite, Leading Directly to epi-Hydroxy Compounds. Carbohydr. Res 1983, 118, C5–C6. [Google Scholar]
- (97).Dong H; Rahm M; Thot N; Deng L; Brinck T; Ramström O Control of the Ambident Reactivity of the Nitrite Ion. Org. Biomol. Chem 2013, 11, 648–653. [DOI] [PubMed] [Google Scholar]
- (98).Binkley RW; Ambrose MG Synthesis and Reactions of Carbohydrate Trifluoromethanesulfonates. J. Carbohydr. Chem 1984, 3, 1–49. [Google Scholar]
- (99).Czarniecki MF; Thornton ER Carbon-13 Nuclear Magnetic Resonance Spin-Lattice Relaxation in the N-Acylneuraminic Acids. Probes for Internal Dynamics and Conformational Analysis. J. Am. Chem. Soc 1977, 99, 8273–8279. [Google Scholar]
- (100).Hori H; Nakajima T; Nishida Y; Ohrui H; Meguro H A Simple Method to Determine the Anomeric Configuration of Sialic Acid and its Derivatives by 13C-NMR,. Tetrahedron Lett 1988, 29, 6317–6320. [Google Scholar]
- (101).Haverkamp J; Spoormaker T; Dorland L; Vliegenthart JFG; Schauer R Determination of the β-Anomeric Configuration of Cytidine 5’-Monophospho-N-acetylneuraminic Acid by Carbon-13 NMR Spectroscopy. J. Am. Chem. Soc 1979, 101, 4851–4853. [Google Scholar]
- (102).Prytulla S; Lauterwein J; Klessinger M; Thiem J Configurational Assignment of N-Acetylneuraminic Acid and Analogs via the Vicinal Carbon Hydrogen Coupling Constants. Carbohydr. Res 1991, 215, 345–349. [Google Scholar]
- (103).Knirel YA; Vinogradov EV; Shashkov AS; Dmitriev BA; Kochetkov NK; Stanislavsky EV; Mashilova GM Somatic Antigens of Pseudomonas aeruginosa. The Structure of O-Specific Polysaccharide Chains of P. aeruginosa O10 (Lanyi) Lipopolysaccharides. Eur. J. Biochem 1986, 157, 129–138. [DOI] [PubMed] [Google Scholar]
- (104).Kononov LO Chemical Reactivity and Solution Structure: on the Way to a Paradigm Shift? RSC Adv 2015, 5, 46718–46734. [Google Scholar]
- (105).Yasomanee JP; Demchenko AV Hydrogen-Bond-Mediated Aglycone Delivery (HAD): A Highly Stereoselective Synthesis of 1,2-cis α-D-Glucosides from Common Glycosyl Donors in the Presence of Bromine. Chem. Eur. J 2015, 21, 6572–6581. [DOI] [PubMed] [Google Scholar]
- (106).Escopy S; Geringer SA; De Meo C Combined Effect of the Picoloyl Protecting Group and Triflic Acid in Sialylation. Org. Lett 2017, 19, 2638–2641. [DOI] [PubMed] [Google Scholar]
- (107).Rakotomanomana N; Lacombe J-M; Pavia A Réduction-Acetylation Sélective des Azido-Sucres par le Melange Acide Thioacétique-Thioacétate de Potassium. Carbohydr. Res 1990, 197, 318–323. [Google Scholar]
- (108).Qin C; Schumann B; Zou X; Pereira CL; Tian G; Hu J; Seeberger PH; Yin J Total Synthesis of a Densely Functionalized Plesiomonas shigelloides Serotype 51 Aminoglycoside Trisaccharide Antigen. J. Am. Chem. Soc 2018, 140, 3120–3127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.