

Abstract
Transcription at enhancers is a widespread phenomenon which produces so-called enhancer RNA (eRNA) and occurs in an activity dependent manner. However, the role of eRNA and its utility in exploring disease-associated changes in enhancer function, and the downstream coding transcripts that they regulate, is not well established. We used transcriptomic and epigenomic data to interrogate the relationship of eRNA transcription to disease status and how genetic variants alter enhancer transcriptional activity in the human brain. We combined RNA-seq data from 537 post mortem brain samples from the CommonMind Consortium with cap analysis of gene expression and enhancer identification, using the assay for transposase-accessible chromatin followed by sequencing (ATACseq). We find 118 differentially transcribed eRNAs in schizophrenia and identify schizophrenia-associated gene/eRNA co-expression modules. Perturbations of a key module are associated with the polygenic risk scores. Furthermore, we identify genetic variants affecting expression of 927 enhancers, which we refer to as enhancer expression quantitative loci or eeQTLs. Enhancer expression patterns are consistent across studies, including differentially expressed eRNAs and eeQTLs. Combining eeQTLs with a genome-wide association study of schizophrenia identifies a genetic variant that alters enhancer function and expression of its target gene, GOLPH3L. Our novel approach to analyzing enhancer transcription is adaptable to other large-scale, non-poly-A depleted, RNA-seq studies.
Introduction
The majority of identified common variation affecting risk for schizophrenia (SCZ) falls outside of genes1, where it presumably induces much of the dysregulation in gene expression associated with the disorder2. Instead of directly affecting protein structure, SCZ-associated genetic variants are thought to alter protein abundance by disrupting microRNA3, lncRNA4, and proximal as well as distal enhancer function. Studying these aspects of transcription might, therefore, broaden our understanding of SCZ and mechanistically elucidate the underlying dysregulation of disease associated protein coding genes.
Enhancers are small segments of promoter-distal regulatory DNA elements that increase the expression of target genes. In 2010, Kim et al.5 sequenced rRNA-depleted total RNAs from mouse cortical neurons and discovered bidirectional RNA transcription at enhancers producing so-called enhancer RNA (eRNA). This eRNA was produced in proportion to the activity of the enhancer. Because of the aforementioned regulatory nature of SCZ risk variants, it is plausible that a fraction of these affect transcriptional activity of eRNAs, leading to downstream changes in gene expression.
Spatiotemporal orchestration of gene expression is of critical importance for cellular differentiation and homeostasis, both of which are likely altered in SCZ. Very little is known, however, about the mechanism underlying biogenesis and regulation of eRNAs, or the role they play in gene regulation. Increasing evidence supports the idea that eRNA interaction with chromosomal looping factors alters the 3-dimensional structure of the genome to positively influence enhancer–promoter looping and gene transcription6. The FANTOM5 Consortium examined enhancer function by measuring eRNA transcription through cap end gene expression (CAGE) in a broad set of functional contexts7, 8. Although widespread, only a subset of enhancers identified by chromatin modifications has been found to produce eRNAs. Such enhancers, however, have the highest rate of functional validation7.
To the best of our knowledge, eRNA has not yet been used to examine how genetic variation affects enhancer function and the impact of eRNA on disease is rarely assessed6. This can be attributed, in part, to the fact that RNA libraries are often generated using poly‐A selection, which depletes un-adenylated eRNA transcripts5. We recently performed a large-scale transcriptomic analysis using total RNA-seq without poly-A selection, from 537 post mortem samples diagnosed with SCZ (n=258) and controls (n=279). These samples were part of the collection from the CommonMind consortium (CMC)2. Here, we expand the scope of the CMC study to interrogate enhancer function in SCZ, to examine how genetic variation affects enhancers, and to evaluate specific effects of previously identified SCZ risk variants on enhancer and gene expression.
Materials and Methods
Study population
Total RNA-seq data from the dorsolateral prefrontal cortex and genotyping of 258 patients with SCZ and 279 controls were obtained from the CommonMind consortium (CMC; www.synapse.org/cmc) (Supplementary Figure 1, Supplementary Table 1, Supplementary Methods). The dorsolateral prefrontal cortex was selected based on the transcriptional vulnerability9, neuroimaging studies10 and relevance to cognitive and psychotic symptoms11, which are among the core symptoms of schizophrenia. To validate eRNA quantification, we compared our results with a study of Alzheimer’s disease (AD) (187 AD cases and 73 controls). A flowchart describing our analytical approach is outlined in Supplementary Figure 2.
Identification of enhancer RNA
Enhancers from the FANTOM58 and regions of open chromatin from neuronal/non-neuronal cells were used to interrogate eRNA transcription. We generated cell type-specific maps of open chromatin regions in postmortem brain tissue using ATAC-seq in 8 control CMC samples (Supplementary Methods). Due to the low expression of eRNAs compared to (pre-)mRNAs, we excluded enhancers overlapping exons or introns of Gencode 19 genes as well as enhancers that did not show levels of transcription above the local background (Supplementary Methods). We note that ATAC-seq detects other cis regulatory elements besides enhancers, including promoters and insulators. We removed ATAC-seq non-enhancer elements, by retaining intergenic open chromatin regions, with robust RNA expression.
Quantification of reads and differential expression
Read counts were obtained using the Rsubread package12 for all Ensembl genes and the identified eRNAs. We subsequently retained only genes and eRNAs showing >0.5 Transcripts Per Kilobase Million (TPM) in 50% or more of the individuals. To model the identified expression, we constructed a linear model with disease, known and hidden covariates, as well as ancestry, similarly to Fromer et al. using the voom/limma package13 (Supplementary Methods). This model was used to identify genes and eRNAs showing differential expression between cases and controls, and to obtain expression data adjusted for technical covariates, which was used in downstream analyses. We further conducted gene set enrichment analyses on the genes showing differential expression in SCZ using the GOSeq14 and GSVA15 packages.
Gene co-expression analyses
To further explore the identified SCZ associated genes and eRNAs, we conducted a gene co-expression analysis using the weighted gene co-expression network analysis (WGCNA)16 and coexpp (https://bitbucket.org/multiscale/coexpp) packages. We explored the resultant modules by examining the overlap with the identified differentially expressed genes, gene sets derived from previous SCZ genetic findings, cell type-specific studies or co-expression analyses (hypothesis-driven gene set) and gene sets derived from widely used databases for functional gene classification (hypothesis-free gene set) (Supplementary Methods). In addition, changes in the co-expression structure were interrogated using the sparse-Leading-Eigenvalue-Driven (sLED) package17, which evaluates the difference matrix D between the covariance (or correlation) matrices derived from gene expression in cases and controls. sLED identifies the genes driving the large entries in D using methods from the sparse principal component literature.
Genetic variants affecting gene and eRNA expression
Expression quantitative trait loci were identified with MatrixEQTL18 for both genes (geQTLs) and eRNA (eeQTLs) using a 1MB and 40KB cis-window, respectively. The QTL analysis was performed in a subsample of 415 individuals with Caucasian ancestry. For genetic variants affecting both gene and eRNA expression, we used the Causal Interference Test (CIT)19 to assay if the eRNA regulates gene expression or vice versa. Furthermore, we used our QTLs to explore how genetic variants identified by genome wide association study (GWAS) of SCZ20 affect gene and enhancer expression using the summary data-based Mendelian randomization (SMR) method21.
Validation of eRNA expression and function
To corroborate the RNA-seq based eRNA expression, qPCR was used to validate differentially expressed eRNA and eeQTLs in individuals from the CMC cohort as well as an independent cohort (Supplementary Methods). We further assayed the function of a SCZ associated eRNA using a luciferase assay and its effect on an adjacent gene, through short interfering RNA (siRNA) knock-down.
Results
Differential transcriptional activity of enhancers in schizophrenia
As an investigative step, we first examined the average expression at enhancers from CAGE and ATAC-seq and found a pattern of reads consistent with bidirectional transcription, in keeping with the mode of eRNA transcription (Supplementary Figure 3). Overall, a higher density of RNA-seq reads mapped in FANTOM5 brain-specific enhancers (Supplementary Figure 4), which is expected given the tissue-specificity of enhancer sequences. To assess the robustness of the eRNA quantification, we compared the CMC expression data with that of an independent RNA-seq cohort of post mortem brain samples (AD cohort, see Supplementary Methods) and found a high correlation between the two (Pearson’s r=0.97; Supplementary Figure 6).
After read count quantification and data normalization, 1,387 eRNAs and 21,312 Ensembl genes were expressed at levels sufficient for analysis (Supplementary Figure 5). Considering these transcripts, we investigated how clinical (diagnosis, sex, age at death and genetic ancestry) and technical (post-mortem interval, RNA integrity number [RIN], library batch and institution) covariates correlated with expression. Covariates jointly explained 40% of the variance in gene/eRNA expression and were thus employed to adjust the expression values for all downstream analyses (Supplementary Figure 7).
Comparing expression in SCZ to controls, 1,647 Ensembl genes and 118 eRNAs were expressed differentially after correction for multiple testing at FDR ≤ 5% (Supplementary Data Table 1). Unsupervised hierarchical clustering of the differentially expressed transcripts (DETs) showed case–control distinctions that were independent of institution, PMI, age at death, RIN, and gender (Figure 1a). DETs had modest fold changes, with a mean of 1.09 (range 1.03–1.45) for Ensembl genes and 1.15 (range 1.05–1.34) for eRNAs (Figure 1b). Using an elastic net model for classification, we robustly identified case-control differences between the different CMC brain banks (median area under the receiver operator characteristic curve = 0.86; Supplementary Figure 8). Our Ensembl DETs show strong replication with previous studies (Supplementary Figure 9). Finally, we found high reproducibility of the differentially expressed eRNAs, using qPCR-based quantification for 7 eRNAs in two SCZ cohorts and controls (Supplementary Figure 10).
Figure 1. Differential expression between schizophrenia cases and controls in the DLPFC.
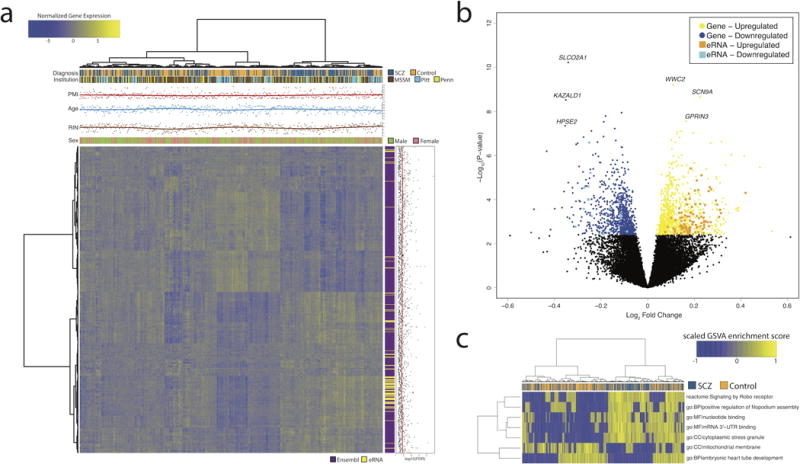
(a) Bivariate clustering of individuals (columns) and genes (rows) depicting the case-vs.-control differences of the 1,647 genes and 118 eRNAs that were differentially expressed. Bars and scatterplots on top show disease status, brain bank (MSSM, Mount Sinai brain bank; Pitt, University of Pittsburgh brain bank; Penn, University of Pennsylvania brain bank), postmortem interval (PMI), age at death (Age), RNA integrity number (RIN), and gender. The vertical color bar and scatter plot illustrate the transcript type and -log10(FDR) values from the differential expression analysis, respectively. (b) Volcano plot illustrating the distribution of log2 fold-changes and -log10 P-values of transcripts in the differential expression analysis. Coloring indicates differentially expressed genes and eRNAs. (c) Bivariate clustering of individuals (columns) and gene sets (rows) based on their GSVA enrichment score for the 7 significant gene sets (Bonferroni-adjusted P ≤ 0.05). The GSVA score indicates whether genes in a pathway are concordantly activated in one direction, either over-expressed (yellow) or under-expressed (blue) relative to the overall population. The color bar indicates disease status.
Using several curated gene sets, we next explored whether the DETs share common pathways or functional categories (Supplementary Methods). After multiple testing corrections, we detected enrichment for 7 gene sets (Figure 1c; Supplementary Data Table 2). The most enriched pathway was signaling by the Round-About (Robo) receptors (combined P = 4.8×10−8; Bonferroni-adjusted P = 1.9×10−4). Nine out of the 26 genes found in the signaling by Robo receptors pathway were DETs, including ABL2, ENAH, GPC1, HGF, ROBO1, ROBO2, SLIT2, SOS1 and SRGAP2. The axonal wiring molecule SLIT and its ROBO receptors are conserved regulators of nerve cord patterning that contribute to wiring brain circuits, cytoskeletal remodeling related to axonal and dendritic branching, and neurogenesis22.
Brain co-expression networks capture SCZ associations
To further explore the transcriptional dysregulation in SCZ, we examined whether eRNAs and genes clustered in similar expression modules based on weighted gene co-expression network analysis (WGCNA). The co-expression network generated from the controls consisted of 15 modules, each containing between 30 and 3,915 transcripts (Supplementary Data Table 3). The eRNAs clustered within specific modules (median count per module = 7) with genes (median count per module = 485), pointing to a putative effect on the regulation of transcription.
We subsequently prioritized modules for association with SCZ by conducting three different analyses: first, enrichment with DETs (Supplementary Table 2); second, enrichment with SCZ candidate genes (Supplementary Table 3); and, third, determining differences in the co-regulation of transcripts among patients with SCZ and controls using a sparse-Leading-Eigenvalue-Driven (sLED) test. In the sLED test, changes in co-expression structure were assessed and drivers of such changes identified by showing high “leverage” (a high leverage gene is one for which the gene-gene co-regulation differs markedly between case and control samples, Supplementary Figure 11). We combined results across all three analyses and the top finding (green) was the only module that had significant support from all three different analyses and survived multiple testing corrections (combined P = 1.7 × 10−4; Bonferroni adjusted P = 2.5 × 10−3) (Figure 2a). More specifically, the green module showed association with DETs (odds ratio = 3.4, P = 2.4 × 10−49), prior genetic associations with SCZ – including genes in GWAS loci (fold-enrichment (FE) = 1.46, P = 0.025) and rare nonsynonymous variants (FE = 1.07, P = 0.008) – as well as differences in the co-regulation patterns in SCZ. Based on the sLED test, we identified 179 out of 1,275 transcripts in the green module as the top genes that have non-zero leverage. These include (i) a primary set of 62 transcripts that account for 99% of the leverage and containing 4 eRNAs, neu41344, enh37929, enh11818 and neu45495; and (ii) a secondary set that includes the remaining 117 transcripts (including 8 eRNAs: gli45010, gli10291, enh19944, gli18022, neu10536, gli26834, gli64554 and gli66753) (Figure 2b). The most notable differences among controls and patients with SCZ arise in the correlation between primary and secondary genes (Figure 2c) and decreased co-expression between these genes in subjects with SCZ (Figure 2d), indicating a decrease of eRNA/gene co-regulation in SCZ.
Figure 2. Co-expression network analysis.
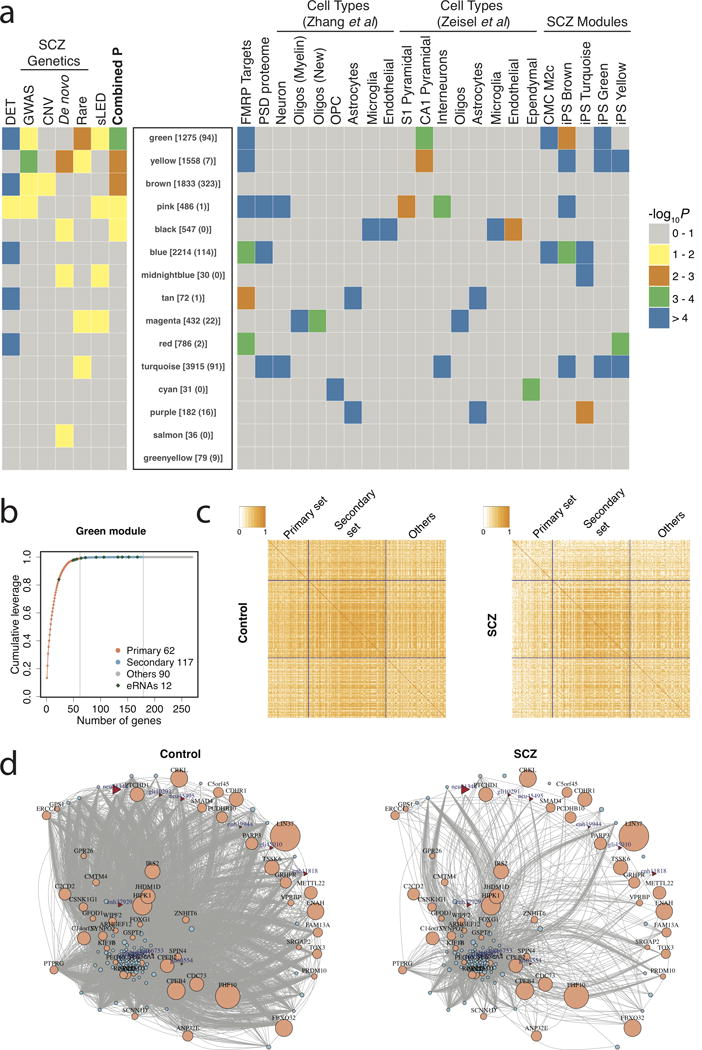
(a) Rank of modules based on a combined P-value including differentially expressed transcripts (DET), prior SCZ genetic associations (GWAS, copy number variants [CNVs], de novo mutations, rare nonsynonymous mutations), and differences in the co-regulation of transcripts among patients with SCZ and controls, using a sparse-Leading-Eigenvalue-Driven (sLED) test. The number of total transcripts and eRNAs in each module is given in brackets and parentheses, respectively. The enrichment of each module with fragile X mental retardation protein (FMRP) targets, postsynaptic density proteins, cell type-specific markers, and SCZ associated modules from prior studies, is depicted at right. (b) Scree plot of sLED leverage in the green module, in which 179 transcripts are detected to have non-zero leverage, including the primary set with 62 transcripts that account for 99% of the leverage, and the secondary set with the remaining 117 transcripts that account for 1% of the leverage. “Others” consist of 90 additional randomly selected transcripts. (c) Absolute correlation matrices among transcript categories in control and SCZ samples. (d) Gene co-expression networks of top genes in the green module in control and SCZ samples. Edges represent absolute correlation |rij| ≥ 0.5 between gene pairs. Ensembl and eRNA transcripts are indicated with circles and triangles, respectively. The size of the nodes indicates the sLED leverage of each transcript. Ensembl transcripts without annotated gene symbols and unconnected transcripts were excluded.
The green module was enriched for multiple pathways and biological processes, including zinc ion binding, Wnt signaling, postsynaptic membrane, and nervous system development (Supplementary Data Table 4). Gene sets identified in prior genetic and co-expression studies that highlighted select neurobiological functions were also enriched in the green module, including targets of fragile X mental retardation protein (FMRP), postsynaptic density proteins, neuronal markers and co-expression modules previously associated with SCZ (Figure 2a and Supplementary Data Table 4). Jointly, these data show that eRNAs are co-expressed with Ensembl transcripts and, for a neuronal module that was enriched in DETs and prior SCZ genetic signals, we found dysregulation of eRNAs to be an important component of the transcriptomic perturbation in SCZ.
Given the green module’s strong association with SCZ (Figure 2a), we wondered if we could determine how genetic variation affected co-expression of the eRNAs and genes within the module. As we have shown previously, experimentally demonstrating causal links from specific genetic variation to DET is not possible due to limited power2. This lack of power extends to questions related to genetic drivers of co-expression. As an alternative approach, we explored whether a composite score of variation (specifically, increased polygenic risk score (PRS) for SCZ), explains dysregulation in the co-expression patterns among cases and controls in this network. To assess the per subject perturbation we used the joint distribution of gene expression in control subjects to impute the expected gene expression for the 167 top Ensembl genes identified by sLED in the green module. For each case subject, we next evaluated the deviance between its actual and expected expression levels (Supplementary Methods). We found that the correlation between PRS and the deviance was 0.11 across case subjects, which was significantly greater than zero by the Pearson correlation test (P = 0.046). We conclude that SCZ patients with higher PRS tend to have stronger dysregulation of the green module, which could affect neuronal and synaptic function.
Generation of gene and eRNA QTLs
To explore how genetic variants affect gene and eRNA expression, we performed gene-level QTL (gene expression QTL or geQTL) and eRNA-level QTL (here termed enhancer expression QTL or eeQTL) analyses using a subset of individuals from our cohort of European descent (N = 415). For generation of geQTLs and eeQTLs, we adjusted for known and hidden confounders (Supplementary Methods). We identified 2,269,239 significant cis-geQTL, (cis window defined at 1Mb) at FDR ≤ 5%, for 15,629 (73%) of 21,312 Ensembl genes (Supplementary Table 4). We found a high concordance with the previously reported CMC geQTLs2 with a proportion of non-null-hypotheses (π1) estimate of 1 and a concordance in the direction of allelic effect of 99.7% (Supplementary Figure 12). For the eeQTLs, we chose to use a smaller cis window of 40 Kb, based on an exploratory analysis (Supplementary Table 4), and in concordance with previous studies23,24. We identified 58,140 significant cis-eeQTL at FDR ≤ 5%, for 927 (67%) of 1,387 eRNAs. The majority of significant eeQTLs are in the immediate proximity of the enhancer sequences (Figure 3a). The 58,140 SNP–eRNA pairs encompassed 50,022 unique SNPs from the 205,814 found within 40 Kb of at least one eRNA (24.3%) and 14% of the eeQTL SNPs (eeSNPs) predicted expression of more than one eRNA.
Figure 3. Association of eRNA-level QTL (enhancer expression QTL or eeQTL) with gene-level QTL (gene expression QTL or geQTL).
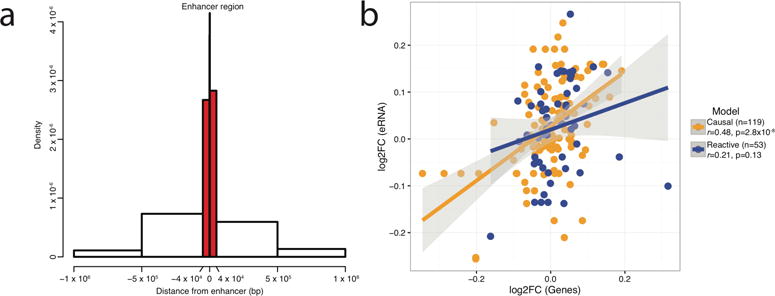
(a) Distribution of cis-eeQTL location relative to the center of the enhancer. The majority of eeQTL SNPs (eeSNPs) were located within the enhancer region (1.5 kb upstream or downstream from the center of the eRNA) or within 40 kb upstream or downstream from the center of the eRNA (highlighted in red). For each eRNA, only the most significant cis-eeQTL was used for this analysis. (b) Correlation scatterplot for log2 fold-changes (log2FC) among cases with SCZ and controls for eRNA-gene pairs that have support for causal (n = 119), or reactive (n = 53) interactions. The correlation was significant only for eRNA-genes that support the causal model.
Prior experiments have reported that, at least for some enhancers, sequence-specific eRNA transcripts contribute to enhancer-mediated transcriptional activation of neighboring coding genes25. To further explore this, we used a causal inference test (CIT)19 to quantify the effect of eRNA regulation on gene expression and identified potential eRNA-gene pairs. CIT assesses eeQTL-geQTL pairs and identifies causal (SNP → eRNA → gene) or reactive (SNP → gene → eRNA) interactions. We examined 60,739 interactions (SNP, eRNA and gene interactions) and found more support for the causal (n = 2,772) than for the reactive (n = 198) model at FDR ≤ 0.05 (Exact binomial test: P < 2.2 × 10−16). This included an excess number of unique eRNA-gene pairs that have support from at least one significant interaction for the causal (n = 119), compared to the reactive (n = 53) model (Exact binomial test: P < 5.3 × 10−7). Linked eRNA-genes based on the CIT causal model show similar differential expression changes in SCZ compared to controls (Pearson’s r = 0.48, P = 2.8×10−8, empirical P < 0.001; Figure 3b), pointing to a potential upstream dysregulation of eRNAs that drives downstream effects of gene expression in SCZ. No significant correlation is observed for eRNA-gene pairs that have support from the reactive model (Pearson’s r = 0.21, P = 0.13, empirical P = 0.37).
Using brain geQTLs and eeQTLs to analyze genetic risk variants
To identify genes and eRNAs with altered expression in SCZ, we combined our geQTLs and eeQTLs with summary statistics from SCZ GWAS using the Summary-based-results Mendelian Randomization (SMR)26 approach. SMR utilizes Mendelian randomization to test for a joint association in GWAS and geQTL/eeQTL data and it compares the profile of association for nearby co-inherited variants in the GWAS and geQTL/eeQTL analyses to assess if the signals are dissimilar in a heterogeneity in dependent instruments (HEIDI) test. If the HEIDI test is significant, then the profiles are dissimilar and the identified GWAS and eQTL signals are less likely to be driven by the same genetic variant; i.e., the overlap can be incidental due to linkage. Applying SMR to SCZ GWAS identified 81 Ensembl genes that were significant at FDR < 0.05 and survived the HEIDI test (PHEIDI > 0.05) (Figure 4a and Supplementary Figure 13a). Among the SCZ genes, 18 were previously reported2 using a different approach and included FURIN, CLCN3, and SNAP91. Using the same statistical criteria, we also identified 2 eRNAs in SCZ (enh3256 and gli10409) (Figure 4a and Supplementary Figure 13b).
Figure 4. Overlap of enhancer and gene expression QTLs with GWAS of schizophrenia.
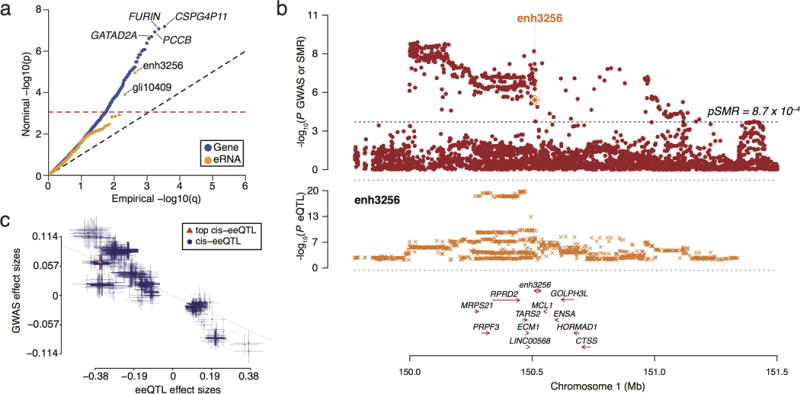
(a) Quantile-quantile plot of the Summary-based-results Mendelian Randomization (SMR) P-values for association of eRNA-level QTLs (eeQTL) and gene-level QTLs (geQTL) with risk variants of schizophrenia (SCZ). The dashed horizontal line indicates SMR significance at FDR < 0.05. (b) Association of enh3256 eeQTLs with SCZ. The top plot shows P-values from the SCZ and SMR P-values for enh3256 (orange diamond) that were significant at FDR < 0.05. The dashed line indicates SMR significance at FDR < 0.05. The bottom plot shows the P-values from the eeQTL analysis of enh3256. (c) The effect sizes and standard errors (error bars) of SCZ GWAS SNPs used for the HEIDI test are plotted against enh3256 eeSNPs. The dashed line represents the SMR estimate of bxy at the top cis-eeQTL (red triangle). Notice that the index eeQTL SNP (top cis-eeQTL in the legend) is associated with increased risk for SCZ and lower expression of enh3256.
The most significant SCZ eRNA, enh3256, was located in a locus that reached genome-wide significance (chr1:150,510,569-150,510,713 [hg19]; index SNP rs140505938; Figure 4b and 4c). We note that the eeSNP with the largest effect size for enh3256 is located within the enhancer sequence (rs72700813; chr1: 150,509,544 [hg19]). Interestingly, there was support from the CIT causal model that enh3256 regulates the GOLPH3L gene (Pcausal=0.031, Preactive=0.063, Pperm=0.009, FDRperm=0.044). GOLPH3L is localized to the Golgi apparatus and is required for efficient anterograde trafficking27. This finding is biologically plausible as the Golgi apparatus is crucial for proper forward trafficking of ion channels, receptors and other signaling molecules in neurons. These functions are known to be dysregulated in SCZ28. The second most significant eRNA, gli10409, was also within a SCZ associated region20 (chr11:109,463,594-109,464,321 [hg19]; index SNP rs12421382). Here, based on the CIT analysis, no association with any Ensembl transcripts was found. We validated both of these eeQTLs in a subset of 70 cases with SCZ and 104 controls using qPCR and an independent cohort of 21 patients with SCZ and 62 controls (Supplementary Figure 14).
Functional validation of regulatory role for enh3256 on GOLPH3L
We next assessed the enhancer activity of the SCZ-associated eRNA enh3256 in vitro and found a significant effect in a luciferase assay using a construct that included the full 145bp enhancer sequence (t-test: t = 42.73, df = 51, P = 4.2 × 10−28; Figure 5a). Subsequently, we examined the activity of smaller 75bp overlapping enhancer fragment sequences and mapped the activity to one such fragment (t-test: t = 49.81, df = 42, P = 5.5 × 10−39; Figure 5a). Both the full length and the active fragment of the enhancer resulted in more than 600% increased luciferase activity compared to empty pGL4.24 vector. To investigate the potential regulatory role of enh3256 eRNA on the adjacent GOLPH3L encoding gene, we designed two specific short interfering RNAs (siRNAs) directed against the active fragment of the enhancer. The effect of a siRNA-mediated knockdown was subsequently determined by qPCR quantification of eRNA and GOLPH3L with two unique Taqman probes per transcript. This revealed that the induction of both enh3256 eRNA and of the adjacent GOLPH3L coding gene was significantly inhibited in the presence of siRNAs, 48 hours after transfection (Figure 5b). Overall, these findings confirmed the enhancer activity of enh3256 eRNA and validated a regulatory role for enh3256 in GOLPH3L gene expression.
Figure 5. Examining the enhancer activity and regulatory role of enh3256 in GOLPH3L expression.
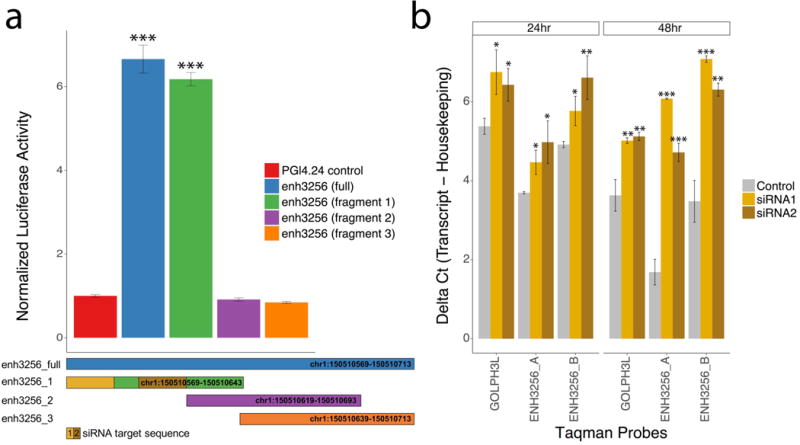
(a) Examining luciferase expression driven by full length (145 bp) and smaller, overlapping, 75 bp fragments of the enh3256 sequence in HEK293 cells. The full-length construct and fragment 1 resulted in increased luciferase activity compared to empty pGL4.24 vector and fragments 2 and 3. (b) qPCR analysis of enh3256 eRNA and GOLPH3L mRNA for HEK293 cells transfected with control and two different siRNAs targeting enh3256 (siRNA1 and siRNA2). qPCR quantification was performed using Taqman probes measuring enh3256 (ENH3256_A and ENH3256_B) and GOLPH3L, 24 and 48 hours after transfection. Higher delta Ct values indicate lower relative expression for each Taqman probe. Taqman probe expression was normalized to b-2-microglobulin in all cases. Expression of luciferase activity was normalized to Renilla Luciferase. Data represent mean ± standard deviation. Statistical significance was determined by two-tailed Student’s t-test. *P < 0.01; **P < 0.05; and ***P < 0.005 versus control.
Discussion
In complex genetic traits like SCZ, most genetic risk variants are non-coding and, as such, are believed to affect gene regulation and, thus, protein abundance rather than protein structure and function2, 29. Therefore, in order to further our understanding of complex genetic traits, a broader understanding of the regulation of gene expression is desirable. Here, we used a novel approach to analyze enhancer transcription using existing, large-scale RNA-seq data from the CommonMind Consortium2. Our analyses had three major goals: first, to detect differences in the transcript levels of eRNA and coding genes; second, to identify perturbations in the eRNA/gene co-regulation driven by the polygenic risk score for SCZ; and, third, to integrate transcript levels with genetics as a means to describe associations of SCZ risk variants with enhancer transcription.
We found replicable differences in the expression levels of coding genes and transcribed eRNAs in cases with SCZ compared to controls. These changes affected a large number of transcripts (1,765 after multiple testing corrections) and were subtle (average fold changes of 1.1), which is consistent with the polygenic nature of genetic risk20 and transcriptome dysregulation2 underlying SCZ. Differentially expressed transcripts are not randomly distributed but, instead, converge to common biological processes, including the Round-About (Robo) receptors pathway, which is involved in cytoskeletal remodeling related to axonal and dendritic branching, and neurogenesis22 during early development. While our study uses postmortem brain tissue from adult cases with SCZ, enrichment of differentially expressed transcripts with the Round-About (Robo) receptors pathway has previously been reported in neurons derived from human induced pluripotent stem cells (hiPSCs) of cases with SCZ compared to controls30. Because gene expression profiles of hiPSC-derived neurons more closely resemble fetal brain tissue31, this provides additional evidence for dysregulation of the Round-About (Robo) receptors pathway in earlier developmental stages.
Coordinated expression of genes is an essential feature of the development and maintenance of cells in the human brain32. We show that one subnetwork of co-expressed genes, dubbed the green module, shows far less correlation structure in the DLPFC of SCZ subjects compared to controls. Intriguingly, we show that in patients with SCZ, perturbation of predicted expression of key genes in the green module – predicted on the basis of co-expression patterns in controls – is positively associated with increased polygenic risk score. This result has potentially important implications for the etiology of SCZ. It is now commonly accepted that liability to SCZ typically emerges from polygenic inheritance, the combined effect of thousands of risk alleles, each with only a small impact on liability20. It remains a mystery, however, why subjects, each representing a random draw of myriad risk alleles, present with the constellation of symptoms we recognize as SCZ. Our results suggest that increased risk score, regardless of what alleles contribute to that score, leads to increased perturbation of the green module. If this module is a driver of liability for SCZ, as we suspect, this could be a mechanism for how polygenic risk translates in to SCZ associated features. It is worth noting that the relationship between the PRS and perturbation of gene expression in the green module is modest, as we might expect for a variety of reasons, including noisy measurements of gene expression and the limited predictive power of the PRS. Moreover, it will be critical to determine if this pattern can be replicated across other studies. If it can, this module could be key to understanding the etiology of, and treatment for, SCZ.
By integrating genetics with eRNA transcription, we generated the first QTL map of eRNAs that we further leveraged to address two questions: (1) Do eRNA transcripts contribute to enhancer-mediated transcriptional activation of neighboring coding genes? (2) Are eRNA transcripts affected by SCZ risk variants? To address the first question, we applied the causal inference test and found more support for the SNP → eRNA → gene, compared to the SNP → gene → eRNA model. This result is consistent with the current notion of eRNA regulatory effects on gene25. We then integrated geQTLs and eeQTLs with summary statistics from a SCZ GWAS using the SMR approach to identify genes and eRNAs with altered expression in SCZ. This analysis identified a genetic variant that, through altered transcription of enh3256, affects expression of GOLPH3L. Experimental manipulation of enh3256 replicated the impact on GOLPH3L expression in vitro.
An important benefit to our approach is that it can be applied to any total RNA-seq experiment to extract information about enhancer activity at little or no additional cost. There are, however, several shortcomings to the approach. Some eRNAs are too unstable and/or expressed at levels too low to be interrogated, unless very deep sequencing or a more targeted approach is used. In addition, eRNAs overlapping introns and exons had to be excluded, as it was impossible to tell which reads belonged to the enhancer and which to the (pre-)mRNA of the gene. If the more expensive and, thus, less frequently employed stranded total RNA-seq approach were used, then more enhancers could be interrogated by taking into account the strand from which the reads originated. Finally, while we have shown the utility of studying eRNAs in SCZ, our study does not address the relative importance of genetic variants affecting different families of regulatory RNA molecules such as miRNA and lncRNA. A direct comparison of the association of each RNA species with SCZ can be addressed in future studies and will require the presence of high-dimensional datasets in the same individuals, quantifying coding genes, eRNAs, miRNAs and lncRNAs.
As enhancer derived RNAs are generally less well characterized, interpreting the biological importance of a trait-associated enhancer is often less straightforward than that for a protein-coding gene. Overall, our study addressed this by examining enhancer and gene co-expression, by using causal inference to link eRNA and genes, by co-localizing eeQTL with SCZ risk variants and by validating the effect of a schizophrenia-associated eRNA on a target gene, GOLPH3L, using siRNA knock-down. Large-scale studies conducted as part of the PsychENCODE Project33 will examine how genetic variants affect histone modification, chromatin accessibility, and other epigenomics features that could further our understanding of the gene regulatory mechanisms implicated in SCZ.
Supplementary Material
Consortia.
The CommonMind Consortium includes: Menachem Fromer, Douglas M Ruderfer, Hardik R Shah, Lambertus L Klei, Kristen K Dang, Thanneer M Perumal, Benjamin A Logsdon, Milind C Mahajan, Lara M Mangravite, Laurent Essioux, Hiroyoshi Toyoshiba, Raquel E Gur, Chang-Gyu Hahn, David A Lewis, Vahram Haroutunian, Barbara K Lipska, Joseph D Buxbaum, Keisuke Hirai.
Acknowledgments
This work was supported by the National Institutes of Health – R01AG050986 (Roussos), R01MH109677 (Roussos), R37MH057881 (Devlin, Roeder) and R01MH109900 (Roeder) –, Brain Behavior Research Foundation (20540 Roussos), Alzheimer’s Association (NIRG-340998 Roussos) and the Veterans Affairs (Merit grant BX002395 Roussos). This study was additionally funded by The Lundbeck Foundation, Denmark (Grant number R102-A9118). Further, this work was supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai. This paper is dedicated to the memory of Pamela Sklar.
The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
RNA-seq and genotyping data were generated as part of the CommonMind Consortium supported by funding from Takeda Pharmaceuticals Company Limited, F. Hoffman-La Roche Ltd and NIH grants R01MH085542, R01MH093725, P50MH066392, P50MH080405, R01MH097276, R01-MH-075916, P50M096891, P50MH084053S1, R37MH057881 and R37MH057881S1, HHSN271201300031C, AG02219, AG05138 and MH06692. Brain tissue for the study was obtained from the following brain bank collections: the Mount Sinai NIH Brain and Tissue Repository, the University of Pennsylvania Alzheimer’s Disease Core Center, the University of Pittsburgh NeuroBioBank and Brain and Tissue Repositories and the NIMH Human Brain Collection Core. CMC Leadership: Pamela Sklar, Joseph Buxbaum (Icahn School of Medicine at Mount Sinai), Bernie Devlin, David Lewis (University of Pittsburgh), Raquel Gur, Chang-Gyu Hahn (University of Pennsylvania), Keisuke Hirai, Hiroyoshi Toyoshiba (Takeda Pharmaceuticals Company Limited), Enrico Domenici, Laurent Essioux (F. Hoffman-La Roche Ltd), Lara Mangravite, Mette Peters (Sage Bionetworks), Thomas Lehner, Barbara Lipska (NIMH).
Footnotes
Conflict of Interest
The authors declare no competing financial interests.
References
- 1.Roussos P, Mitchell Amanda C, Voloudakis G, Fullard John F, Pothula Venu M, Tsang J, et al. A Role for Noncoding Variation in Schizophrenia. Cell Reports. 2014;9(4):1417–1429. doi: 10.1016/j.celrep.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nature neuroscience. 2016;19(11):1442–1453. doi: 10.1038/nn.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hauberg ME, Roussos P, Grove J, Børglum AD, Mattheisen M. Analyzing the Role of MicroRNAs in Schizophrenia in the Context of Common Genetic Risk Variants. JAMA psychiatry. 2016;73(4):369–377. doi: 10.1001/jamapsychiatry.2015.3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu J, Xu J, Pang L, Zhao H, Li F, Deng Y, et al. Systematically characterizing dysfunctional long intergenic non-coding RNAs in multiple brain regions of major psychosis. Oncotarget. 2016;7(44):71087–71098. doi: 10.18632/oncotarget.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim T-K, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465(7295):182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nature reviews Genetics. 2016;17(4):207–223. doi: 10.1038/nrg.2016.4. [DOI] [PubMed] [Google Scholar]
- 7.Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507(7493):455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arner E, Daub CO, Vitting-Seerup K, Andersson R, Lilje B, Drabløs F, et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science. 2015;347(6225):1010–1014. doi: 10.1126/science.1259418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roussos P, Katsel P, Davis KL, Siever LJ, Haroutunian V. A system-level transcriptomic analysis of schizophrenia using postmortem brain tissue samples. Archives of general psychiatry. 2012;69(12):1205–1213. doi: 10.1001/archgenpsychiatry.2012.704. [DOI] [PubMed] [Google Scholar]
- 10.Bora E, Fornito A, Radua J, Walterfang M, Seal M, Wood SJ, et al. Neuroanatomical abnormalities in schizophrenia: a multimodal voxelwise meta-analysis and meta-regression analysis. Schizophrenia research. 2011;127(1-3):46–57. doi: 10.1016/j.schres.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 11.Barch DM, Sheffield JM. Cognitive impairments in psychotic disorders: common mechanisms and measurement. World Psychiatry. 2014;13(3):224–232. doi: 10.1002/wps.20145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic acids research. 2013;41(10):e108–e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biology. 2010;11(2):R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC bioinformatics. 2013;14(1):7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Statistical applications in genetics and molecular biology. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
- 17.Zhu L, Lei J, Devlin B, Roeder K. Testing high-dimensional covariance matrices, with application to detecting schizophrenia risk genes. Ann Appl Stat. 2017;11(3):1810–1831. doi: 10.1214/17-AOAS1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28(10):1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Millstein J, Zhang B, Zhu J, Schadt EE. Disentangling molecular relationships with a causal inference test. BMC genetics. 2009;10:23. doi: 10.1186/1471-2156-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.PGC-SCZ. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–487. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 22.Blockus H, Chédotal A. Slit-Robo signaling. Development. 2016;143(17):3037–3044. doi: 10.1242/dev.132829. [DOI] [PubMed] [Google Scholar]
- 23.Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nature neuroscience. 2016;19(1):40–47. doi: 10.1038/nn.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482(7385):390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lam MTY, Li W, Rosenfeld MG, Glass CK. Enhancer RNAs and regulated transcriptional programs. Trends in Biochemical Sciences. 2014;39(4):170–182. doi: 10.1016/j.tibs.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nature genetics. 2016 doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 27.Ng MM, Dippold HC, Buschman MD, Noakes CJ, Field SJ. GOLPH3L antagonizes GOLPH3 to determine Golgi morphology. Molecular biology of the cell. 2013;24(6):796–808. doi: 10.1091/mbc.E12-07-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Landek-Salgado MA, Faust TE, Sawa A. Molecular substrates of schizophrenia: homeostatic signaling to connectivity. Molecular psychiatry. 2016;21(1):10–28. doi: 10.1038/mp.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fullard JF, Halene TB, Giambartolomei C, Haroutunian V, Akbarian S, Roussos P. Understanding the genetic liability to schizophrenia through the neuroepigenome. Schizophrenia research. doi: 10.1016/j.schres.2016.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473(7346):221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brennand K, Savas JN, Kim Y, Tran N, Simone A, Hashimoto-Torii K, et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Molecular psychiatry. 2015;20(3):361–368. doi: 10.1038/mp.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478(7370):483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Psych EC, Akbarian S, Liu C, Knowles JA, Vaccarino FM, Farnham PJ, et al. The PsychENCODE project. Nature neuroscience. 2015;18(12):1707–1712. doi: 10.1038/nn.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.