

Abstract
BI 425809 is a potent and selective glycine transporter 1 (GlyT1) inhibitor being developed for the treatment of cognitive impairment in Alzheimer disease and schizophrenia. Translational studies evaluated the effects of BI 425809 on glycine levels in rat and human cerebrospinal fluid (CSF). Oral administration of BI 425809 in rats induced a dose‐dependent increase of glycine CSF levels from 30% (0.2 mg/kg, not significant) to 78% (2 mg/kg, P < 0.01), relative to vehicle. Similarly, oral administration of BI 425809 in healthy volunteers resulted in a dose‐dependent increase in glycine CSF levels at steady state, with a mean 50% increase at doses as low as 10 mg. The peak plasma concentration (Cmax) of BI 425809 was achieved earlier in plasma than in CSF (t max 3–5 vs. 5–8 hours, respectively). Generally, BI 425809 was safe and well tolerated. These data provide evidence of functional target engagement of GlyT1 by BI 425809.
Study Highlights.
What is the current knowledge on this topic?
Schizophrenia and Alzheimer’s disease (AD) are characterized by abnormalities in glutamatergic pathways related to NMDA receptor hypofunction. Inhibition of GlyT1 on the presynaptic membrane or astrocytes is hypothesized to increase glycine levels within the synapse. The NMDA receptor function may be enhanced by increasing levels of its co‐agonist, glycine, within the synaptic cleft, which may lead to improvements in cognitive function.
What question did this study address?
What are the effects of administration of BI 425809, a novel GlyT1 inhibitor, on glycine levels in both rat and human CSF?
What does this study add to our knowledge?
A dose‐dependent increase of glycine levels in CSF was demonstrated in both rats and healthy volunteers. These data provide evidence for functional target engagement for BI 425809.
How might this change clinical pharmacology or translational science?
These data provide evidence for GlyT1 inhibition by BI 425809 in the human brain, and, therefore, a potential therapeutic mechanism for cognitive improvement in AD and schizophrenia. These findings also support the selection of BI 425809 dosing regimens for future studies.
Schizophrenia and Alzheimer’s disease (AD) are characterized by abnormalities in glutamatergic pathways related to N‐methyl‐D‐aspartate (NMDA) receptor hypofunction.1, 2 These abnormalities have been associated with cognitive impairment in both disorders.3 NMDA receptors are activated by glutamate as well as by glycine, which acts as a necessary co‐agonist at the glycine modulatory site.4 The inhibition of glycine transporter 1 (GlyT1), a transporter that functions to regulate the synaptic levels of glycine, may, therefore, improve NMDA receptor hypofunction by elevating the levels of glycine in the synaptic cleft.4 It is hypothesized that increased NMDA receptor signaling results in an increase in long‐term potentiation and synaptic plasticity, which may improve cognitive function and memory.5
BI 425809 is a novel potent and selective GlyT1 inhibitor being developed for the treatment of cognitive impairment associated with schizophrenia and AD.6, 7 It has previously been observed that the half‐life value of BI 425809 is ~40 hours, consistent with attainment of steady state within 8 days after administration.6, 8 The objective of the preclinical study described here was to characterize the potency and selectivity of BI 425809, and its effects on glycine concentration in rat cerebrospinal fluid (CSF). To translate this preclinical study into humans, BI 425809 was further evaluated in a phase I study with healthy volunteers. The objectives of this phase I study were threefold: to explore the pharmacokinetics (PKs) of BI 425809 in CSF and plasma following multiple dosing over a period of 14 days, to investigate its pharmacodynamics (PDs) by means of measuring glycine levels in CSF, and to evaluate safety and tolerability of BI 425809.
Methods
Preclinical study design and methodology
Animals
Procedures involving animals and their care were conducted in conformity with institutional and European Union guidelines (EEC Council Directive 86/609) and were approved by the Ethical Committee of the responsible regional council (Tübingen).
Adult male Wistar rats (Janvier, Le Genest Saint Isle, France), weighing 200–220 g at day of arrival, were housed in groups of four per cage, with a controlled temperature (22 ± 1°C) and a 12‐hour light‐dark cycle (lights on at 6 am). Standard rodent food and tap water were freely available. The rats were left undisturbed for a minimum of 5 days to allow them to adjust to a new environment until the beginning of the experiment.
Determination of potency and selectivity
The molecular potency of BI 425809 for GlyT1 was determined by inhibition of [3H]‐glycine uptake in human SK‐N‐MC cells and rat primary cortical neurons. Briefly, the SK‐N‐MC cells were grown on scintillating Cytostar‐96‐well microplates and were incubated for 20 minutes. The inhibitor was diluted to concentrations between 0.9 and 3000 nM in Hank's Balanced Salt Solution buffer, containing 5 mM of alanine. The uptake of glycine was initiated by the addition of 250 nM [3H]‐labelled glycine (0.0125 mCi/mL) and scintillation counting was started immediately over a period of 57 minutes using a MicroBeta Trilux scintillation counter. The neurons used for these experiments were prepared according to standard protocols from rat cortex (E18) and had been differentiated in vitro for 7, 11, or 18 days.9 The assay was run as described for the SK‐N‐MC cells with the exception of using 200 nM [3H]‐labelled glycine (0.01 mCi/mL). The mean half maximal inhibitory concentration (IC50) was calculated from the IC50 obtained from each experiment individually; further details on statistical analysis are given in the respective paragraph below.
Selectivity against GlyT2 was evaluated by inhibition of [3H]‐glycine uptake in HEK cells overexpressing human GlyT2 following the same protocol as described above for the SK‐N‐MC cells. Selectivity of BI 425809 against other off‐targets (using receptor binding assays against 102 different targets at 10 μM) was evaluated at MDS Pharma Services (Taipei, Taiwan).
Collection of CSF from rats and determination of glycine in CSF
Glycine in CSF was measured using a modified method of Perry et al.10. Briefly, rats were placed in an experimental room for 1 hour prior to drug administration. Subsequently, the rats were administered orally with a single dose of vehicle (0.5% Natrosol, containing 0.01% Tween‐80; n = 8), 0.2 mg/kg (n = 8), 0.6 mg/kg (n = 8), or 2 mg/kg (n = 8) of BI 425809, with an application volume of 5 mL/kg body weight. After 90 minutes, the animals were fixed in a stereotaxic apparatus under anesthesia (with subcutaneous inactin 0.85 mL/kg plus ketavet 0.75 mL/kg), and directly thereafter, blood was collected, and CSF was collected via puncture of the cisterna magna. Plasma samples were generated from blood, then frozen in individual tubes and stored at −20°C prior to determination of compound levels. Collected CSF samples (50–100 μl) were frozen in individual tubes in liquid nitrogen and stored at −70°C prior to being used for the determination of glycine and compound levels. Compound levels were determined by high performance liquid chromatography‐tandem mass spectrometry (HPLC‐MS/MS). Glycine was quantified in CSF according to the method described by Voehringer et al.11
Clinical study design and methodology
Study design
This nonrandomized, open‐label, sequential‐group, multiple‐dose phase I study was conducted at a single site in Belgium in 25 healthy male volunteers in accordance with the International Conference for Harmonization (ICH) of Technical Requirements for Pharmaceuticals for Human Use, Good Clinical Practice (GCP), and local legislation in accordance with the principles of the Declaration of Helsinki (ClinicalTrials.gov identifier: NCT02362516). The study was reviewed and approved by the independent ethics committee at the study center. Subjects provided their written informed consent prior to enrollment in the study.
Treatments
Subjects were assigned to four sequential dose groups (BI 425809 5, 10, 25, or 50 mg) with six subjects planned per group. The dose range for this trial was selected based on the data obtained in the first‐in‐human single rising dose trial.12 The 10‐ and 25‐mg doses were expected to reflect a potential human therapeutic dose, the 5‐mg dose was expected to be subtherapeutic, and the 50‐mg dose was expected to be in the supratherapeutic range. Each subject received the respective dose of BI 425809, once daily for 14 consecutive days in the morning. The dose groups were assessed consecutively in ascending order of doses, with a time period of at least 7 days between the first drug administration in the previous dose group and the first drug administration of the subsequent dose group. The decision to proceed to the next dose group was based on the safety and tolerability data of the preceding groups.
CSF was serially collected through a lumbar catheter for 14 hours after the first dose and through single lumbar puncture before the last dose on day 14. On day 1, three predose samples were taken within 2 hours of catheter placement at ~30‐minute intervals. Subsequent CSF samples were taken over the course of day 1 at ~1‐hour intervals. Plasma samples were collected over the course of day 1 at ~1‐hour intervals, at 24‐hour intervals on days 2–4, and at 48‐hour intervals on days 6–12. On day 14, plasma samples were collected over the course of the day at ~1‐hour intervals.
Subjects
Subjects included in this study were healthy male volunteers, between the ages of 18 and 55 years, with a body mass index (BMI) of 18.5–29.9 kg/m2. Subjects were deemed healthy according to the investigator's assessment, which was based on a complete medical history, including a physical examination, visual tests (fundoscopy) and vital signs (blood pressure and pulse rate), 12‐lead electrocardiogram (ECG), and clinical laboratory tests.
Subjects were excluded if they showed evidence of a concomitant disease upon examination or there were abnormal findings from the medical examination, vital signs assessment, ECG, or laboratory tests, deemed clinically relevant by the investigator. Other exclusion criteria included: gastrointestinal (GI); hepatic, renal; respiratory; cardiovascular; metabolic; immunological; or hormonal disorders; diseases of the central nervous system (CNS); other neurological disorders or psychiatric disorders; history of relevant orthostatic hypotension; fainting spells or blackouts; chronic or relevant acute infections; and a history of relevant allergy/hypersensitivity.
Additionally, criteria for removal of individual subjects included: an adverse event (AE) or clinically significant laboratory change or abnormality that warranted discontinuation of treatment, as judged by the investigator; withdrawal of consent; and suicidal behavior or suicidal ideation.
Study end points
Pharmacokinetics
The primary PK end points, which were determined for BI 425809 after the first dose, included: area under the concentration‐time curve (AUC) of the analyte in plasma and CSF over the time interval from 0–14 hours (AUC0–14) and the maximum measured concentration of the analyte in plasma and CSF (Cmax). The primary PK end point, before the last dose, was the concentration of the analyte in plasma and CSF at the time point 312 hours (C 312), meaning 312 hours after the first administration of BI 425809, which corresponds to the hypothesized concentration at steady state. Concentrations of BI 425809 in plasma and CSF were determined by a validated HPLC‐MS/MS method (Covance, Salt Lake City, UT). Noncompartmental analysis of plasma concentration‐time data was performed using Phoenix WinNonlin (professional Network version 5.2, Pharsight Corporation, Cary, NC).
Additional end points of interest, after the first dose, included, among others: C max ratio of BI 425809 in CSF compared with plasma and the time from dosing to maximum measured concentration of the analyte in plasma and CSF (t max).
Pharmacodynamics
All PD end points, except for time to maximum glycine concentration and area under the biomarker effect vs. time curve ratio from time point 0 hours to 14 hours (AUECdiff, 0–14)), were expressed as percent change from baseline in glycine in CSF. The PD end points determined for glycine, after the first dose, included: time to maximum glycine concentration (t max); maximum increase in glycine from baseline (E max); increase in glycine from baseline at the time point 14 hours (E14); and the ratio between the area under the biomarker effect vs. time curve on treatment from time point 0 hours to 14 hours (AUECtreatment) and the baseline area under the biomarker effect versus time curve from time point 0 hours to 14 hours (AUECbase; AUECdiff, 0–14). The PD end point before the last BI 425809 dose was the increase in glycine from baseline 312 hours after the first dose (E312). Glycine concentrations in CSF were determined by a validated HPLC‐MS/MS method (Boehringer Ingelheim International GmbH, Drug Metabolism and Pharmacokinetics, Biberach, Germany). The assay used is an absolute quantitative assay for exploratory biomarkers using the Marfey's derivative of glycine as analyte.13
Safety
Safety and tolerability of BI 425809 was assessed by qualifying and quantifying the number of subjects with drug‐related AEs.
Statistical analysis
Preclinical
The IC50 values were calculated using a nonlinear fit log (inhibitor) vs. response (four parameter analysis). Comparisons between vehicle and compound‐treated groups of animals were performed by one‐way analysis of variance (ANOVA) followed by the Dunnett's post hoc analysis.
Clinical
The relationship between the exposures of BI 425809 in CSF compared with plasma on day 1 was explored descriptively using a mixed model. Dose proportionality of the primary end points of BI 425809 in plasma and CSF (AUC0‐14, C max, and C 312) was explored using a power model; a 95% confidence interval (CI) was computed for the slope. Attainment of steady state was assessed using the trough plasma concentrations of BI 425809 between days 2 and 14 and the concentrations taken 24 hours after the last dose for each dose level. Pairwise comparisons of concentrations were performed using two‐sided 95% CIs. Safety data were analyzed using descriptive statistics. All statistical analyses were performed using SAS (current version 9.4, SAS Institute, Cary, NC).
Results
Preclinical
The IC50 of BI 425809 was determined to be 5.2 ± 1.2 nM in rat primary neurons (Figure 1) and 5.0 nM in human SK‐N‐MC cells. In addition, no activity of BI 425809 against the related glycine transporter, GlyT2, was demonstrated nor against other off‐targets (IC50 > 10 μM; data not shown).
Figure 1.
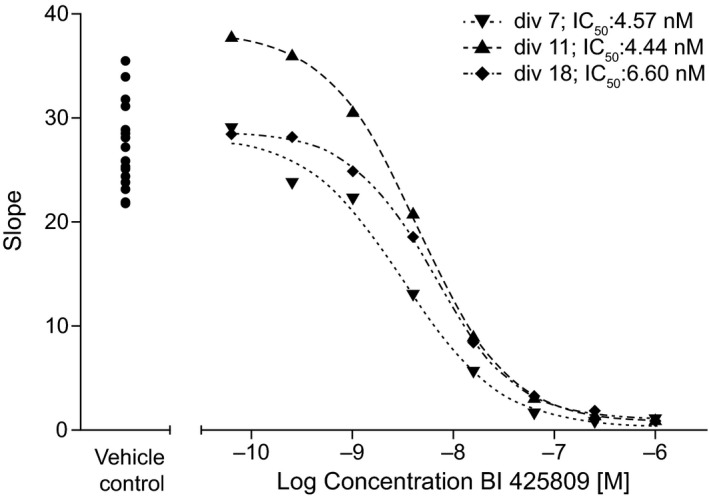
Inhibition of [3H]‐glycine uptake* through GlyT1 by BI 425809 in rat primary cortical neurons. div X, differentiated for X days; GlyT1, glycine transporter 1; IC 50, half maximal inhibitory value. *The velocity of glycine uptake into primary neurons kept for 7, 11, or 18 days in culture (div 7, 11, or 18) is depicted as function of the logarithmic concentration of BI 425809 in the culture media.
As shown in Figure 2, single oral administration of BI 425809 induced a dose‐dependent increase of glycine CSF levels. Increases of glycine reached on average 30% at the dose of 0.2 mg/kg (not significant), 61% at 0.6 mg/kg (P < 0.05), and 78% at 2 mg/kg (P < 0.01), relative to vehicle. At the time point of plasma/CSF sampling, average plasma exposure levels of BI 425809 were 79, 217, and 723 nM at 0.2, 0.6, and 2 mg/kg, respectively. The CSF to plasma ratio was 2% for all dose groups.
Figure 2.
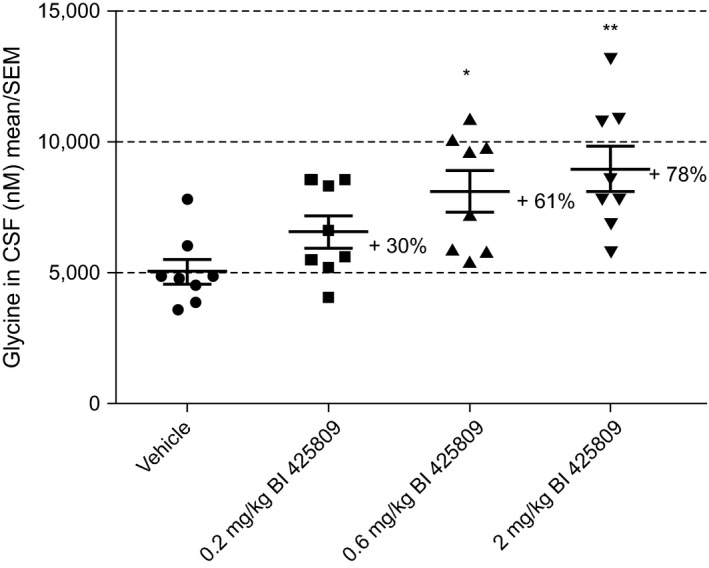
Effects of BI 425809 on glycine levels in the rat cerebrospinal fluid (CSF). Data are expressed as mean ± SEM of eight animals per group; *P < 0.05, **P < 0.01 as compared with vehicle, Dunnett's post hoc analyses.
Clinical
Study population
Of the 25 subjects who entered the trial, 22 (88.0%) completed the planned observation time according to the protocol. Two subjects (8.0%) discontinued due to AEs (moderate headache, nausea, vomiting (5‐mg dose group), and moderate procedural headache (10‐mg dose group)). These AEs were of moderate intensity and were not considered drug‐related by the investigator. One subject (4.0%) in the 10‐mg dose group withdrew consent for further participation due to personal circumstances.
The treated subjects comprised 24 white and 1 Asian healthy male volunteers. The median (minimum, maximum) age was 46 years (20 and 56 years) and the mean (SD) BMI was 25.3 (2.8) kg/m2. The demographic characteristics of the subjects are summarized in Table S1.
Pharmacokinetics
The PK parameters in plasma and CSF are summarized in Table 1. Plasma concentrations of BI 425809 increased dose‐dependently over the dose range tested. After reaching C max, plasma concentrations of BI 425809 decreased in a biphasic manner (Figure S1). Following dosing, BI 425809 was absorbed in plasma with median t max values of 3–5 hours in all dose groups. Steady state was achieved by day 6. In CSF, the maximum concentrations were observed in a median t max range of 5–8 hours. Upon reaching the maximum concentration of BI 425809 in CSF, the concentration remained relatively constant until the last sampling time point (14 hours postdose; Figure 3).
Table 1.
Summary of BI 425809 pharmacokinetic parameters in plasma and CSF
| Geometric mean (% gCV), unless otherwise stated | ||||
|---|---|---|---|---|
| BI 425809 5 mg (n = 6) | BI 425809 10 mg (n = 6) | BI 425809 25 mg (n = 8) | BI 425809 50 mg (n = 5) | |
| Plasma | ||||
| AUC0–14, nM | 421 (17.6) | 829 (18.9) | 1,840 (24.2)d | 3,300 (29.3) |
| C max, nM | 42.3 (23.2) | 85.6 (19.5) | 176.0 (22.6)d | 328.0 (37.5) |
| C 312, nM | 66.7 (40.9)b | 107.0 (48.2)c | 266.0 (38.4) | 445.0 (50.3) |
| t max, ha | 5.0 (2.0–8.0) | 4.5 (2.0–5.0) | 3.0 (3.0–10.0)d | 5.0 (3.1–12.0) |
| CSF | ||||
| AUC0–14, nM | 29.9 (15.4) | 59.8 (19.3) | 124.0 (27.2)e | 260.0 (29.1) |
| C max, nM | 2.9 (21.3) | 5.6 (17.5) | 12.2 (26.4)d | 25.0 (37.5) |
| C 312, nM | 5.6 (51.8)b | 9.7 (41.9)c | 21.7 (33.2)e | 42.4 (36.7) |
| t max, ha | 8.0 (3.0–10.0) | 5.5 (5.0–6.0) | 6.1 (4.0–8.1)d | 8.1 (6.0–12.0) |
aMedian (minimum‐maximum); b n = 5; c n = 4; d n = 7; e n = 6.
AUC0–14, area under the concentration‐time curve of BI 425809 in plasma and CSF over the time interval from 0 to 14 hour; C 312, concentration of BI 425809 in plasma and CSF at the time point 312 hour; C max, maximum measured concentration of BI 425809 in plasma and CSF after first dose; CSF, cerebrospinal fluid; gCV, geometric coefficient of variation; t max, time from first dosing to maximum measured concentration of BI 425809 in plasma and CSF.
Figure 3.
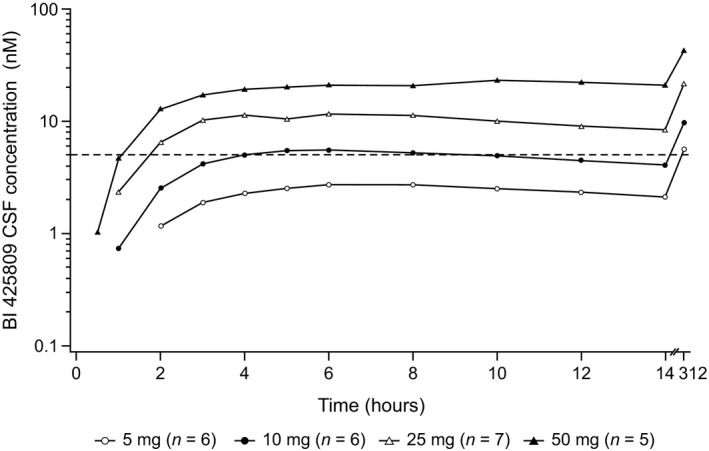
Geometric mean concentration‐time profiles of BI 425809 in cerebrospinal fluid (CSF), after single and multiple dosing in healthy male subjects. The dashed line in the figure represents the half maximal inhibitory concentration (IC 50) value of glycine transporter 1 (GlyT1).
There was no apparent difference in the dose‐normalized parameters in CSF between the dose groups, indicating a dose‐proportional increase in the exposure to BI 425809. In all dose groups, multiple dosing led to accumulation of BI 425809 in plasma and CSF. A high correlation (Spearman's correlation coefficient of 0.97) between CSF and plasma trough concentration at steady state (C 312) of BI 425809 was observed when all treatment groups were pooled (Figure S2); similar results were observed for AUC0–14 and C max. Concentrations of BI 425809 in CSF were markedly lower than in plasma. However, the CSF to plasma ratio was constant over time ranging from 7% to 9% over all dose groups.
Pharmacodynamics
Following single oral doses of BI 425809, CSF glycine levels increased continuously and slowly in all but one dose group (25 mg) and reached their maximum at 6–10 hours (Table 2). The 25‐mg dose group revealed a zig zag pattern (Figure 4 a). The increase in glycine after single doses of BI 425809 was dose‐dependent from 5–25 mg as indicated by the AUECdiff, 0–14 and E max values (Table 2), whereas the increase for the 50‐mg dose group did not clearly differ from the 25‐mg dose group.
Table 2.
Pharmacodynamic parameters of glycine in CSF
| PD parameter | N | BI 425809 5 mg | N | BI 425809 10 mg | N | BI 425809 25 mg | N | BI 425809 50 mg |
|---|---|---|---|---|---|---|---|---|
| Ratio of AUECdiff, 0–14, %a | 6 | 16.0 (17.4) | 6 | 37.0 (12.1) | 6 | 53.0 (30.5) | 5 | 47.0 (35.3) |
| E max, % | 6 | 35.3 (26.4) | 6 | 62.3 (15.3) | 8 | 136 (107) | 5 | 91.9 (89.9) |
| t max, hb | 6 | 6.5 (2.0–14.0) | 6 | 10.0 (6.0–14.0) | 8 | 6.2 (3.1–10.7) | 5 | 8.1 (6.0–14.1) |
| E 14, % | 6 | 18.0 (32.1) | 6 | 39.8 (14.5) | 6 | 60.9 (60.7) | 5 | 50.6 (33.8) |
| E 312, % | 4 | 1.17 (24.3) | 4 | 57.0 (48.3) | 6 | 37.9 (45.8) | 5 | 123 (52.5) |
All values are mean (SD) unless otherwise stated. AUECdiff, 0–14, ratio between AUECtreatment (area under the biomarker effect vs. time curve on treatment during the given interval) and AUECbase (baseline area under the biomarker effect vs. time curve during the given interval); CSF, cerebrospinal fluid; E 14, effect of glycine at the time point 14 hours after the first dose; E 312, effect of glycine at the time point 312 hours before the last dose is given; E max, maximum effect of glycine after the first dose; t max, time from dosing to maximum measured concentration of BI 425809; PD, pharmacodynamic.
aThe mean of the three glycine CSF pre‐dose levels was taken as a baseline value for the calculation of the AUECdiff, 0–14 parameter up to the last sampling point; bmedian (minimum‐maximum).
Figure 4.
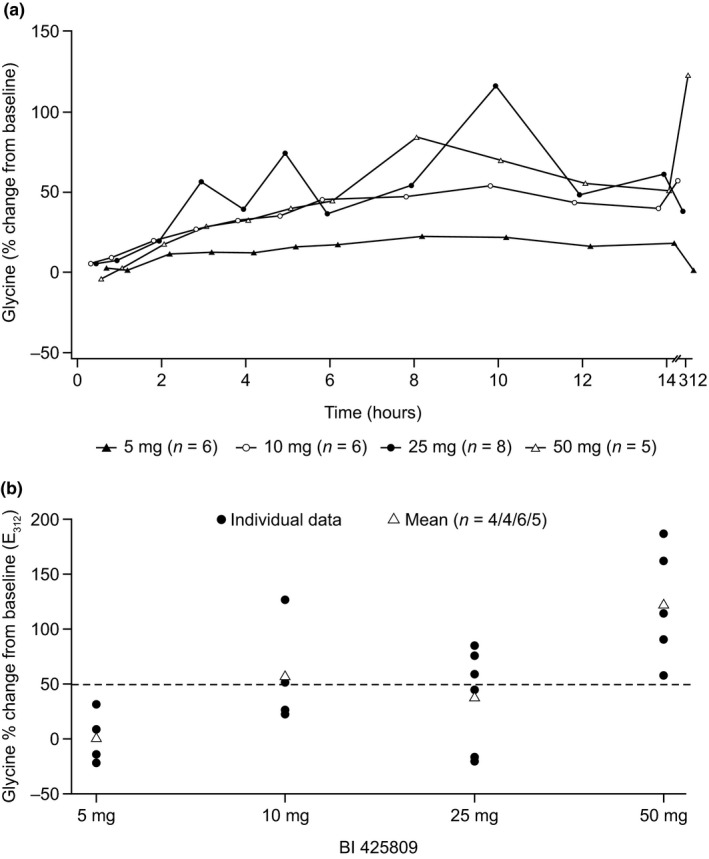
(a) Arithmetic mean effect‐time profiles of glycine (% change from baseline) in cerebrospinal fluid (CSF) after single and arithmetic mean E 312 (% change from baseline) after multiple oral administrations of 5–50 mg BI 425809 to healthy male subjects. E 312, effect of glycine at the time point 312 hours before the last dose is given. (b) Percentage change from baseline in individual and mean E 312 of glycine in CSF after multiple dosing of BI 425809. The dotted line represents a 50% increase from baseline in CSF glycine concentration. E 312, effect of glycine at the time point 312 hours before the last dose is given.
After multiple dosing of BI 425809, steady state mean trough glycine CSF concentrations (E 312 h) increased with increasing doses from 1% (5 mg) to 123% (50 mg) compared with baseline (Table 2 and Figure 4 b). At steady state, there was a moderate correlation (Spearman's correlation coefficient of 0.66) between glycine change from baseline and BI 425809 concentration in CSF (Figure S3).
Safety
Of the 25 subjects treated, 22 (88.0%) reported at least one treatment‐emergent AE. There was no apparent dose‐dependency in terms of the frequency of observed AEs, with AEs most frequently reported by subjects in the 25‐mg dose group (Table 3). The most frequently reported AEs were procedural headache (16 subjects; 64.0%) followed by back pain (8 subjects; 32.0%) related to the lumbar catheter placement, and reported in a similar frequency across all dose groups. Overall, the frequency of drug‐related AEs was low; one subject (4.0%) in the 25‐mg dose group experienced mild nausea, abdominal pain, and upper abdominal pain. These AEs were of mild intensity and spontaneously resolved. There were no protocol‐specified AEs of special interest, deaths, or other serious AEs reported on‐treatment.
Table 3.
Overall summary of subjects with treatment‐emergent AEs in ≥5% subjects in any one system organ class
| Preferred term, n (%) | BI 425809 5 mg (n = 6) | BI 425809 10 mg (n = 6) | BI 425809 25 mg (n = 8) | BI 425809 50 mg (n = 5) | Total on‐treatment (n = 25) |
|---|---|---|---|---|---|
| Total with AEsa | 5 (83.3) | 5 (83.3) | 8 (100.0) | 4 (80.0) | 22 (88.0) |
| Procedural headache | 4 (66.7) | 4 (66.7) | 5 (62.5) | 3 (60.0) | 16 (64.0) |
| Back pain | 3 (50.0) | 0 | 4 (50.0) | 1 (20.0) | 8 (32.0) |
| Nausea | 1 (16.7) | 2 (33.3) | 2 (25.0) | 0 | 5 (20.0) |
| Vomiting | 1 (16.7) | 1 (16.7) | 2 (25.0) | 0 | 4 (16.0) |
| Neck pain | 0 | 1 (16.7) | 1 (12.5) | 1 (20.0) | 3 (12.0) |
| Fatigue | 0 | 0 | 1 (12.5) | 1 (20.0) | 2 (8.0) |
| Dizziness | 0 | 1 (16.7) | 1 (12.5) | 0 | 2 (8.0) |
| Muscle spasms | 1 (16.7) | 0 | 0 | 0 | 1 (4.0) |
| Paraesthesia | 1 (16.7) | 0 | 0 | 0 | 1 (4.0) |
| Somnolence | 1 (16.7) | 0 | 0 | 0 | 1 (4.0) |
| Catheter site pain | 1 (16.7) | 0 | 0 | 0 | 1 (4.0) |
| Wound | 0 | 1 (16.7) | 0 | 0 | 1 (4.0) |
| Gingivitis | 0 | 1 (16.7) | 0 | 0 | 1 (4.0) |
| Procedural pain | 0 | 0 | 1 (12.5) | 0 | 1 (4.0) |
| Abdominal pain | 0 | 0 | 1 (12.5) | 0 | 1 (4.0) |
| Upper abdominal pain | 0 | 0 | 1 (12.5) | 0 | 1 (4.0) |
| Diarrhea | 0 | 0 | 1 (12.5) | 0 | 1 (4.0) |
| Dry mouth | 0 | 0 | 1 (12.5) | 0 | 1 (4.0) |
| Musculoskeletal stiffness | 0 | 0 | 1 (12.5) | 0 | 1 (4.0) |
| Tension headache | 0 | 0 | 1 (12.5) | 0 | 1 (4.0) |
| Nasopharyngitis | 0 | 0 | 0 | 1 (20.0) | 1 (4.0) |
AEs, adverse events.
A subject could have reported more than one AE.
Discussion
The preclinical study presented here evaluated the potency and selectivity of the novel GlyT1 inhibitor BI 425809, and its effects on glycine concentration in rat CSF following a single systemic administration. To translate this preclinical study into humans, the clinical study presented here assessed the PK of BI 425809 in CSF and plasma, investigated its PD by measuring glycine levels in CSF, and evaluated the safety and tolerability of BI 425809 following multiple oral doses. The results from the clinical study suggest that BI 425809 had a favorable PK profile and was generally well tolerated with no dose‐dependent relationship in the occurrence of AEs. Thus, functional target engagement of GlyT1 by BI 425809 was demonstrated preclinically in rats and in humans by a dose‐dependent increase of glycine levels in CSF.
In rats, single oral administration of BI 425809 resulted in a dose‐dependent increase in glycine CSF levels demonstrating functional target engagement of GlyT1 in the brain. This shows that glycine levels in CSF can be used to assess GlyT1 inhibition centrally, thereby supporting the use of CSF glycine analysis for confirming central target engagement in humans.
The clinical PK data from the present study show that multiple dosing led to an accumulation of BI 425809 in plasma and CSF (across all dose groups). The IC50 value of GlyT1 (5.0 nM) in CSF was reached with the BI 425809 5 mg dose at steady state. A Spearman's correlation coefficient of 0.97 indicated a strong correlation between the CSF and plasma trough concentrations at steady state (C 312) of BI 425809. Therefore, for future studies, CSF exposure of BI 425809 can be predicted by measuring BI 425809 plasma concentration.
Steady state dosing of BI 425809 (once daily) led to an overall dose‐dependent increase in glycine CSF levels. In the 5‐mg dose group, a mean increase of 1.2% from baseline in CSF glycine levels was observed. However, in the 10‐mg, 25‐mg, and 50‐mg dose groups, mean increases of 57%, 38%, and, 123%, respectively, in CSF glycine levels were reported. Taken together, these data clearly demonstrate central functional target engagement by BI 425809 in humans. However, it should be noted that mean exposure of the 25‐mg dose group was influenced by two subjects for which CSF glycine exposure was unchanged in comparison to baseline. Both subjects showed a relatively high glycine increase after the first dose. This may be due to the high intra‐individual variability in the observed CSF glycine levels or due to a lack of response to BI 425809 treatment. It should be taken into consideration that this steady state assessment was based on a single measurement, and it can, therefore, be difficult to distinguish between an outlying observation and a lack of response.
Due to the high intra‐individual variability in CSF glycine levels and the low number of subjects in each dose group, it was difficult to establish a quantitative dose‐response relationship. In line with this, the PK/PD correlation was moderate (Spearman's correlation coefficient of 0.66) between glycine change from baseline and BI 425809 concentration in CSF. Although human CSF BI 425809 exposure at the 5‐mg dose was already in the range of IC50 for in vitro GlyT1 inhibition, there was no obvious effect on CSF glycine levels at this dose level.
In this study, one subject reported drug‐related AEs of mild nausea, abdominal pain, and upper abdominal pain reported in the 25‐mg dose group. The most frequently reported AE was procedural headache (16 subjects; 64.0%), which was reported in a similar frequency across all dose groups. Overall, there were no relevant dose group differences in the frequency of subjects reporting AEs. The AEs reported were in general agreement with recent reports on GlyT1 inhibitors.14, 15, 16 Generally, BI 425809 was well tolerated, with no indication of safety concerns in a healthy male population.
The presented clinical study has several limitations; the sample size was small and the study population consisted of male subjects only. The safety evaluation may be limited as the most frequently reported AEs in this trial, such as procedural headache and back pain, are likely due to the lumbar puncture or lumbar catheter placement rather than as a result of treatment, per se. Additionally, the observed high variability in CSF glycine levels, which is in line with data from the preclinical study described here (Figure 2) and a previous study with the GlyT1 inhibitor bitopertin in humans,17 limits the quantitative analysis and interpretation of the data.
Conclusions
The preclinical data show that BI 425809 is a potent and selective GlyT1 inhibitor and that systemic administration of BI 425809 leads to a dose‐dependent increase in glycine levels in rat CSF demonstrating indirect functional target engagement. This shows that glycine levels in CSF can be used to assess GlyT1 inhibition centrally.
The clinical data show that BI 425809 demonstrated a clear dose‐dependent increase in CSF exposure and a linear CSF/plasma correlation over dose levels, enabling prediction of CSF exposure from plasma levels. Consistent with the preclinical data, glycine increase at 25 mg was as high as for 10 mg in healthy males, indicating indirect functional target engagement for BI 425809.
Multiple oral doses of BI 425809 ranging from 5 to 50 mg for 14 days were generally well tolerated in healthy volunteers.
These data will aid in the selection of BI 425809 dosing regimens for future studies.
Funding
This work was funded by Boehringer Ingelheim International GmbH. The sponsor was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
Conflict of Interest
H.R., M.D., O.K., C.D.C., B.S., S.K., C.S., V.M., S.G., K.H.L., R.G., G.W., and S.W. are employees of Boehringer Ingelheim International GmbH. G.F. was an employee of Boehringer Ingelheim International GmbH at the time of the study and is a current employee of Novartis. S.R. is an employee of SGS.
Author Contributions
H.R., M.D., O.K., C.D.C., B.S., S.K., C.S., V.M., S.G., K.H.L., G.F., R.G., S.R., G.W., and S.W. wrote the manuscript. H.R., M.D., O.K., C.D.C., B.S., C.S., V.M., S.G., K.H.L., G.F., R.G., G.W., and S.W. designed the research. H.R., M.D., O.K., C.D.C., B.S., S.K., C.S., V.M., S.G., S.R., G.W., and S.W. performed the research. H.R., M.D., O.K., C.D.C., B.S., S.K., C.S., V.M., S.G., S.R., G.W., and S.W. analyzed the data.
Supporting information
Figure S1.Geometric mean concentration‐time profiles of BI 425809 in plasma, after single and multiple dosing in healthy male subjects.
Figure S2.Correlation of BI 425809 concentrations in CSF versus plasma at trough in steady state (C312; all treatment groups pooled).
Figure S3.Percentage change from baseline in individual E312 of glycine in CSF versus plasma at trough in steady state (C312) of BI 425809 in CSF (all treatment groups pooled).
Table S1.Demographic characteristics of all subjects.
Acknowledgments
We gratefully acknowledge the contributions of Nancy Kötteritzsch, Sandra Schwäble, Jakub Pekarek, and Jasmin Link from Boehringer Ingelheim International GmbH, as well as Sun Young (Angela) Yum, a previous employee of Boehringer Ingelheim International GmbH, Jos Leempoels from SGS Antwerpen, and the editorial support of Fishawack Communications. Editorial support in the form of initial preparation of the outline based on input from all authors, and collation and incorporation of author feedback to develop subsequent drafts, assembling tables and figures, copyediting, and referencing was provided by Lisa Auker, PhD, of Fishawack Communications, and was funded by Boehringer Ingelheim International GmbH.
References
- 1. Goff, D.C. & Coyle, J.T. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am. J. Psychiatry 158(9), 1367–1377 (2001). [DOI] [PubMed] [Google Scholar]
- 2. Hu, N.W. , Ondrejcak, T. & Rowan, M.J. Glutamate receptors in preclinical research on Alzheimer's disease: Update on recent advances. Pharmacol. Biochem. Behav. 100(4), 855–862 (2012). [DOI] [PubMed] [Google Scholar]
- 3. Lakhan, S.E. , Caro, M. & Hadzimichalis, N. NMDA receptor activity in neuropsychiatric disorders. Front. Psychiatry 4, 52 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hashimoto, K. Glycine transport inhibitors for the treatment of schizophrenia. Open Med. Chem. J. 4, 10–19 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rezvani, A. H . Animal Models of Cognitive Impairment. Boca Raton (FL): CRC Press/Taylor & Francis; 2006. <http://www.ncbi.nlm.nih.gov/books/NBK2532/>. [Google Scholar]
- 6. Moschetti, V. et al Safety, tolerability and pharmacokinetics of oral BI 425809, a glycine transporter 1 inhibitor, in healthy male volunteers: a partially randomised, Single‐blind, placebo‐controlled, first‐in‐human study. Eur. J. Drug Metab. Pharmacokinet. 43, 239–249 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rosenbrock, H. et al Improving cognitive function in rodents via increasing glycine levels in brain by the novel glycine transporter‐1 inhibitor BI 425809. Alzheimer's Dement. 12(7), P1018 (2016). [Google Scholar]
- 8. Moschetti, V. et al Multiple rising doses of oral BI 425809, a GlyT1 inhibitor, in young and elderly healthy volunteers: a randomised, double‐blind, phase I study investigating safety and pharmacokinetics. Clin. Drug Invest. 70, 1–4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Banker G, Goslin K . Culturing Nerve Cells. Cambridge, MA, USA: The MIT Press; 1991. [Google Scholar]
- 10. Perry, K.W. et al Neurochemical and behavioral profiling of the selective GlyT1 inhibitors ALX5407 and LY2365109 indicate a preferential action in caudal vs. cortical brain areas. Neuropharmacology 55(5), 743–754 (2008). [DOI] [PubMed] [Google Scholar]
- 11. Voehringer, P. , Fuertig, R. & Ferger, B. A novel liquid chromatography/tandem mass spectrometry method for the quantification of glycine as biomarker in brain microdialysis and cerebrospinal fluid samples within 5 min. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 939, 92–97 (2013). [DOI] [PubMed] [Google Scholar]
- 12. Moschetti, V. et al Safety, tolerability and pharmacokinetics of single rising doses of BI 425809 given orally to healthy male volunteers: a partially randomised (within dose groups), single‐blind, placebo‐controlled, phase I study (abstract T64). NPJ: Schizophr. 2, 16007 (2016). [Google Scholar]
- 13. Bhushan, R. & Bruckner, H. Marfey's reagent for chiral amino acid analysis: a review. Amino Acids 27(3–4), 231–247 (2004). [DOI] [PubMed] [Google Scholar]
- 14. D'Souza, D.C. et al Glycine transporter inhibitor attenuates the psychotomimetic effects of ketamine in healthy males: preliminary evidence. Neuropsychopharmacology 37(4), 1036–1046 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hirayasu, Y. et al A double‐blind randomized study assessing safety and efficacy following one‐year adjunctive treatment with bitopertin, a glycine reuptake inhibitor, in Japanese patients with schizophrenia. BMC Psychiatry 16(1), 66 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Umbricht, D. et al Effect of bitopertin, a glycine reuptake inhibitor, on negative symptoms of schizophrenia: a randomized, double‐blind, proof‐of‐concept study. JAMA Psychiatry 71, 637–646 (2014). [DOI] [PubMed] [Google Scholar]
- 17. Hofmann, C. et al Effects of the glycine reuptake inhibitors bitopertin and RG7118 on glycine in cerebrospinal fluid: Results of two proofs of mechanism studies in healthy volunteers. Psychopharmacology 233(13), 2429–2439 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.Geometric mean concentration‐time profiles of BI 425809 in plasma, after single and multiple dosing in healthy male subjects.
Figure S2.Correlation of BI 425809 concentrations in CSF versus plasma at trough in steady state (C312; all treatment groups pooled).
Figure S3.Percentage change from baseline in individual E312 of glycine in CSF versus plasma at trough in steady state (C312) of BI 425809 in CSF (all treatment groups pooled).
Table S1.Demographic characteristics of all subjects.