

Abstract
It remains unclear whether PAX6 acts as a crucial transcription factor for lung cancer stem cell (CSC) traits. We demonstrate that PAX6 acts as an oncogene responsible for induction of cancer stemness properties in lung adenocarcinoma (LUAD). Mechanistically, PAX6 promotes GLI transcription, resulting in SOX2 upregulation directly by the binding of GLI to the proximal promoter region of the SOX2 gene. The overexpressed SOX2 enhances the expression of key pluripotent factors (OCT4 and NANOG) and suppresses differentiation lineage factors (HOPX and NKX2-1), driving cancer cells toward a stem-like state. In contrast, in the differentiated non-CSCs, PAX6 is transcriptionally silenced by its promoter methylation. In human lung cancer tissues, the positive linear correlations of PAX6 expression with GLI and SOX2 expression and its negative correlations with HOPX and NKX2-1 expression were observed. Therapeutically, the blockade of the PAX6-GLI-SOX2 signaling axis elicits a long-lasting therapeutic efficacy by limiting CSC expansion following chemotherapy. Furthermore, a methylation panel including the PAX6 gene yielded a sensitivity of 79.1% and specificity of 83.3% for cancer detection using serum DNA from stage IA LUAD. Our findings provide a rationale for targeting the PAX6-GLI-SOX2 signaling axis with chemotherapy as an effective therapeutic strategy and support the clinical utility of PAX6 gene promoter methylation as a biomarker for early lung cancer detection.
Keywords: lung cancer, cancer stem cell, PAX6, SOX2, GLI
Introduction
Cancer cells display a broad spectrum of functional and morphological heterogeneity even within a single tumor lesion. This phenomenon could be explained by the cancer stem cell (CSC) model, where a limited number of CSCs can divide asymmetrically and give rise to more differentiated progeny that represents most of the tumor cell population 1. Furthermore, undifferentiated CSCs are resistant to conventional chemotherapies that kills differentiated cancer cells, and they are responsible for subsequent tumor progression or recurrence 2. As CSCs contribute to the driving force of tumorigenesis, metastasis, and treatment failure 2, the elimination of CSCs is crucial for achieving long-term therapeutic efficacy. However, an incomplete understanding of the molecular pathways critical to lung CSC generation and expansion has hindered the development of therapeutic strategies targeting CSCs.
Paired box 6 (PAX6), which belongs to homeobox gene superfamily, is an essential transcription factor for embryonic development 3, 4, and the dysregulation of PAX6 expression results in developmental disorders and formation of tumors 5–7. In neural stem cells, PAX6 controls the balance between self-renewal and differentiation by regulating the cellular networks involved in brain patterning, neuronal migration, and neural circuit formation 4, 8. In human cancer, the expression and biological role of PAX6 differs depending on cancer types. PAX6 is downregulated in tumors compared with normal tissues and acts as a tumor suppressor gene in glioblastoma and prostate cancer 9–11, whereas it is overexpressed and acts as an oncogene that facilitates cell growth and suppresses terminal differentiation in pancreas cancer 12, 13. The study of PAX6 in non-small cell lung cancer (NSCLC) is very limited. A recent study reported that PAX6 promotes proliferation and cell cycle progression of human NSCLC cell lines 14, and other studies found that the CpG island promoter region of PAX6 gene is frequently methylated 15, 16, which generally acts as a regulatory mechanism for its transcriptional silencing 17. Further studies are needed to understand the role of PAX6 gene in the pathogenesis of NSCLC.
NSCLC has three major histopathological subtypes: lung adenocarcinoma (LUAD), the most common lung cancer; lung squamous cell carcinoma (LUSC); and large cell carcinoma. Due to the unspecific nature and the late onset of symptoms, approximately two-thirds of NSCLC patients are diagnosed at an advanced stage, implying a very poor rate of cure 18. Therefore, identifying biomarkers to detect cancer at an early stage is needed in clinical practice. Promoter methylation (PM) is one of the most common epigenetic alterations, and aberrant PM of candidate genes can be an early event in cancer progression, indicating its potential as a biomarker for early cancer detection 17. In addition, assessing PM in serum DNA may be a promising, minimally invasive approach 19. Although frequent PM of the PAX6 gene has been reported in NSCLC 15, 16, its potential as a minimally invasive early lung cancer detection biomarker using serum samples is still unexplored.
CSCs retain substantial characteristics of embryonic stem cells (ESCs) through the common molecular signaling pathways and stemness-related factors, such as the Hedgehog (Hh)-GLI pathway and pluripotency-determinant molecule SOX2 20–23. As PAX6 is an indispensable factor for ESC traits 8, we hypothesized that it may contribute to CSC traits. To test this hypothesis, this study was designed to investigate 1) the contribution of PAX6 to LUAD-associated CSC (LUAD CSC) generation and expansion, 2) the relevance of PAX6 PM in regulating LUAD CSCs, and 3) the potential of early detection by testing PM of PAX6 and other two homeobox genes (HOXA9 and UNCX) using serum samples from stage IA LUAD. Our study provides a rationale for targeting PAX6-GLI-SOX2 signaling axis and reveal the clinical utility of PAX6 PM as a serum biomarker for early lung cancer detection.
Results
PAX6 is a critical oncogene responsible for cancer stemness properties via SOX2 in LUAD
Given the reported PAX6 PM in a small cohort of LUAD 15 and the crucial role of PAX6 in ESC traits 8, we hypothesized that epigenetic alteration and the expression of PAX6 may have a role in the regulation and maintenance of LUAD CSCs. We first screened the methylation status of PAX6 promoter region of eight primary LUAD tumors and the adjacent matched normal samples. The promoter CpG islands were frequently methylated in the tumor samples compared with matched normal samples. Furthermore, PAX6 PM inversely correlated with its expression (Supplementary Fig. S1A–B). We confirmed this correlation in the LUAD cohort of The Cancer Genome Atlas (TCGA) (Supplementary Fig. S1C). In all the NSCLC cell lines with PAX6 PM, the expression levels of PAX6 were mostly absent (Supplementary Fig. S1A and S1D). To determine the association of PAX6 PM with its transcriptional silencing, we treated 4 LUAD cell lines with a demethylating agent (5-Aza-dC) and found the robust reactivation of PAX6 in the cell lines with PM (Fig. 1A). This finding suggests that PAX6 PM is a potential regulatory mechanism for its transcriptional silencing. Interestingly, we observed lower PM values and upregulated expression of PAX6 in spheroid LUAD cells compared with parental cells (Fig. 1B–C).
Figure 1.
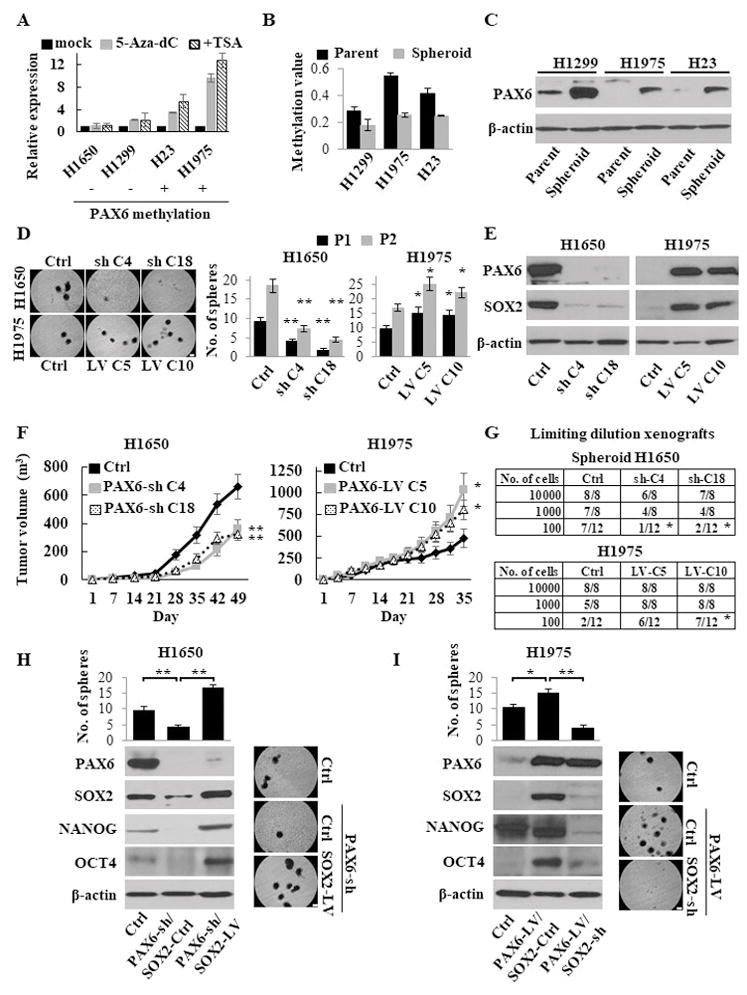
PAX6-induced cancer stemness properties via SOX2. (A) Re-expression of the PAX6 gene after treatment of LUAD cells with 5-Aza-dC ± Trichostatin A (TSA), as determined by Q-RT-PCR. The relative expression levels were calculated as a ratio of the values of 5-Aza-dC ± TSA treatment relative to the value of mock treated cells considered as 1.0. Plus (+) and minus (-) marks represent cell lines with and without methylation of the PAX6 gene, respectively. (B) The PM values of the PAX6 gene in isogeneic parental and spheroid LUAD cells, as measured by Q-MSP. (C) Western blotting analysis of PAX6 in isogeneic parental and spheroid cells. (D) Sphere formation and self-renewal assays through the second (P2) passage from the first passage (P1) in stable PAX6 knockdown (PAX6-sh) NCI-H1650 and overexpressed (PAX6-LV) NCI-H1975 cells compared with control (PAX6-Ctrl) cells. Left, representative images of sphere formation (scale bars, 200 μm); Right, number of the spheres over 100 μm. (E) Western blotting analysis of PAX6 and SOX2 expression in stable PAX6-sh NCI-H1650 and PAX6-LV NCI-H1975 cells. (F) The in vivo tumorigenesis after xenotransplantation of stable PAX6-sh NCI-H1650 (left) and PAX6-LV NCI-H1975 (right) cells (four mice per group). (G) Limiting dilution xenograft assays in stable PAX6-sh NCI-H1650 spheroid (upper) and PAX6-LV NCI-H1975 (lower) cells. Tumor-initiating capacity is shown as the numbers of tumors / the number of injections. (H) Sphere formation assay (upper left) and western blotting analysis (lower left) in PAX6-sh or PAX6-Ctrl NCI-H1650 cells transduced with SOX2-LV or SOX2-Ctrl (PAX6-Ctrl/SOX2-Ctrl, PAX6-sh/SOX2-Ctrl, and PAX6-sh/SOX2-LV). Right, representative images of sphere formation (scale bars, 200 μm). The spheres over 100 μm were counted. (I) Sphere formation assay (upper left) and western blotting analysis (lower left) of PAX6-LV or PAX6-Ctrl NCI-H1975 cells transduced with SOX2-sh or SOX2-Ctrl (PAX6-Ctrl/SOX2-Ctrl, PAX6-LV/SOX2-Ctrl, and PAX6-LV/SOX2-sh). Right, representative images of sphere formation (scale bars, 200 μm).
Each error bar indicates mean ± SEM. *, P <0.05; **, P <0.01 (Wilcoxon–Mann–Whitney test [D and F] Chi-squared test [G], and Kruskal–Wallis with post-hoc test [H and I]). See also Fig. S1–S3.
To understand the role of PAX6 in LUAD CSC maintenance and expansion, we constructed the lentiviral-based stable PAX6 overexpressed (PAX6-LV) NCI-H1975 and NCI-H23 cells, and PAX6 knockdown (PAX6-sh) NCI-H1650 and NCI-H1299 cells. PAX6-LV cells enhanced sphere-forming, self-renewal, migratory, and invasive abilities, while PAX6-sh cells showed the opposite effects (Fig. 1D and Supplementary Fig. S2A–C). Of note, the PAX6-sh spheroid cells no longer exhibited functions comparable to the control (PAX6-Ctrl) spheroid cells in terms of stemness properties such as invasion, chemoresistance, and anti-apoptosis, while the PAX6-LV spheroid cells showed the enhanced stemness properties (Supplementary Fig. S2D–F). In addition, PAX6 expression status was associated with the expression of numerous stemness-related molecules, including the key pluripotent factors SOX2, OCT4, and NANOG (Fig. 1E and Supplementary Fig. S3A–B). To examine the role of PAX6 in tumorigenesis, an in vivo tumor formation assay was performed using PAX6-sh and PAX6-LV cells. A remarkable reduction in tumor volume was observed in mice injected with PAX6-sh cells compared with PAX6-Ctrl cells, while enhanced tumorigenesis was observed in PAX6-LV cells (Fig. 1F and Supplementary Fig. S3C), indicating that PAX6 acts as an oncogene in LUAD. Because a limiting dilution xenograft assay is one of the important features of CSCs, serially diluted PAX6-sh spheroid or PAX6-LV cells were subcutaneously injected into NSG mice. As expected, the PAX6-sh spheroid cells showed significantly low tumor initiation ability, while the PAX6-LV cells exhibited a more aggressive ability (Fig. 1G). Thus, PAX6 plays an indispensable role in LUAD CSC maintenance and progression.
Among the numerous stemness-related molecules associated with PAX6 expression (Supplementary Fig. S3A), we focused on SOX2, as it is a master regulator for CSC traits in various cancer types 23, 24. To understand the interaction of PAX6 with SOX2 in LUAD CSCs, we genetically induced SOX2 into PAX6-sh NCI-H1650 cells. As expected, the forced expression of SOX2 restored the stemness abilities, including sphere formation, chemoresistance, anti-apoptosis, and expression of the key pluripotent factors (OCT4 and NANOG) that were suppressed due to PAX6 knockdown (Fig. 1H and Supplementary Fig. S3D–E). In contrast, SOX2 knockdown attenuated the PAX6-induced stemness properties in PAX6-LV NCI-H1975 cells (Fig. 1I and Supplementary Fig. S3D–E). These findings suggest that PAX6 promotes a stem-like state via SOX2.
PAX6 activates GLI-SOX2 signaling axis in LUAD CSC maintenance
To elucidate the intrinsic signaling pathways driven by PAX6 in LUAD, we performed gene set enrichment analyses (GSEA) for identifying oncogenic signatures in the groups with high and low expression of PAX6 in the TCGA LUAD cohort. The Hedgehog (Hh) pathway significantly enriched in the group with high PAX6 expression compared with the group with low expression (Fig. 2A). Similar to our TCGA cohort analysis, PAX6-LV cells showed the overexpression of GLI1 and GLI2 that are downstream effectors of the Hh pathway, while PAX6-sh cells showed a decreased expression of GLI1 and GLI2 (Fig. 2B and Supplementary Fig. S3B and S4A). In addition, we confirmed the direct binding of GLI1 and GLI2 to the proximal promoter region of the SOX2 gene using a chromatin immunoprecipitation assay in this cellular models (Fig. 2C), consistent with previous reports showing direct regulation of SOX2 by GLI 25, 26. As expected, we observed the concomitantly enhanced expression of GLI and SOX2 in the LUAD spheroid cells compared with parental cells (Supplementary Fig. S4B). To further confirm the role of the PAX6-GLI-SOX2 signaling axis in LUAD CSCs, GLI was genetically inhibited in PAX6-LV NCI-H1975 cells with high stemness properties. The dual knockdown of GLI1 and GLI2 resulted in a dramatic inhibition of sphere formation and SOX2 expression, and the forced expression of SOX2 rescued the GLI knockdown-attenuated sphere-forming ability (Fig. 2D). In contrast, the stimulation of PAX6-sh NCI-H1650 cells with the recombinant Sonic hedgehog ligand (Shh) enhanced sphere formation and expression of GLI and SOX2, but knockdown of SOX2 abolished the effect (Fig. 2E). These accumulated findings strengthen the relevance of PAX6-GLI-SOX2 axis in maintaining LUAD CSCs.
Figure 2.
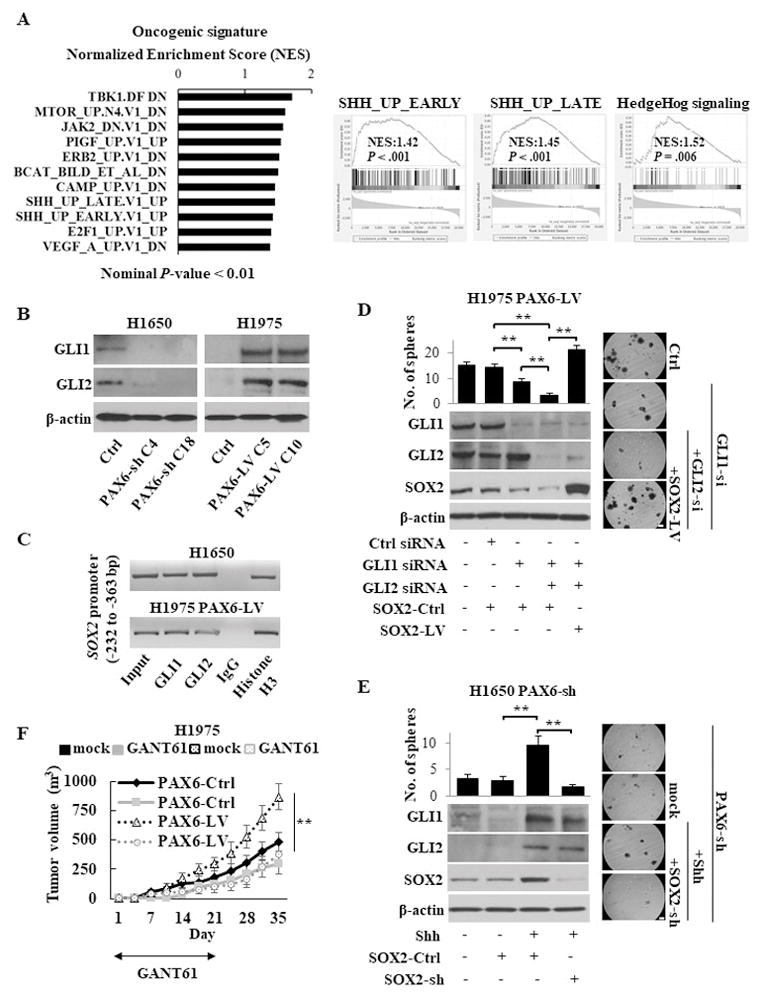
The PAX6-GLI-SOX2 signaling axis in LUAD CSCs. (A) Gene set enrichment analysis (GSEA) related to the oncogenic signatures and hallmarks in the groups with high and low expression of PAX6 in the TCGA LUAD cohort. Left, the enhanced oncogenic pathways in group with high PAX6 expression, as determined by a normalized enrichment score (NES); Right, the enrichment of Hedgehog pathway in group with high PAX6 expression. The GSEAs from both oncogenic signatures gene set (left and middle panels) and hallmarks gene set (right panel) showed the enriched Hedgehog pathway. SHH; Sonic hedgehog. (B) Western blotting analysis of GLI in stable PAX6-sh NCI-H1650 or PAX6-LV NCI-H1975 cells. (C) ChIP assays conducted on the proximal promoter region of the SOX2 gene using the indicated antibodies in NCI-H1650 or PAX6-LV NCI-H1975 cells. Histone H3 and normal IgG were used as the positive and negative control, respectively. (D) Sphere formation assay (upper left) and western blotting analysis of indicated molecules (lower left) of PAX6-LV NCI-H1975 cells transfected with GLI siRNA ± SOX2-LV. Right, representative images of sphere formation (scale bars, 200 μm). The spheres over 100 μm were counted. (E) Sphere formation assay (upper left) and western blotting analysis (lower left) of PAX6-sh NCI-H1650 cells stimulated with recombinant human Sonic Hedgehog ligand (Shh; 1μg /ml) after transfection with SOX2-sh or SOX2-Ctrl. Right, representative images of sphere formation (scale bars, 200 μm). (F) The in vivo tumorigenesis of stable PAX6-LV or PAX6-Ctrl NCI-H1975 cells in the presence or absence of the GLI antagonist GANT61 (50 mg/kg) treatment for 21 days (five mice per group).
Each error bar indicates mean ± SEM. **, P <0.01 (Kruskal–Wallis with post-hoc test [D, E, and F]). See also Fig. S4.
There are no available inhibitors for PAX6 and SOX2 because of their undruggable nature 27, 28. Therefore, pharmacological inhibition of Hh pathway components, such as Smoothened (SMO) and GLI may be a clinically relevant approach to block the PAX6-GLI-SOX2 signaling axis. Consistent with the findings of GLI genetic knockdown, the direct and specific GLI1 and GLI2 antagonist GANT61 treatment resulted in a concentration-dependent reduction of GLI and SOX2 expression and suppressed sphere formation and in vivo tumorigenicity (Supplementary Fig. S4C–F). Furthermore, treatment with GANT61 attenuated PAX6-promoted tumorigenicity (Fig. 2F). In contrast, treatment with the SMO antagonist cyclopamine showed a minor inhibitory effect, despite inhibition of SMO expression (Supplementary Fig. S4C and S4G), indicating that PAX6 activates Hh signaling in a SMO-independent manner.
Correlations of PAX6 with stemness-related factors and differentiation lineage factors in human samples
LUAD can be stratified into two molecular subtypes: the distal airway stem cell (DASC)-like subtype and the alveolar-like subtype. The DASC-like subtype is similar to undifferentiated distal airway stem cells, and the alveolar-like subtype represents histologically more differentiated tumors 29. To test whether the expression status of PAX6 is associated with these molecular subtypes, we assessed molecular subtype-related genes using their individual enrichment scores from results of GSEA in the groups with high and low PAX6 expression in the TCGA LUAD cohort (Supplementary Table S1). The group with low PAX6 expression significantly enriched genes associated with alveolar-like subtype, especially HOPX and NKX2-1 (Fig. 3A).
Figure 3.
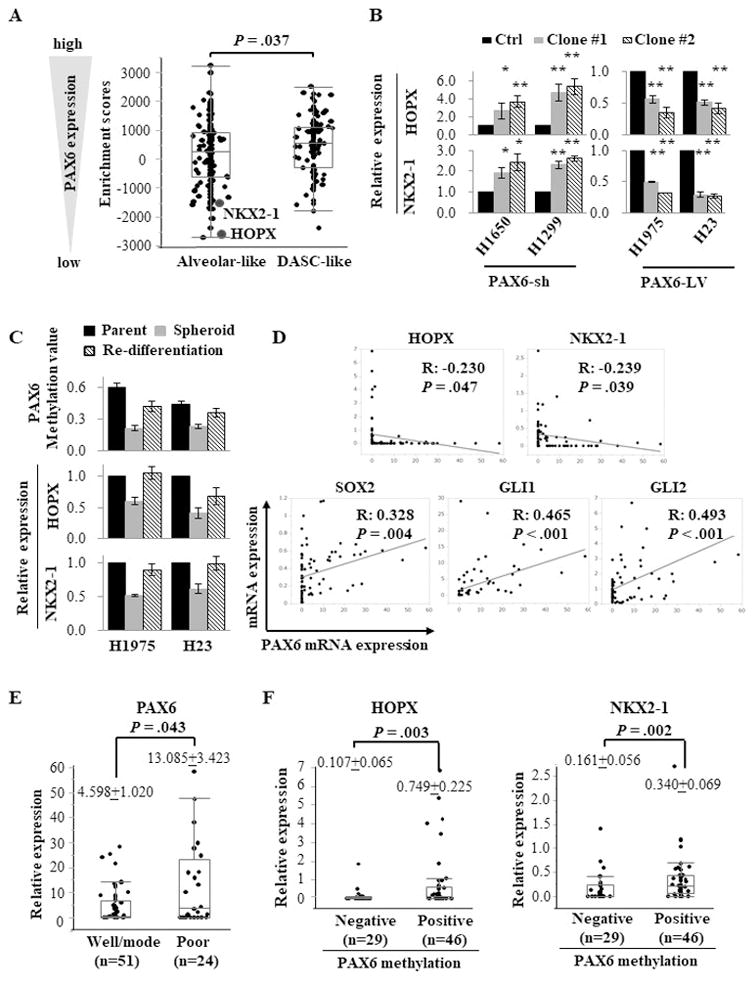
PAX6-regulated differentiation lineage factors (HOPX and NKX2-1) and stemness-related factors (SOX2 and GLI) in LUAD. (A) The enrichment scores of molecular subtype-related genes from GSEA in the groups with high and low expression of PAX6 in the TCGA LUAD cohort. The enrichment score of each gene was summarized in Supplementary Table S1. The alveolar-related genes, especially HOPX and NKX2-1, were enriched in group with low PAX6 expression compared with high group. DASC, distal airway stem cell. (B) The relative expression levels of HOPX and NKX2-1 in PAX6-sh (NCI-H1650 and NCI-H1299) and PAX6-LV (NCI-H1975 and NCI-H23) cells compared with PAX6-Ctrl cells, as measured by Q-RT-PCR. (C) The promoter methylation levels of the PAX6 gene and expression levels of HOPX and NKX2-1 in re-differentiated cells compared with spheroid cells. When spheroid cells were cultured under standard conditions containing the fetal bovine serum (FBS), the floating spheroid cells could adhere and acquire epithelial morphology similar to parental cells. The cells showed higher expression of HOPX and NKX2-1 along with re-methylation of the PAX6 gene compared with corresponding spheroid cells, as measured by Q-RT-PCR and Q-MSP. (D) A linear correlation analysis of PAX6 with SOX2, GLI1, GLI2, HOPX, and NKX2-1 in 75 human lung cancer tissues. The relative mRNA expression levels were calculated as the Q-RT-PCR values of each molecules vs. β-actin (The values of PAX6, GLI1, and HOPX were multiplied by 1000 for easy tabulation). The extent of the correlation is indicated by the R coefficient. (E) The association between PAX6 expression and the status of histological differentiation (well/moderate vs. poor differentiation). Scatter plots show the distribution of relative mRNA expression values of PAX6 according to the differentiation status. (F) The association of PAX6 promoter methylation with the expression levels of HOPX and NKX2-1.
Each error bar indicates mean ± SEM. *, P <0.05; **, P <0.01 (Wilcoxon–Mann–Whitney test [A, B, E, and F]). See also Fig. S5 and S6.
Because HOPX and NKX2-1 are central differentiation lineage transcription factors in the alveolar-like subtype 29, 30, we examined the association of PAX6 with HOPX and NKX2-1 in PAX6-sh and PAX6-LV cells. As expected, PAX6-sh cells exhibited a higher expression of HOPX and NKX2-1 compared with PAX6-Ctrl cells, while PAX6-LV cells showed a lower expression (Fig. 3B). To determine whether HOPX and NKX2-1 are negatively regulated through the PAX6-GLI-SOX2 signaling axis, GLI was genetically inhibited in the PAX6-LV NCI-H1975 cells. GLI1 knockdown induced the expression of HOPX and NKX2-1, and the dual knockdown of GLI1 and GLI2 further upregulated their expression. However, SOX2 induction reduced the GLI knockdown-induced expression of HOPX and NKX2-1 (Supplementary Fig. S5A). In contrast, SOX2 knockdown restored Shh-reduced expression in PAX6-sh NCI-H1650 cells (Supplementary Fig. S5B). In addition, when spheroid cells cultured under standard conditions, the re-differentiated cells showed the restored expression of HOPX and NKX2-1 along with the PAX6 re-methylation, similar to parental cells (Fig. 3C). Collectively, PAX6 not only enhanced the key pluripotent factors but also repressed differentiation lineage factors via SOX2-GLI signaling. On the other hand, in the differentiated non-CSCs tumor cell population, PAX6 is transcriptionally silenced by its PM, resulting in an increased expression of HOPX and NKX2-1 through the suppression of the PAX6-GLI-SOX2 signaling axis.
To confirm the association of PAX6 with differentiation lineage factors (HOPX and NKX2-1) and stemness-related factors (SOX2 and GLI), we analyzed their mRNA expression levels by Q-RT-PCR in 75 human lung tumor tissues. PAX6 showed inverse linear correlations with HOPX and NKX2-1 expression and positive linear correlations with SOX2 and GLI expression (Fig. 3D). Similar findings were confirmed in the TCGA LUAD cohort (Supplementary Fig. S5C). Notably, the histologically differentiated tumors showed a lower expression of PAX6 than those with poorly differentiated tumors (Fig. 3E). Furthermore, we observed that tumors with PAX6 PM have higher expression levels of HOPX and NKX2-1 and lower levels of SOX2 and GLI than those without PM (Fig. 3F and Supplementary Fig. S6A). Although it was not significant, PAX6 PM showed a trend toward a histologically differentiated status (Supplementary Table S2 and Fig. S6B).
Blocking the PAX6-GLI-SOX2 axis attenuates CSC expansion following chemotherapy
CSCs are selectively enriched following chemotherapy through the enhanced survival or repopulation of residual tumors, leading to tumor regrowth 2, 31. To understand the underlying mechanisms of CSC generation and expansion due to chemotherapy, we treated NCI-H1975 and NCI-H23 tumor-bearing mice with a combination chemotherapy of pemetrexed and cisplatin (Pem-Cis) that is a standard regimen for LUAD in clinical practice. In this xenograft models, we observed increased sphere formation and an activated PAX6-GLI-SOX2 signaling axis following Pem-Cis chemotherapy (Supplementary Fig. S7A–B). To further assess the role of PAX6 in CSC expansion following chemotherapy, PAX6-sh or PAX6-Ctrl tumor-bearing mice were treated with Pem-Cis chemotherapy. In contrast to PAX6-Ctrl tumors, PAX6-sh tumors retained their low sphere-forming ability along with reduced PAX6-GLI-SOX2 signaling, even after treatment with Pem-Cis chemotherapy (Fig. 4A–B). Pharmacologically, treatment with GLI antagonist GANT61 also suppressed CSC expansion following Pem-Cis chemotherapy in both PAX6-Ctrl and PAX6-LV tumors (Fig. 4C). Interestingly, we observed a demethylation of the PAX6 promoter region, consistent with the upregulation of PAX6, in tumors treated with Pem-Cis chemotherapy compared with those with mock treatment (Fig. 4D). Thus, the chemotherapy-induced demethylation of the PAX6 promoter may expand LUAD CSCs via the activation of PAX6-GLI-SOX2 signaling.
Figure 4.
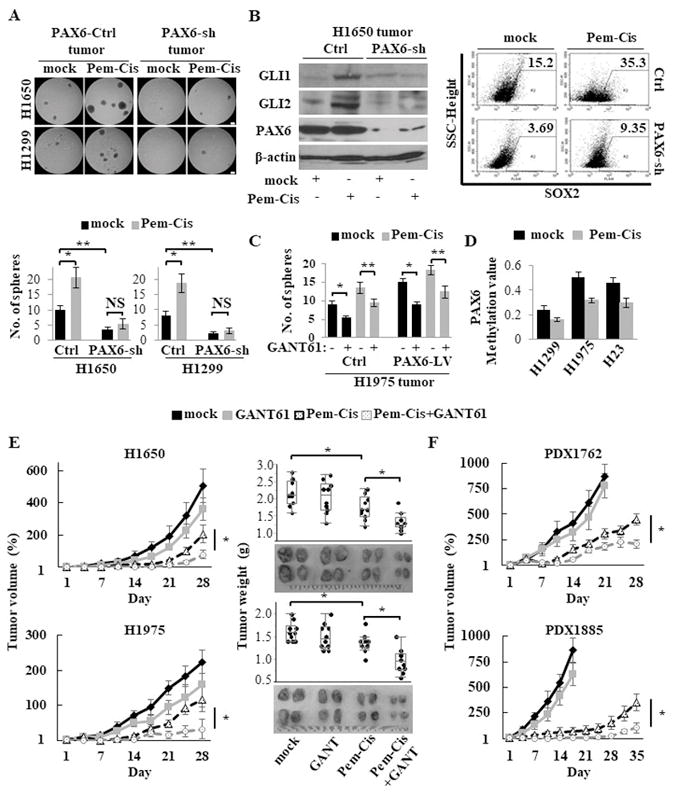
CSC expansion following chemotherapy abrogated by targeting the PAX6-GLI-SOX2 axis in xenograft models. (A) Sphere formation assay of cancer cells isolated from xenografted tumor tissues after treatment with pemetrexed (75 mg/kg, once daily, 5 days on and 2 days off) plus cisplatin (4 mg/kg, once every 7 days; Pem-Cis) chemotherapy for 14 days in PAX6-sh or PAX6-Ctrl xenograft models (NCI-H1650 and NCI-H1299 cells). Upper, representative images of sphere formation (scale bars, 200 μm); Lower, number of spheres over 100 μm. (B) The expression levels of GLI, PAX6, and SOX2 of xenografted tumor tissues after treatment with Pem-Cis chemotherapy for 14 days in PAX6-sh or PAX6-Ctrl NCI-H1650 xenograft models, as measured by western blotting analysis for GLI and PAX6 (left) and flow cytometry for SOX2 (right). (C) Sphere formation assay of cancer cells isolated from xenografted tumor tissues after treatment with Pem-Cis chemotherapy in the presence or absence of the GLI antagonist GANT61 (50 mg/kg) treatment for 14 days in PAX6-LV or PAX6-Ctrl NCI-H1975 xenograft models. (D) The promoter methylation levels of the PAX6 gene of xenografted tumor tissues after treatment with Pem-Cis chemotherapy for 14 days in xenograft models, as measured by Q-MSP. (E) The in vivo therapeutic efficacy of the combination of Pem-Cis chemotherapy for 14 days with GANT61 (50 mg/kg) for 21 days in NCI-H1650 and NCI-H1975 xenograft models (five mice per group). Left, tumor growth curve; Right, tumor weight from both flanks of five mice per group. Growth curves were calculated by comparing the tumor size before any treatment with the size at different time points of therapy. (F) The in vivo therapeutic efficacy of the combination of Pem-Cis chemotherapy with GANT61 (50 mg/kg) in patient derived xenograft (PDX) models (five mice per group).
Each error bar indicates mean ± SEM. *, P <0.05; **, P <0.01 (Wilcoxon–Mann–Whitney test [A and C] and Kruskal–Wallis with post-hoc test [E and F]). See also Fig. S7.
Given the crucial role of the PAX6-GLI-SOX2 signaling axis in LUAD CSC expansion following chemotherapy, we hypothesized that a combination of chemotherapy with a treatment to block this axis might be more effective than chemotherapy alone. To test our hypothesis, the tumor-bearing mice randomly divided into four groups: mock, GANT61, Pem-Cis chemotherapy, or Pem-Cis chemotherapy plus GANT61 treatment. The combination treatment with Pem-Cis chemotherapy plus GANT61 demonstrated significantly continuous suppression of tumor growth rate and tumor weight compared with either treatment alone (Fig. 4E). Finally, we confirmed the therapeutic efficacy of the same regimens in the most clinically relevant LUAD patient-derived xenograft (PDX) models (Fig. 4F).
PAX6 promoter methylation (PM) is a frequent and cancer-specific event in lung cancer
We observed that PAX6 expression significantly downregulated in tumor tissues compared with the adjacent histologically normal tissues in the TCGA LUAD cohort (Supplementary Fig. S8A). To test the cancer specificity of the PM event of the PAX6 gene, Q-MSP assay conducted on 25 primary LUAD tumors and matched adjacent normal samples. PAX6 showed significantly higher PM values in tumors compared with their corresponding adjacent normal samples (Fig. 5A). To determine the broader idea of PM prevalence of PAX6, we performed Q-MSP assay on an additional 67 tumor samples that lead to a total of 92 tumor samples (25+67) in a training cohort. Again, higher PM values were observed in the tumor samples (Fig. 5B). The optimal cutoff value for distinguishing between tumors (n=92) and normal samples (n=25) was calculated using receiver operating characteristic (ROC) analysis. By using the optimal methylation cutoff value, the observed sensitivity and specificity for cancer detection were 66.3% (61/92) and 88.0% (22/25), respectively (Supplementary Table S3). As expected, the expression levels of PAX6 were significantly higher in subjects without PM than in those with PM (Fig. 5C).
Figure 5.
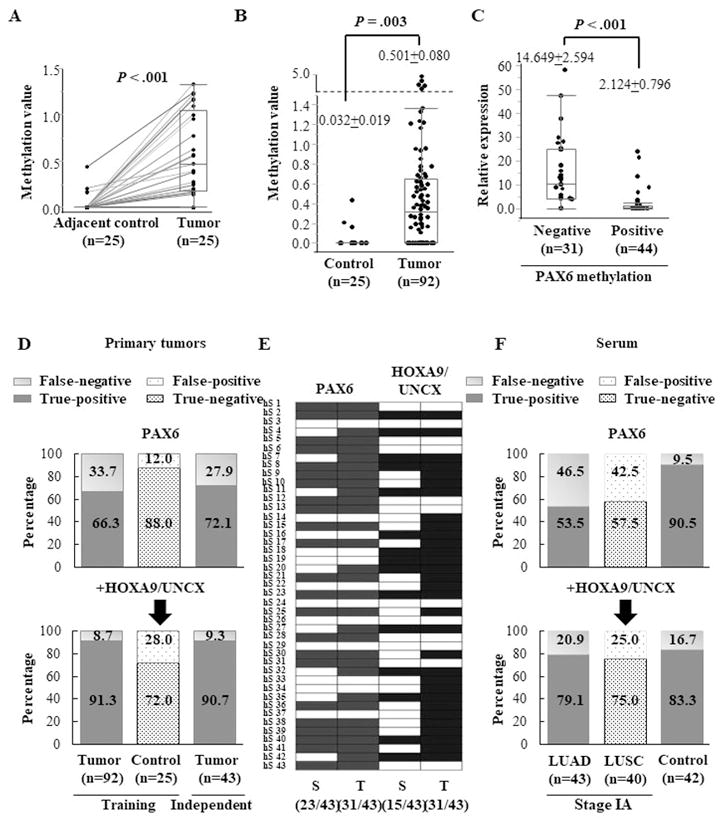
The potential of a methylation panel (PAX6, HOXA9, and UNCX) as a biomarker for early cancer detection using serum samples from subjects with LUAD. (A) The promoter methylation levels of the PAX6 gene measured by Q-MSP in 25 primary LUAD tumors and matched adjacent normal tissues. The promoter methylation values of tumors and the matched adjacent tissues were connected with a line. (B) Box plot of the promoter methylation values of the PAX6 gene measured by Q-MSP in primary lung tumors (n=92) and adjacent normal tissues (n=25). (C) The relative expression levels of PAX6 according to the status of its PM. The relative mRNA expression levels were calculated by multiplying the Q-RT-PCR values (normalized by β-actin) with 1000 for easy tabulation. PAX6 PM was determined by Q-MSP. (D) The sensitivity and specificity of methylation gene panel in the training cohort (n=92) and independent stage IA LUAD cohort (n=43). The schematic representation shows true positives (red), false negatives (pale red), true negatives (blue), and false positive (pale blue) detected by the PAX6 gene alone or the panel of three genes (PAX6, HOXA9, and UNCX). (E) The promoter methylation patterns of the PAX6 gene and HOXA9/UNCX genes in 43 primary tumors and matched serum samples from patients with stage IA LUAD. Cells in color represent the presence of methylation in the corresponding gene in tumor (T) and the matched serum (S) samples. The frequency of positive methylation is shown in the parenthesis. (F) The sensitivity and specificity of the methylation gene panel for cancer detection in serum samples from stage IA LUAD (n=43) and stage IA LUSC (n=40). See also Fig. S8.
Each error bar indicates mean ± SEM. The paired t-test (A) and Wilcoxon–Mann–Whitney test (B and C) were performed. See also Fig. S8.
To confirm the PM frequency in an independent cohort, we tested 43 stage IA LUAD primary tissues. Using the same cutoff as of training set, 31 of 43 subjects (72.1%) were PM positive for the PAX6 gene, indicating a potential for detecting neoplastic changes at a very early stage LUAD (Fig. 5D). Collectively, these findings suggest that PM of the PAX6 gene is a frequent and cancer-specific event.
PAX6 gene PM has a potential as a biomarker for early lung cancer detection
Previous report suggest that robust and frequent PM of a homeobox gene superfamily occurs in early-stage lung cancer 16, and the homeodomain-containing PAX6 belongs to the homeobox gene superfamily 32. We recently reported a candidate PM panel of six genes as a biomarker for early lung cancer detection in the same cohort used in this study, among which HOXA9 and UNCX also belong to the homeobox gene superfamily 19. Therefore, we assessed the cancer detection accuracy of a combination panel of three homeobox genes (PAX6, HOXA9, and UNCX). Notably, the combination panel yielded a sensitivity of 91.3% (84/92) and a specificity of 72.0% (18/25) in the training cohort and a sensitivity of 90.7% (39/43) in an independent set of stage IA LUAD cohort (Fig. 5D).
Given the high positive PM frequency of our gene panel in early stage primary LUAD samples, we next assessed the potential for minimally invasive early cancer detection using serum samples from 43 stage IA LUAD, 40 stage IA LUSC, and 42 population-matched control subjects from the New York University Lung Cancer Screening Cohort 33. The mean methylation value of the PAX6 gene was significantly higher in cancer subjects compared with controls (Supplementary Fig. S8B). By determining the optimal cutoff using ROC curves in 43 stage IA LUAD and 42 control samples, the sensitivity and specificity of PAX6 PM for LUAD detection were 53.5% (23/43) and 90.5% (38/42), respectively (Supplementary Table S3). In the 43 primary tumors and the matched serum samples from stage IA LUAD, the PM status of the PAX6 gene in serum DNA was always concordant with the primary tumor DNA, and the concordance rate was 74.2% (23/31) (Fig. 5E). Notably, our PM panel of three genes yielded a sensitivity of 79.1% (34/43) and specificity of 83.3% (35/42) in serum DNA from stage IA LUAD and detected 30 (75.0%) of the 40 serum samples from stage IA LUSC using the same cutoff (Fig. 5F).
Since malignant pleural effusion (MPE) is a common complication of NSCLC 34, we also assessed the feasibility of our methylation gene panel for detecting cancer using 70 pleural effusion (PE) samples (Supplementary Table S3). PM of the PAX6 gene was significantly higher in MPE than in PE with negative cytology, and our combination panel showed a sensitivity of 78.4% and a specificity of 75.8% (Supplementary Fig. S8C–D). Similar diagnostic accuracy observed in the ascites samples, despite the fact that the malignant ascites were collected from subjects with various types of cancer (Supplementary Fig. S8E–F).
Discussion
Lung cancer is the second most common cancer and the leading cause of cancer deaths worldwide 18. Despite advances in treatment, such as chemotherapy and molecular targeted therapy, the five-year survival rate is less than 15% in lung cancer 18. The poor outcome is largely due to the advanced stage at the time of diagnosis and the emergence of treatment resistance. Thus, development of novel therapeutic strategies and biomarkers for early cancer detection are major clinical challenges to improving the prognosis of lung cancer patients. Given the central role of CSCs governing at the top of the cellular hierarchy in tumor initiation, progression, and therapeutic resistance 2, a better understanding of the molecular mechanisms crucial to CSC maintenance and expansion may shed light on the improvement of clinical management. Here, we provide a rationale for targeting the PAX6-GLI-SOX2 signaling axis and support the clinical utility of PAX6 PM as a serum biomarker for early cancer detection.
The functional role of PAX6 is cellular context-dependent in cancer. Our findings suggest that PAX6 acts as a critical oncogene responsible for cancer stemness properties via GLI-SOX2 signaling, accelerating CSC expansion in LUAD. SOX2 establishes a continuum between tumor initiation and progression via the direct regulation of key genes that control cancer stemness properties 24. Indeed, we found that SOX2 regulates the expressions of pluripotent transcription factors (OCT4 and NANOG) in line with previous reports 23, 25. Furthermore, the association of SOX2 with poor outcomes is reported in LUAD 26. Interestingly, PAX6 forms a molecular complex with SOX2, leading to synergistic gene activation during lens development 35. In addition, SOX2 is required for maintenance of PAX6 expression 35. Thus, PAX6-induced SOX2 acts as a master regulator to govern and maintain LUAD CSCs, and the regulatory circuitry between PAX6 and SOX2 may further stabilize a stemness-like state.
Normal lung epithelial differentiation is coordinated by a complex network of lineage-specific transcription factors 36. Aberrant activation of cell lineage-restricted pathways is required for oncogenic transformation, whereas the activation of differentiation programs restrains metastasis and invasion 29. Notably, HOPX and NKX2-1 are essential differentiation lineage transcription factors for both normal lung maturation and the LUAD alveolar-like subtype 29, 30, 37, and PAX6 represses the expression of HOPX and NKX2-1. In addition, we observed the inverse correlation of PAX6 expression with HOPX and NKX2-1 expression, as well as a positive correlation of PAX6 expression with SOX2 and GLI expression in both our and the TCGA LUAD cohorts. Thus, PAX6 not only enhanced the key pluripotent factors but also repressed the differentiation lineage factors via SOX2, driving lung cancer cells toward a stem-like state in LUAD. On the other hand, in the differentiated non-CSCs that represent most of the tumor cell population, PAX6 is transcriptionally silenced by its PM. It is generally believed that methylation is a dynamic epigenetic alteration during ESC differentiation, which contributes to the turn-on and turn-off of particular genes that are related to pluripotency and self-renewal 38, 39. Indeed, when spheroid cells were converted into a differentiated state, the demethylated promoter region of the PAX6 gene was re-methylated along with the restored expression of HOPX and NKX2-1. In addition, the histologically differentiated tumors showed a lower PAX6 expression and a trend toward a higher PM frequency compared with poorly differentiated tumors in the human samples. Collectively, the relative amount of PAX6 may determine cell fate by switching toward a stem-like or differentiated state in LUAD. Further research is needed to determine in which contexts the epigenetic alteration of PAX6 regulates the switch toward a stem-like or differentiated state of LUAD cells.
Hh signaling is a stemness-related pathway tightly regulated during development, and its aberrant activation plays a critical role in maintaining CSCs in several cancer types 22. In LUAD, the aberrant activation of this pathway is observed 26, 40, consistent with our findings showing SMO-independent overexpression of GLI in lung CSCs. Hh signaling can involve canonical and noncanonical pathways. The canonical pathway follows the Patched (PTCH)–SMO–GLI axis, whereas noncanonical pathways can facilitate SMO-independent GLI activation 20, 40. The activated GLI promotes the expression of Hh target genes, among which SOX2 is upregulated directly by the binding of GLI to the proximal promoter region of the SOX2 gene. Furthermore, the GLI gene has PAX6 binding sites within its transcribed regions, indicating a possible target gene for PAX6 41. Thus, our findings suggest that PAX6 promotes the activation of the Hh signaling pathway in the noncanonical manner and subsequent SOX2 overexpression.
Therapeutically, the blockade of the PAX6-GLI-SOX2 signaling axis elicits a long-lasting therapeutic efficacy by limiting CSC expansion following chemotherapy, indicating these transcription factors are promising target molecules for LUAD CSCs. However, most transcription factors, including PAX6 and SOX2, are generally considered as undruggable targets due to the lack of small molecule binding pockets and the highly charged surface 27, 28. Thus, the pharmacological inhibition of Hh pathway components may be a clinically appropriate approach to block this axis. Importantly, GLI is pharmacologically targetable and the final effector of the Hh pathway, even if this pathway is activated in canonical and/or noncanonical manners 27. Indeed, the GLI antagonist, but not the SMO antagonist, attenuated PAX6-promoted tumorigenicity and GLI expression. Therefore, GLI may be the most preferable target in blocking the PAX6-GLI-SOX2 axis for the eradication of LUAD CSCs.
Cancer detection at an early stage increases survival rates. While low-dose spiral computed tomography (CT) can be a reliable screening tool for the early detection of lung cancer and decreases the mortality rate 42, it has poor specificity and high false positive rate leading to surgical resections for benign nodules 42–44. Therefore, novel, minimally invasive biomarkers are ideal to increase diagnostic accuracy and decrease unnecessary invasive diagnostic procedures. DNA methylation analysis in blood samples is one of the most promising approaches because it is a minimally invasive alternative compared to the more invasive routine procedures required for diagnosis. In NSCLC, a large fraction of the methylated CpG islands are mapped to homeobox genes in the genome, suggesting a common epigenetic pathway involving the homeobox gene network 16. Our combination panel of three homeobox genes (PAX6, HOXA9, and UNCX) showed comparable sensitivity (79.1% vs. 71.1%) and superior specificity (83.3% vs. 62.7%) in serum samples from stage IA LUAD compared with the CT screening cohort 42, 43. Furthermore, the diagnostic accuracy maintain comparable sensitivity with superior specificity compared to previous studies using blood samples 19, 45, indicating the potential for its use as a serum biomarker for early lung cancer detection. In addition, our gene panel successfully detected not only MPE but also malignant ascites, suggesting the utility for cancer detection from various bodily fluids. Further studies are required to determine the clinical feasibility in a larger cohort.
In summary, we demonstrate that PAX6 drives cancer cells toward a stem-like state via the GLI-SOX2 signaling axis and the blockade of this axis suppresses CSC expansion following chemotherapy in LUAD. In addition, a methylation panel including PAX6 can potentially serve as a minimally invasive serum biomarker for early cancer detection. Our findings provide a clinical rationale for targeting the PAX6-GLI-SOX2 signaling axis and demonstrate the clinical utility of PAX6 PM as a biomarker for early cancer detection.
Materials and Methods
Compounds and reagents
Cisplatin, demethylating agent 5-Aza-2′-deoxycytidine (5-Aza-dC), histone deacetylase inhibitor Trichostatin A (TSA) were purchased from Sigma-Aldrich (St. Louis, USA). Pemetrexed and GLI antagonist GANT61 were purchased from MedKoo Bioscences (Morrisville, USA). The SMO antagonist cyclopamine and Recombinant human Shh ligand were purchased from STEMCELL Technologies (Cambridge, USA) and R&D Systems (Minneapolis USA), respectively. Recombinant human epidermal growth factor (EGF) and the fibroblast growth factors (FGF)-basic were purchased from PeproTech (New Jersey, USA).
Cell lines and tissue samples
LUAD cell lines NCI-H1650, NCI-H1975, NCI-H1299, and NCI-H23 were obtained from and propagated according to the recommendation of the American Type Culture Collection (Manassas, VA, USA). Re-authentification of cells was performed using PowerPlex 16 HS for short tandem repeats analysis at Genetic resource core facility, the Johns Hopkins University School of Medicine (JHUSOM), Institute of Genetic Medicine, and all cell lines have been confirmed as authentic.
This study included human samples, including 92 primary NSCLC, 43 primary tumor tissues with matched serum samples from stage IA LUAD, 40 serum samples from stage IA LUSC, 42 population-matched control serum samples, 70 pleural effusions (PEs), and 49 ascites samples. The demographic and clinical characteristics of all the cohorts were summarized previously 19. Informed consent was obtained from all patients before collecting samples. Approval for research on human subjects was obtained from the Johns Hopkins University Institutional Review Boards. This study qualified for an exemption under the U.S. Department of Health and Human Services policy for the protection of human subjects [45 CFR 46.101(b)] in accordance with U.S. Common Rule.
TCGA analysis
The gene expression data of 515 primary LUAD samples and 58 tumor adjacent, histologically normal samples in the TCGA LUAD cohort 46 was downloaded from the Broad GDAC Firehose. Gene Set Enrichment Analysis (GSEA) was performed to identify signatures enriched by PAX6-high group in Oncogenic and Hallmarks gene set collections from the Molecular Signatures Database (MSigDB; Broad Institute) using the GSEAPreranked tool.
RNA extraction and quantitative reverse transcription real time PCR (Q-RT-PCR)
Total RNA from cell lines and formaldehyde fixed-paraffin embedded human tissues was isolated using the RNeasy Plus Mini Kit (Qiagen) and the RecoverAll™ Total Nucleic Acid Isolation Kit (Ambion), respectively. Q-RT-PCR was performed using the Fast SYBR Green Master Mix (Thermo Fisher Scientific).
Western blotting analysis
Whole cell lysates were extracted using RIPA buffer (Thermo Scientific) supplemented with 10 μL/mL Halt™ Protease Inhibitor Cocktail Kit (Life Technologies) and 30 μL/mL Halt™ Phosphatase Inhibitor Cocktail Kit (Life Technologies). The protein concentrations were determined using a Pierce™ BCA Protein Assay Kit (Life Technologies), and the protein were separated on NuPAGE® 4–12% Bis-Tris Gel (Life Technologies) according to the manufacturer’s protocol. NANOG (D73G4), OCT4 (D7O5Z), and GLI1 (C68H3) antibodies were obtained from Cell Signaling Technology, except for SOX2 (EPR3131; Abcam, Cambridge, USA), GLI2 (AF3635; Novus Biologicals), PAX6 (ab5790; Abcam), and β-actin (A2228; Sigma-Aldrich). Secondary horseradish peroxidase (HRP)–conjugated antibodies were obtained from Cell Signaling Technology, and chemiluminescent detection of HRP-labeled antibodies was performed using Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare, Piscataway, USA). As loading control, β-actin was used.
DNA extraction and quantitative methylation-specific polymerase chain reaction (Q-MSP)
DNA was extracted using the standard phenol-chloroform extraction protocol as described previously 19. Bisulfite treatment was conducted with an EpiTect Bisulfite Kit (Qiagen). For 5-Aza-2′-deoxycytidine (5-Aza-dC) treatment, cells were treated with 5 μmol/L of the 5-Aza-dC, as described previously 47. For Q-MSP, amplification reactions were done in a 7900HT Fast Real-Time PCR System (Life Technologies). Results were analyzed by Sequence Detector System (SDS) software (Applied Biosystems, Foster City, CA, USA).
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were conducted using SimpleChIP Enzymatic Chromatin IP Kit (Cell signaling Technology). GLI1 and GLI2 antibodies were obtained from Cell Signaling Technology and Novus Biologicals, respectively.
Gene silencing and expression
PAX6 siRNA Lentiviral Particles (Cat # sc-36195-v) was used for the knockdown of the gene expression (PAX6-sh; Santa Cruz Biotechnology, Dallas, USA). This particles include a pool of 3 different siRNA duplexes (sc-36195A, sc36195B, and sc-36195C) as follows: sense; CCAACGGAUGUGUGAGUAATT, antisense; UUACUCACACAUCCGUUGGTT (sc-36195A), sense; GAAGCUGCAAAGAAAUAGATT, antisense; UCUAUUUCUUUGCAGCUUCTT (sc-36195B), and sense: CUACCAACCAAUUCCACAATT, antisense: UUGUGGAAUUGGUUGGUAGTT (sc-36195C). Control shRNA Lentiviral Particles (Cat # sc-108080) was used as a control (PAX6-Ctrl; Santa Cruz Biotechnology). Pax-6 Lentiviral Activation Particles (Cat # sc-418357-LAC) for PAX6 induction (PAX6-LV) and Control Lentiviral Activation Particles (Cat # sc-437282) for a control (PAX6-Ctrl) were used (Santa Cruz Biotechnology). EF1A-Human-SOX2 lentivirus (Cat # PLV-10013) for SOX2 induction (SOX2-LV) and EF1A-vector control lentivirus (Cat # PLV-10074) for a control (SOX2-Ctrl) were purchased from Cellomics Technology (Rockville, USA). SOX2 shRNA pGFP-C-shLenti Vector (Cat # TL309173) for SOX2 knockdown (SOX2-sh) and Non-effective 29-mer scrambled shRNA pGFP-C-shLenti Vector (Cat # TR30021) for a control (SOX2-Ctrl) were purchased from Origene (Rockville, USA). SOX2-sh lentiviral vector includes 4 unique 29mer shRNA constructs as follows; AACATGATGGAGACGGAGCTGAAGCCGCC, CAGTACAACTCCATGACCAGCTCGCAGAC, CGTTCATCGACGAGGCTAAGCGGCTGCGA, and TTTACTCCATTATGCACAGTTTGAGATAA. Lentiviral particles were produced by cotransfection of each lentiviral vector with the Lenti-vpak Packaging Kit (Origene) into 293 cells according to the manufacturer’s protocol. Stable cells were established by optimal antibiotic selection. For the knockdown of GLI1 and GLI2, cells were transfected with GLI1 Silencer Select siRNA (GLI1-siRNA; Cat # 4392420 s5815; Thermo Fisher Scientific) and GLI2 Silencer Select siRNA (GLI2-siRNA; Cat # # 4392420 s5819; Thermo Fisher Scientific) at the final concentration of 10 nM by forward transfection using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s protocol. The target sequences for GLI1 and GLI2 are as follows: GLI1-siRNA sense; CCAACUUGCCCAAUCACAATT, GLI1-siRNA antisense; UUGUGAUUGGGCAAGUUGGGT, GLI2-siRNA sense; AGAUCCACAUGUACGAACATT, and GLI2-siRNA antisense; UGUUCGUACAUGUGGAUCUGG. Silencer Select Negative Control No. 1 siRNA (Thermo Fisher Scientific; Cat # 4390843) was used as a control for nonspecific effect. To verify the knockdown, western blotting analysis was performed 72 hours after transfection.
In vivo xenograft assay
Mice were maintained in accordance with the American Association of Laboratory Animal Care guidelines. Patient-derived xenograft (PDX) tumor tissues (1762 and 1885) were obtained from Champion Oncology (Maryland, USA). Athymic (nu+/nu+) mice and NOD/SCID/IL2Rγ−/− (NSG) mice were obtained from Harlan Laboratories and the JHUSOM animal care facility, respectively. When tumors reached a volume of 100–200 mm2, tumor-bearing mice were randomly assigned into experimental groups (five mice per group). For GANT61 treatment, GANT61 in solvent (50 mg/kg) or solvent only were administered every other day for 21 days. For pemetrexed plus cisplatin (Pem-Cis) treatment, pemetrexed (75 mg/kg) for 5 days followed by a 2 day holiday plus cisplatin (4 mg/kg) every 7 days were administered for 14 days. No blinding was done. All experiments using mice were approved by the JHUSOM Animal Care and Use Committee, and the mice were maintained in accordance with the American Association of Laboratory Animal Care guidelines.
Statistical analysis
In each set of data analyses, the estimate variation is indicated in each figure as a standard error of mean (SEM). The two groups were compared with the Wilcoxon–Mann–Whitney test. A comparison between the multiple groups was performed using the Kruskal–Wallis with post-hoc test (Dwass-Steel test) for non-parametrically continuous variables. Categorical variables were analyzed using Fisher’s exact test or a Chi-squared test. The level of statistical significance was set at P < 0.05. All statistical analyses were conducted using the JMP 12 software package (SAS Institute).
Supplementary Material
Acknowledgments
Financial support: This work was funded by the Flight Attendant Medical Research Institute Clinical Innovative Award 103015 (M.O. Hoque), NCI R01CA206027 (M.O. Hoque), the Career Development award from SPORE in Cervical Cancer Grants P50 CA098252 (M.O. Hoque).
Footnotes
Detailed materials and methods are provided in the Supplemental Information.
Conflict of interest: The authors declare no potential conflicts of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Eramo A, Haas TL, De Maria R. Lung cancer stem cells: tools and targets to fight lung cancer. Oncogene. 2010;29:4625–35. doi: 10.1038/onc.2010.207. [DOI] [PubMed] [Google Scholar]
- 2.Beck B, Blanpain C. Unravelling cancer stem cell potential. Nature reviews Cancer. 2013;13:727–38. doi: 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- 3.Asami M, Pilz GA, Ninkovic J, Godinho L, Schroeder T, Huttner WB, et al. The role of Pax6 in regulating the orientation and mode of cell division of progenitors in the mouse cerebral cortex. Development (Cambridge, England) 2011;138:5067–78. doi: 10.1242/dev.074591. [DOI] [PubMed] [Google Scholar]
- 4.Sansom SN, Griffiths DS, Faedo A, Kleinjan DJ, Ruan Y, Smith J, et al. The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet. 2009;5:e1000511. doi: 10.1371/journal.pgen.1000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lang D, Powell SK, Plummer RS, Young KP, Ruggeri BA. PAX genes: roles in development, pathophysiology, and cancer. Biochemical pharmacology. 2007;73:1–14. doi: 10.1016/j.bcp.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 6.Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell. 2001;105:43–55. doi: 10.1016/s0092-8674(01)00295-1. [DOI] [PubMed] [Google Scholar]
- 7.Yamaoka T, Yano M, Yamada T, Matsushita T, Moritani M, Ii S, et al. Diabetes and pancreatic tumours in transgenic mice expressing Pa x 6. Diabetologia. 2000;43:332–9. doi: 10.1007/s001250050051. [DOI] [PubMed] [Google Scholar]
- 8.Osumi N, Shinohara H, Numayama-Tsuruta K, Maekawa M. Concise review: Pax6 transcription factor contributes to both embryonic and adult neurogenesis as a multifunctional regulator. Stem cells (Dayton, Ohio) 2008;26:1663–72. doi: 10.1634/stemcells.2007-0884. [DOI] [PubMed] [Google Scholar]
- 9.Hu B, Wang Q, Wang YA, Hua S, Sauve CG, Ong D, et al. Epigenetic Activation of WNT5A Drives Glioblastoma Stem Cell Differentiation and Invasive Growth. Cell. 2016;167:1281–95. e18. doi: 10.1016/j.cell.2016.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shyr CR, Tsai MY, Yeh S, Kang HY, Chang YC, Wong PL, et al. Tumor suppressor PAX6 functions as androgen receptor co-repressor to inhibit prostate cancer growth. The Prostate. 2010;70:190–9. doi: 10.1002/pros.21052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayes DA, Hu Y, Teng Y, Siegel E, Wu X, Panda K, et al. PAX6 suppresses the invasiveness of glioblastoma cells and the expression of the matrix metalloproteinase-2 gene. Cancer research. 2006;66:9809–17. doi: 10.1158/0008-5472.CAN-05-3877. [DOI] [PubMed] [Google Scholar]
- 12.Lang D, Mascarenhas JB, Powell SK, Halegoua J, Nelson M, Ruggeri BA. PAX6 is expressed in pancreatic adenocarcinoma and is downregulated during induction of terminal differentiation. Molecular carcinogenesis. 2008;47:148–56. doi: 10.1002/mc.20375. [DOI] [PubMed] [Google Scholar]
- 13.Mascarenhas JB, Young KP, Littlejohn EL, Yoo BK, Salgia R, Lang D. PAX6 is expressed in pancreatic cancer and actively participates in cancer progression through activation of the MET tyrosine kinase receptor gene. The Journal of biological chemistry. 2009;284:27524–32. doi: 10.1074/jbc.M109.047209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao X, Yue W, Zhang L, Ma L, Jia W, Qian Z, et al. Downregulation of PAX6 by shRNA inhibits proliferation and cell cycle progression of human non-small cell lung cancer cell lines. PloS one. 2014;9:e85738. doi: 10.1371/journal.pone.0085738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shiraishi M, Sekiguchi A, Terry MJ, Oates AJ, Miyamoto Y, Chuu YH, et al. A comprehensive catalog of CpG islands methylated in human lung adenocarcinomas for the identification of tumor suppressor genes. Oncogene. 2002;21:3804–13. doi: 10.1038/sj.onc.1205454. [DOI] [PubMed] [Google Scholar]
- 16.Rauch TA, Wang Z, Wu X, Kernstine KH, Riggs AD, Pfeifer GP. DNA methylation biomarkers for lung cancer. Tumour Biol. 2012;33:287–96. doi: 10.1007/s13277-011-0282-2. [DOI] [PubMed] [Google Scholar]
- 17.Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nature reviews Cancer. 2004;4:707–17. doi: 10.1038/nrc1432. [DOI] [PubMed] [Google Scholar]
- 18.Leon G, MacDonagh L, Finn SP, Cuffe S, Barr MP. Cancer stem cells in drug resistant lung cancer: Targeting cell surface markers and signaling pathways. Pharmacology & therapeutics. 2016;158:71–90. doi: 10.1016/j.pharmthera.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Ooki A, Maleki Z, Tsay JJ, Goparaju C, Brait M, Turaga N, et al. A Panel of Novel Detection and Prognostic Methylated DNA Markers in Primary Non-Small Cell Lung Cancer and Serum DNA. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23:7141–52. doi: 10.1158/1078-0432.CCR-17-1222. [DOI] [PubMed] [Google Scholar]
- 20.Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nature reviews Clinical oncology. 2015;12:445–64. doi: 10.1038/nrclinonc.2015.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masui S, Nakatake Y, Toyooka Y, Shimosato D, Yagi R, Takahashi K, et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nature cell biology. 2007;9:625–35. doi: 10.1038/ncb1589. [DOI] [PubMed] [Google Scholar]
- 22.Justilien V, Fields AP. Molecular pathways: novel approaches for improved therapeutic targeting of Hedgehog signaling in cancer stem cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21:505–13. doi: 10.1158/1078-0432.CCR-14-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ooki A, Del Carmen Rodriguez Pena M, Marchionni L, Dinalankara W, Begum A, Hahn NM, et al. YAP1 and COX2 Coordinately Regulate Urothelial Cancer Stem-like Cells. Cancer research. 2018;78:168–81. doi: 10.1158/0008-5472.CAN-17-0836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014;511:246–50. doi: 10.1038/nature13305. [DOI] [PubMed] [Google Scholar]
- 25.Santini R, Pietrobono S, Pandolfi S, Montagnani V, D’Amico M, Penachioni JY, et al. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene. 2014;33:4697–708. doi: 10.1038/onc.2014.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bora-Singhal N, Perumal D, Nguyen J, Chellappan S. Gli1-Mediated Regulation of Sox2 Facilitates Self-Renewal of Stem-Like Cells and Confers Resistance to EGFR Inhibitors in Non-Small Cell Lung Cancer. Neoplasia (New York, NY) 2015;17:538–51. doi: 10.1016/j.neo.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Infante P, Alfonsi R, Botta B, Mori M, Di Marcotullio L. Targeting GLI factors to inhibit the Hedgehog pathway. Trends in pharmacological sciences. 2015;36:547–58. doi: 10.1016/j.tips.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Stolzenburg S, Rots MG, Beltran AS, Rivenbark AG, Yuan X, Qian H, et al. Targeted silencing of the oncogenic transcription factor SOX2 in breast cancer. Nucleic acids research. 2012;40:6725–40. doi: 10.1093/nar/gks360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheung WK, Zhao M, Liu Z, Stevens LE, Cao PD, Fang JE, et al. Control of alveolar differentiation by the lineage transcription factors GATA6 and HOPX inhibits lung adenocarcinoma metastasis. Cancer cell. 2013;23:725–38. doi: 10.1016/j.ccr.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watanabe H, Meyerson M. Hopping between differentiation states in lung adenocarcinoma. Cancer cell. 2013;23:707–9. doi: 10.1016/j.ccr.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu YP, Yang CJ, Huang MS, Yeh CT, Wu AT, Lee YC, et al. Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer research. 2013;73:406–16. doi: 10.1158/0008-5472.CAN-12-1733. [DOI] [PubMed] [Google Scholar]
- 32.Holland PW, Booth HA, Bruford EA. Classification and nomenclature of all human homeobox genes. BMC Biol. 2007;5:47. doi: 10.1186/1741-7007-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenberg AK, Lu F, Goldberg JD, Eylers E, Tsay JC, Yie TA, et al. CT scan screening for lung cancer: risk factors for nodules and malignancy in a high-risk urban cohort. PloS one. 2012;7:e39403. doi: 10.1371/journal.pone.0039403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nam HS. Malignant pleural effusion: medical approaches for diagnosis and management. Tuberc Respir Dis (Seoul) 2014;76:211–7. doi: 10.4046/trd.2014.76.5.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamachi Y, Uchikawa M, Tanouchi A, Sekido R, Kondoh H. Pax6 and SOX2 form a co-DNA-binding partner complex that regulates initiation of lens development. Genes & development. 2001;15:1272–86. doi: 10.1101/gad.887101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maeda Y, Dave V, Whitsett JA. Transcriptional control of lung morphogenesis. Physiological reviews. 2007;87:219–44. doi: 10.1152/physrev.00028.2006. [DOI] [PubMed] [Google Scholar]
- 37.Yin Z, Gonzales L, Kolla V, Rath N, Zhang Y, Lu MM, et al. Hop functions downstream of Nkx2. 1 and GATA6 to mediate HDAC-dependent negative regulation of pulmonary gene expression. American journal of physiology Lung cellular and molecular physiology. 2006;291:L191–9. doi: 10.1152/ajplung.00385.2005. [DOI] [PubMed] [Google Scholar]
- 38.Shen Y, Chow J, Wang Z, Fan G. Abnormal CpG island methylation occurs during in vitro differentiation of human embryonic stem cells. Human molecular genetics. 2006;15:2623–35. doi: 10.1093/hmg/ddl188. [DOI] [PubMed] [Google Scholar]
- 39.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–32. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 40.Po A, Silvano M, Miele E, Capalbo C, Eramo A, Salvati V, et al. Noncanonical GLI1 signaling promotes stemness features and in vivo growth in lung adenocarcinoma. Oncogene. 2017;36:4641–52. doi: 10.1038/onc.2017.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coutinho P, Pavlou S, Bhatia S, Chalmers KJ, Kleinjan DA, van Heyningen V. Discovery and assessment of conserved Pax6 target genes and enhancers. Genome research. 2011;21:1349–59. doi: 10.1101/gr.124115.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aberle DR, Adams AM, Berg CD, Black WC, Clapp JD, Fagerstrom RM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. 2011;365:395–409. doi: 10.1056/NEJMoa1102873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tammemagi MC, Katki HA, Hocking WG, Church TR, Caporaso N, Kvale PA, et al. Selection criteria for lung-cancer screening. N Engl J Med. 2013;368:728–36. doi: 10.1056/NEJMoa1211776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blanchon T, Brechot JM, Grenier PA, Ferretti GR, Lemarie E, Milleron B, et al. Baseline results of the Depiscan study: a French randomized pilot trial of lung cancer screening comparing low dose CT scan (LDCT) and chest X-ray (CXR) Lung Cancer. 2007;58:50–8. doi: 10.1016/j.lungcan.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 45.Begum S, Brait M, Dasgupta S, Ostrow KL, Zahurak M, Carvalho AL, et al. An epigenetic marker panel for detection of lung cancer using cell-free serum DNA. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:4494–503. doi: 10.1158/1078-0432.CCR-10-3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–50. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ooki A, Yamashita K, Kikuchi S, Sakuramoto S, Katada N, Kokubo K, et al. Potential utility of HOP homeobox gene promoter methylation as a marker of tumor aggressiveness in gastric cancer. Oncogene. 2010;29:3263–75. doi: 10.1038/onc.2010.76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.