

Abstract
Sphingolipids, including sphingomyelin (SM) and glucosylceramide (GlcCer), are generated by the addition of a polar head group to ceramide (Cer). Sphingomyelin synthase 1 (SMS1) and glucosylceramide synthase (GCS) are key enzymes that catalyze the conversion of Cer to SM and GlcCer, respectively. GlcCer synthesis has been postulated to occur mainly in cis-Golgi, and SM synthesis is thought to occur in medial/trans-Golgi; however, SMS1 and GCS are known to partially co-localize in cisternae, especially in medial/trans-Golgi. Here, we report that SMS1 and GCS can form a heteromeric complex, in which the N terminus of SMS1 and the C terminus of GCS are in close proximity. Deletion of the N-terminal sterile α-motif of SMS1 reduced the stability of the SMS1–GCS complex, resulting in a significant reduction in SM synthesis in vivo. In contrast, chemical-induced heterodimerization augmented SMS1 activity, depending on an increase in the amount and stability of the complex. Fusion of the SMS1 N terminus to the GCS C terminus via linkers of different lengths increased SM synthesis and decreased GlcCer synthesis in vivo. These results suggest that formation of the SMS1–GCS heteromeric complex increases SM synthesis and decreases GlcCer synthesis. Importantly, this regulation of relative Cer levels by the SMS1–GCS complex was confirmed by CRISPR/Cas9–mediated knockout of SMS1 or GCS combined with pharmacological inhibition of Cer transport protein in HEK293T cells. Our findings suggest that complex formation between SMS1 and GCS is part of a critical mechanism controlling the metabolic fate of Cer in the Golgi.
Keywords: sphingolipid, ceramide, dimerization, Golgi, protein domain, fusion protein, glucosylceramide synthase (GCS), lipid metabolism, sphingomyelin synthase 1 (SMS1), sterile α-motif (SAM) domain
Introduction
Sphingolipids, including sphingomyelin (SM)2 and glucosylceramide (GlcCer), are generated by the addition of a polar head group to ceramide (Cer); the head group is phosphocholine in SM and glucose in GlcCer. GlcCer serves as the core structure of more than 300 glycosphingolipids (GSLs), which are generated by the step-by-step addition of a sugar chain to GlcCer. In contrast, further extension of the polar head group does not occur in SM production. SM and GSLs mainly localize in the external leaflet of the plasma membrane and interact with cholesterol to form lipid microdomains, which play important roles in various cellular functions (1). Given that SM-rich microdomains in the plasma membrane are spatially and functionally distinct from GSL-rich microdomains (2), SM and GlcCer might have different biological functions.
Two enzymes, SM synthase (SMS) and GlcCer synthase (GCS; also termed GlcT-1, UGCG, or CGT) are involved in this metabolic branch point. SMS catalyzes the transfer of phosphocholine from phosphatidylcholine to Cer. SMS has two isoforms in mammals; SMS1 is localized in the Golgi apparatus, whereas SMS2 occurs in both the Golgi apparatus and plasma membranes (3). SMS1 is mainly responsible for the de novo synthesis of SM (4, 5), whereas SMS2 participates in the maintenance of the SM level in the plasma membrane (5, 6) and modulates the interaction between signaling proteins (7, 8). In contrast, only one GCS has been identified (9). GCS catalyzes the transfer of glucose from UDP-glucose to Cer and is also located in the Golgi apparatus (10). Therefore, SMS1 and GCS are key enzymes responsible for Cer metabolism in the Golgi apparatus.
Cer is synthesized de novo in the cytosolic leaflet of the ER bilayer by sequential enzyme reactions that are initiated by the condensation of serine and palmitoyl-CoA (11). Newly synthesized Cer is transported to the trans-Golgi in a Cer transport protein (CERT)–dependent manner for SM synthesis. In contrast, for GlcCer synthesis, Cer is transported from the ER to the cis-Golgi in a CERT-independent manner. The different roles of Cer delivery were confirmed in CERT-deficient cells; the level of SM, but not those of GlcCer and GSLs, was reduced in the mutant cells, indicating that CERT-transported Cer is selectively destined for SM synthesis (12). SMS1 is localized mainly in the medial/trans-Golgi (3, 13), whereas GCS is broadly distributed in the Golgi (10, 13–15). Immunoelectron microscopy and partial separation of Golgi subcompartments revealed that a significant fraction of SMS1 and GCS co-localize in certain Golgi compartments (5, 10, 13, 14), in particular, in medial/trans-Golgi cisternae (13). The question of why CERT-transported Cer in the trans-Golgi is selectively used for SM but not GlcCer synthesis despite the co-localization of SMS1 and GCS has not yet been solved.
The above reports prompted us to examine the mechanism for the alternate usage of Cer by SMS1 and GCS. In the present study, a co-immunoprecipitation assay revealed that SMS1 and GCS form a heteromeric complex. Biomolecular fluorescence complementation (BiFC) revealed that the N terminus of SMS1 was proximal to the C terminus of GCS, suggesting direct binding of the two enzymes. In addition, deletion analysis of SMS1 indicated that the N-terminal sterile α-motif (SAM) of SMS1 was important for stable interaction with GCS. Metabolic labeling experiments revealed that manipulation of the amount and stability of SMS1–GCS resulted in the up-regulation of SMS1 activity and the down-regulation of GCS activity. This regulation of SMS1–GCS was confirmed through knockout (KO) of SMS1 or GCS combined with pharmacological inhibition of CERT. Our findings suggest that the heteromeric complex of SMS1 and GCS is involved in the regulation of Cer metabolism, which may be one of the reasons why CERT-transported Cer is specifically used for SM synthesis.
Results
SMS1 forms a heteromeric complex with GCS
Before investigating the complex formation of SMS1 with GCS, the co-localization of both enzymes was examined. As expected on the basis of previous reports (3, 13), confocal microscopy showed that SMS1 strongly co-localized with GCS and the trans-Golgi network marker, p230, whereas SMS2 only partially co-localized with GCS and p230 (Fig. 1A). The similar results were obtained with the cis-Golgi marker, GM130 (data not shown). Although p230 is the marker of trans-Golgi network, the staining of p230 showed the perinuclear location similar to that of cis-Golgi marker GM130, suggesting that p230 locates near trans-Golgi cisternae, where it is positioned adjacent to the trans-Golgi network. High-resolution microscopy would be needed to discriminate cis-, medial-, and trans-Golgi cisternae and the trans-Golgi network.
Figure 1.
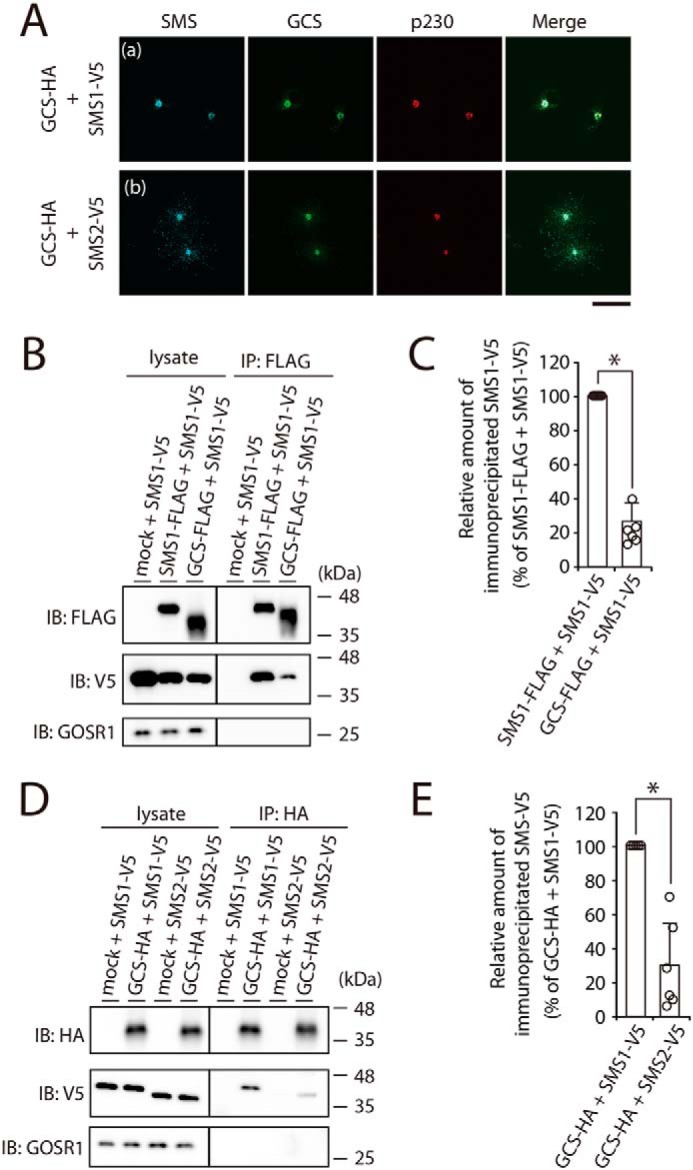
SMS1 forms a heteromeric complex with GCS. A, COS7 cells were transfected with the following combinations of plasmids: HA-tagged GCS and V5-tagged SMS1 or V5-tagged SMS2. At 24 h post-transfection, the cells were co-stained with anti-V5 and anti-HA antibodies, followed by appropriate Alexa Fluor–conjugated secondary antibodies, and analyzed by confocal microscopy. Localization was confirmed by co-staining with antibody against the trans-Golgi network marker p230. Blue, SMS1 and SMS2; green, GCS; red, p230. Scale bar, 50 μm. The results shown are from one experiment representative of three independent experiments. B and C, COS7 cells were transfected with the following combinations of plasmids: FLAG-tagged SMS1, FLAG-tagged GCS, or mock (empty vector) and V5-tagged SMS1. At 24 h post-transfection, the cells were lysed in a buffer containing 1% CHAPS, and immunoprecipitation was performed using an anti-FLAG affinity gel. B, cell lysates and precipitated proteins were analyzed by immunoblotting (IB) with anti-FLAG, anti-V5, or anti-GOSR1 antibody. Left panel, cell lysate; right panel, immunoprecipitate (IP). C, the quantities of V5-tagged SMS1 precipitated with the FLAG-tagged SMS1 or FLAG-tagged GCS are represented as the ratio of the band intensities. The values represent the means ± S.D. from six independent experiments. *, p < 0.01. D and E, COS7 cells were transfected with the indicated combination of plasmids. Proteins from cell lysates were immunoprecipitated using an anti-HA affinity gel. D, cell lysates and precipitated proteins were analyzed by immunoblotting with anti-HA, anti-V5, or anti-GOSR1 antibody. E, the quantities of V5-tagged SMS1 or V5-tagged SMS2 precipitated with HA-tagged GCS were represented as the ratio of the band intensities. The values represent the means ± S.D. from six independent experiments. *, p < 0.01.
Potential interactions between SMS1 and GCS were analyzed by immunoprecipitation and immunoblotting of differential epitope-tagged SMS1 and GCS. COS7 cells were transfected with V5-tagged SMS1 and FLAG-tagged SMS1 or GCS, and cell extracts were immunoprecipitated with anti-FLAG beads. Consistent with our previous study (16), a significant amount of SMS1-V5 was detected with the immunoprecipitated SMS1-FLAG, indicating that SMS1 forms a homomeric complex. Interestingly, a significant amount of SMS1-V5 was precipitated with GCS-FLAG (Fig. 1B, right middle panel). The band intensity of SMS1-V5 precipitated with GCS-FLAG was ∼22% of that of SMS1-V5 precipitated with SMS1-FLAG (Fig. 1C), suggesting that the interaction affinity of the SMS1–GCS complex is lower than that of the SMS1 homomeric complex. However, given that no Golgi-resident membrane protein GOSR1 was precipitated with GCS-FLAG (Fig. 1B, right lower panel), SMS1 specifically interacts with GCS. Immunoprecipitation using SMS2 and GCS generated very low signal as compared with that of SMS1 and GCS (Fig. 1D, right middle panel). The band intensity of SMS2-V5 precipitated with GCS-HA was ∼30% of the intensity of SMS1-V5 precipitated with GCS-HA (Fig. 1E). This may be because there is more SMS1 than SMS2 present in the Golgi complex (Fig. 1A). Therefore, we focused on the SMS1–GCS heteromeric complex in subsequent functional analyses.
The N terminus of SMS1 and the C terminus of GCS are in close proximity within the heteromeric complex
To confirm the interaction between SMS1 and GCS, we employed a BiFC assay using several chimeric proteins in which N- or C-terminal half fragments of fluorescent protein Venus (VN or VC) were fused to the N or C terminus of SMS1 or GCS (Fig. 2A). The BiFC assay is based on VN and VC fragment complementation; when VN and VC in chimeric proteins are in close proximity to each other, the Venus protein emits fluorescence (17, 18).
Figure 2.

The N terminus of SMS1 and the C terminus of GCS are in close proximity within the heteromeric complex. A, schematic representation of the chimeric proteins used for the BiFC assay. The constructs were designed to contain the amino acid sequences of VN (N-terminal residues 1–173 of Venus protein) or VC (C-terminal residues 155–238 of Venus protein), fused to the N or C terminus of SMS1 or GCS. The locations of V5 or FLAG tags are also indicated. B and C, COS7 cells were co-transfected with the indicated combination of plasmids. For confocal microscopy (B), the cells were subjected to immunostaining with anti-V5 and anti-FLAG antibodies followed by appropriate Alexa Fluor–conjugated secondary antibodies and were analyzed by confocal microscopy. Blue, VN-SMS1 and SMS1-VN; red, VC-SMS1, SMS1-VC, VC-GCS, and GCS-VC; green, Venus. Scale bar, 50 μm. The results shown are from one experiment representative of three independent experiments. For flow cytometry (C), the cells were subjected to immunostaining with anti-V5 antibody followed by a PerCP/Cy5.5-conjugated secondary antibody and APC-conjugated anti-FLAG antibody. Venus fluorescence complementation, detected by using the FITC channel, was analyzed (5,000 events) in double-positive cells (PerCP/Cy5.5 + APC) by flow cytometry. The values represent the means ± S.D. from five independent experiments. *, p < 0.01.
BiFC signals were examined with confocal microscopy in COS7 cells co-expressing chimeric proteins (Fig. 2B). When the SMS1 chimeric proteins with VN or VC in the N or C terminus were co-expressed in combinations of VN-SMS1 and VC-SMS1, VN-SMS1 and SMS1-VC, or SMS1-VN and SMS1-VC, BiFC signal was observed in the perinuclear region corresponding to the Golgi complex, where they were co-localized (Fig. 2B, panels a–c). These results indicated that the N- or C-terminal tail of SMS1 is located in close proximity to the N- or C-terminal tail of another SMS1 within the homodimer, which is consistent with our previous results (16). In addition, BiFC signal was also observed when chimeric proteins were co-expressed in combinations of VN-SMS1 and GCS-VC or of SMS1-VN and GCS-VC (Fig. 2B, panels d and e), suggesting that the C terminus of GCS is located in close proximity to the N or C terminus of SMS1. In contrast, complementation did not occur when the chimeric proteins were co-expressed in the combinations of VN-SMS1 and VC-GCS or of SMS1-VN and VC-GCS, although SMS1 and GCS were co-localized in the perinuclear region (Fig. 2B, panels f and g). These results suggested that the N terminus of GCS and the N or C terminus of SMS1 are too far apart for Venus complementation.
We quantified the intensity of Venus fluorescence in cells by flow cytometry (19). Consistent with the immunoprecipitation results (Fig. 1, B and C), significant BiFC signals were observed for the SMS1–GCS heteromeric complex (VN-SMS1 and GCS-VC or SMS1-VN and GCS-VC), although the signals were substantially lower than those for SMS1 homomeric complex (VN-SMS1 and VC-SMS1, VN-SMS1 and SMS1-VC, or SMS1-VN and SMS1-VC) (Fig. 2C, columns d and e versus columns a–c). In contrast, Venus fluorescence was not observed in combinations of VN-SMS1 and VC-GCS or of SMS1-VN and VC-GCS (Fig. 2C, columns f and g), again suggesting that the N terminus of GCS and the N or C terminus of SMS1 are too far apart for Venus complementation. Interestingly, the BiFC signal for the combination of VN-SMS1 and GCS-VC was ∼2.7-fold higher than that for SMS1-VN and GCS-VC (Fig. 2C, column d versus column e). These results indicated that the C terminus of GCS is located closer to the N terminus of SMS1 than to the C terminus of SMS1 within the heteromeric complex. Collectively, these results suggest that the N terminus of SMS1 and the C terminus of GCS are in close proximity within the heteromeric complex.
The N terminus of SMS1 and the C terminus of GCS face the cytosolic side of the Golgi apparatus
To confirm the proximity between the N and C termini of SMS1–GCS, we examined their orientations. The N and C termini of SMS1 have been shown to face the cytosolic side of Golgi membrane (3, 16). The C terminus and a large hydrophilic loop of GCS were previously shown to localize at the cytosolic side by protease sensitivity analysis (20); however, there are no experimental studies on the location of the N terminus of GCS. Hydropathy analysis of GCS has suggested two different models of the GCS N-terminal orientation: a model with luminal N terminus was proposed by Marks et al. (21), whereas another model with a cytosolic N terminus was predicted by SOSUI (http://harrier.nagahama-i-bio.ac.jp/sosui) (54).3 To determine and confirm the orientations of N and C termini of GCS from experimental data, FLAG–GCS–Myc, in which a FLAG tag and a Myc tag were located in the N and C termini of GCS, respectively, was constructed. Immunoblotting revealed that FLAG–GCS–Myc was expressed at almost comparable level to GCS without the tags (Fig. 3A) when nontagged GCS and FLAG–GCS–Myc were transfected in GCS KO#1 cells, as shown below in Fig. 5. In in vitro enzyme assays using C6–NBD–Cer as a substrate, the GCS activity of FLAG–GCS–Myc was similar to that of nontagged GCS, suggesting that the tag does not remarkably affect the topology of GCS (Fig. 3, B and C).
Figure 3.

The N terminus of SMS1 and the C terminus of GCS face the cytosolic side of the Golgi apparatus. A–C, GCS KO#1 cells were transfected with the indicated plasmids. All cells were lysed and analyzed by immunoblotting with anti-GCS antibody. Anti-GAPDH was used as a loading control. The blots are from one experiment representative of four independent experiments. B and C, assays for GCS activity in vitro. Cell lysates were incubated with C6–NBD–Cer in the presence of UDP–Glc. The values represent the means ± S.D. from four independent experiments. NS, not significant. D, COS7 cells were transfected with plasmid encoding FLAG–GCS–Myc. After fixation and permeabilization with 0.1% Triton X-100 (upper panel) or 0.002% digitonin (lower panel), the cells were co-stained with anti-FLAG and anti-Myc antibodies followed by individual Alexa Fluor–conjugated secondary antibodies and were analyzed by confocal microscopy. Green, N-terminal FLAG; red, C-terminal Myc. Scale bar, 50 μm. The results shown are from one experiment representative of three independent experiments. E and F, cartoon representations of the SMS1–GCS heteromer viewed from the cytosolic space (E) and from a section through the Golgi membrane (F). SMS1 and GCS have the following proximity structures: the solid arrows show the relatively close proximal segment between the N terminus of SMS1 and the C terminus of GCS, and the dashed arrows show the relatively long proximal segment between the C terminus of SMS1 and the C terminus of GCS. The transmembrane segments of SMS1 and GCS are shown in black and gray, respectively. N and C termini are represented by blue and red lines, respectively. Numbers 1–6 and I–III show the transmembrane helices. SAM in a box represents the SAM domain.
Figure 5.

Generation of CRISPR/Cas9–based SMS1–GCS DKO, SMS1 KO, and GCS KO cells. A and B, schematic representations of the genomic structures of the human SMS1 locus (A) and human GCS locus (B). Partial genomic structures for SMS1 (NCBI reference sequence NP_671512) and GCS (NCBI reference sequence NP_003349) are shown. The boxes represent exons, and the horizontal lines connecting the exons indicate introns. Open and filled regions in the exons are untranslated and coding regions, respectively. The 20-bp target sequences of gRNA are underlined. The PAM sequences are boxed. C–E, genomic DNA was extracted from SMS1–GCS DKO (C), SMS KO#1 and SMS1 KO#2 (D), or GCS KO#1 and GCS KO#2 (E) cell lines, and a genomic region containing the target sites of the gRNAs was sequenced. The 20-bp target sequences and the PAM sequences are depicted. A bold letter and dashes depict the identified insertion and deletions, respectively. The numbers of insertions and deletions (+, insertions; Δ, deletions) are shown. In GCS KO#1 cells, the 12-bp deletion of GCS caused no frameshift mutation (E); however, the mutant completely lost GCS activity using C6–NBD–Cer in vitro (data not shown). Because the deleted region (Phe14–Val17) seems to be the transmembrane domain, the mutation may cause protein misfolding.
A low concentration of digitonin selectively permeabilizes the plasma membrane, whereas Triton X-100 permeabilizes all cellular membranes (22). When FLAG–GCS–Myc, in which a FLAG tag and a Myc tag were located in the N and C termini of GCS, respectively, was expressed, the fluorescent signal for the FLAG epitope was observed in Triton X-100–treated cells (Fig. 3D, panel a) but not digitonin-treated cells (Fig. 3D, panel c). In contrast, the signal for the Myc epitope was observed in Triton X-100–treated cells (Fig. 3D, panel b), as well as in digitonin-treated cells (Fig. 3D, panel d). These results indicated that the N terminus and the C terminus of GCS are oriented to the luminal side and the cytosolic side of the Golgi membrane, respectively. Given that both the N and C termini of SMS1 are oriented to the cytosolic side of the Golgi membrane, the orientations of N and C termini of the two proteins are consistent with the results of the BiFC assay; the N and C termini of SMS1 are located in proximity to the C terminus of GCS; however, both termini of SMS1 are separated from the N terminus of GCS by the membrane.
Based on the results from the BiFC assay and the antibody accessibility assay, we propose a model in which the distance between the N terminus of SMS1 and the C terminus of GCS is relatively short, as shown by the solid arrows, and the distance between the C terminus of SMS1 and the C terminus of GCS is relatively long, as shown by the dashed arrows (Fig. 3, E and F). In contrast, the N terminus of GCS cannot interact with both the N and C termini of SMS1, because they are segregated by the lipid membrane.
The SAM domain of SMS1 is involved in stable heteromeric complex formation
SMS1 contains a predicted SAM domain composed of amino acids from Tyr6 to Lys68 at its N terminus (Fig. 3, E and F) (4). In general, SAM domains are putative protein interaction modules present in a wide variety of proteins involved in various biological processes (23, 24); however, the function of the SAM domain in SMS1 has not yet been elucidated. To examine whether the N-terminal SAM domain of SMS1 is involved in the heteromeric interaction between SMS1 and GCS, a SAM domain-truncation mutant of SMS1, SMS1–ΔSAM, was employed. Interestingly, SMS1–ΔSAM also precipitated with GCS; however, the precipitated SMS1–ΔSAM was approximately half the level of WT SMS1 (Fig. 4, A, right middle panel, and B), suggesting that deletion of the SAM domain reduces the affinity of SMS1 for GCS.
Figure 4.

The N-terminal SAM domain of SMS1 is important for the stable SMS1–GCS heteromeric complex. A and B, COS7 cells were transfected with the following combinations of plasmids: HA-tagged GCS or mock and V5-tagged SMS1 (WT) or V5-tagged SMS1–ΔSAM. At 24 h post-transfection, proteins from cell lysates were immunoprecipitated using an anti-HA affinity gel. A, cell lysates and precipitated proteins were analyzed by immunoblotting (IB) with anti-HA, anti-V5 antibody, or anti-GOSR1 antibody. Left panel, cell lysate; right panel, immunoprecipitation (IP). B, the quantities of V5-tagged SMS1 or SMS1–ΔSAM precipitated with HA-tagged GCS are represented as the ratio of the band intensities. The values represent the means ± S.D. from 10 independent experiments. *, p < 0.01. C, COS7 cells were co-transfected with indicated plasmids. At 24 h post-transfection, the cells were co-stained with anti-V5 and anti-HA antibodies, followed by appropriate Alexa Fluor–conjugated secondary antibodies and analyzed by confocal microscopy. Localization was confirmed by co-staining with antibody against the trans-Golgi network marker p230. Blue, SMS1 (WT or ΔSAM); green, GCS; red, p230. Scale bar, 50 μm. The results shown are from one experiment representative of three independent experiments. D, COS7 cells were co-transfected with indicated combination of plasmids. At 24 h post-transfection, the cells were subjected to immunostaining with antibodies, and Venus fluorescence complementation was quantified by flow cytometry. The values represent the means ± S.D. from five independent experiments. NS, not significant.
We investigated whether the decreased interaction between SMS1–ΔSAM and GCS was due to altered subcellular localization because of the SAM deletion by using confocal microscopy. SMS1–ΔSAM co-localized with GCS in the Golgi apparatus, showing a similar localization pattern as WT SMS1 (Fig. 4C, panels a and b).
To further examine the influence of SAM domain truncation on the distance between the C terminus of GCS and both termini of SMS1 in the complex, we employed a BiFC assay. The BiFC signal for the combination of VN-SMS1–ΔSAM and GCS-VC was comparable with that of VN-SMS1 and GCS-VC (Fig. 4D, column I versus column II). Similarly, the signal for the combination of SMS1–ΔSAM-VN and GCS-VC was not obviously different from that of SMS1-VN and GCS-VC (Fig. 4D, column III and column IV). Given that SAM truncation had no effect on the proximity between the C terminus of GCS and both termini of SMS1 in the heteromeric complex, SMS1–ΔSAM seems to form a complex with GCS; however, the complex was rather unstable because it dissociated in CHAPS solution in co-immunoprecipitation analysis. These results indicated that the N-terminal SAM domain of SMS1 is important for the interaction affinity and SMS1–GCS complex stability.
SM synthesis by SMS1–ΔSAM is significantly lower than that by WT SMS1 because of complex instability
We attempted to obtain insight into the functions of the SMS1–GCS complex by comparing Cer metabolism by WT SMS1 and SMS1–ΔSAM. To minimize the effect of endogenous SMS1 and/or GCS, we created SMS1–GCS double knockout (DKO) (Fig. 5, A–C), SMS1 KO (Fig. 5, A and D), and GCS KO (Fig. 5, B and E) cells in HEK293T cells via the CRISPR/Cas9 gene-editing system.
We first examined whether SMS and GCS activities in vitro were affected by the SMS1–GCS complex using SMS1–GCS DKO cells. Immunoblotting revealed that each protein was expressed at nearly the same level in SMS1–GCS DKO cells (Fig. 6A), when WT SMS1 and SMS1–ΔSAM were transfected with or without co-transfection of GCS. In in vitro enzyme assays using C6–NBD–Cer as a substrate, no significant difference in SMS activity was observed between sole expression of WT SMS1 and that of SMS1–ΔSAM (Fig. 6B, column I versus column II), consistent with previous reports (16, 25). SMS and GCS activities upon co-expression of SMS1–ΔSAM and GCS were also not significantly different from those upon co-expression of WT SMS1 and GCS (Fig. 6, B, column III versus column IV, and C, column I versus column II). These results suggested that the decreased affinity of SMS1 for GCS by SAM truncation has no effect on their in vitro activities.
Figure 6.

SMS1–ΔSAM, which does not form a stable heteromeric complex with GCS, significantly reduces SM synthesis. SMS1–GCS DKO cells were transfected with the following plasmids: empty vector (mock) or SMS1 (WT or SAM domain-truncation mutant) with or without GCS (WT or catalytically inactive GCS mutant). A and G, cells were lysed and analyzed by immunoblotting (IB) with anti-V5 antibody. Anti-GAPDH antibody was used as a loading control. The blots shown are from one experiment representative of three independent experiments. B and C, the assay for SMS (B) and GCS (C) activities in vitro. All cell lysates were incubated with C6–NBD–Cer in the absence (B) or presence of UDP–Glc (C). The values represent the means ± S.D. from four independent experiments. NS, not significant. D–F, H, and I, assay for SMS (D, E, H, and I) and GCS (D and F) activities in vivo by metabolic labeling. All cells were cultured in medium containing 0.5 μCi of [14C]stearic acid for 3 h at 37 °C. The lipids were extracted, saponified, and separated by TLC. The locations of Cer, stearic acid (SA), GlcCer, SM, and a lipid of unknown identify (shown as X) are indicated (D and H). The values of SM (E and I) and GlcCer (F) are expressed as percentages of radioactive SM or GlcCer to SMS1- or GCS/SMS1-expressing cells and represent the means ± S.D. from five or seven independent experiments. *, p < 0.01; NS, not significant.
To examine SMS and GCS activities more specifically, we carried out metabolic labeling with [14C]stearic acid (Fig. 6D), which allows de novo synthesis of SM and GCS to be measured in the physiological condition. De novo synthesis of SM was markedly increased by the expression of WT SMS1 or SMS1–ΔSAM as compared with mock transfectants. However, the activity of SMS1–ΔSAM was somewhat lower (∼16%) than that of WT SMS1 (Fig. 6E, column I versus column II), suggesting that the SAM domain contributes to the regulation of SMS1 activity within the cells. As expected, the co-expression of WT SMS1 and GCS significantly decreased SM synthesis as compared with the expression of WT SMS1 alone (Fig. 6D), because of competition of SMS1 and GCS for the common substrate Cer.
Next, we examined whether SMS and GCS activities in vivo were affected by the SMS1–GCS complex. Very interestingly, in the condition of GCS co-expression, the activity of SMS1–ΔSAM was markedly reduced to ∼37% of that of WT SMS1 (Fig. 6E, column III versus column IV), whereas de novo synthesis of GlcCer was not significantly different between WT SMS1 and SMS1–ΔSAM (Fig. 6F, column I versus column II). Similar results were obtained upon metabolic labeling with [14C]palmitic acid (Fig. S1, A and B). These results suggested that the reduction in stability of the SMS1–GCS complex by deletion of the SAM domain caused a significant reduction in SM synthesis in the condition of GCS co-expression.
To test whether the interaction of SMS1 with GCS was sufficient to promote SM synthesis, the catalytically inactive GCS mutant GCS-W276A was employed (21). Immunoblotting revealed that each protein was expressed at nearly the same level (Fig. 6G), when WT SMS1 was transfected with or without co-transfection of GCS-W276A. Co-expression of WT SMS1 and GCS-W276A resulted in slightly increased SM synthesis (∼116%) as compared with the expression of WT SMS1 alone (Fig. 6, H and I). These results suggested that the SMS1–GCS complex, but not GlcCer generated by GCS, is involved in the augmentation of SM synthesis.
SMS1 activity is increased upon rapamycin-induced heterodimer complex formation
Based on the above findings (Fig. 6), we hypothesized that SMS1 activity is augmented by complex formation with GCS. Because not all SMS1 and GCS form the heteromeric complex, we employed the rapamycin-induced heterodimerization system (26, 27) to increase the level and stability of the heteromeric complex. A chimeric protein, in which FK506-binding protein (FKBP)–fused SMS1 was connected with FKBP–rapamycin–binding domain (FRB)-fused GCS through a picorna virus-derived 2A autocleavage site (P2A), was constructed (Fig. 7A) (28). In this system, the membrane-permeable chemical compound rapamycin can selectively and rapidly heterodimerize the FKBP-fused and FRB-fused proteins via noncovalent interactions. In an immunoprecipitation assay, GCS–FRB–HA precipitation was greatly enhanced by treatment with rapamycin; ∼13 and ∼54% of total input of GCS–FRB–HA was co-immunoprecipitated with FKBP–SMS1–FLAG in rapamycin-untreated and -treated cells, respectively (Fig. 7B, lower panel). This result suggested that the rapamycin treatment increased the level and stability of intracellular FKBP–SMS1–FLAG/GCS–FRB–HA heterodimer. Confocal microscopy showed that FKBP–SMS1–FLAG and GCS–FRB–HA co-localized in the perinuclear region, and no difference was observed in intracellular localization upon rapamycin treatment (Fig. 7C, panels a and b).
Figure 7.

SM synthesis is increased by rapamycin-induced heterodimerization with FKBP-fused SMS1 and FRB-fused GCS. A, schematic representation of the chimeric proteins engineered for rapamycin-induced heterodimerization. FKBP-fused SMS1 was linked with the FRB-fused GCS through a picorna virus-derived 2A autocleavage site (P2A). The locations of FLAG and HA tags are indicated. B and C, SMS1–GCS DKO cells were transfected with FKBP–SMS1–FLAG–P2A–GCS–FRB–HA. At 24 h post-transfection, the cells were treated with 500 nm rapamycin for 3 h. B, proteins from cell lysates were immunoprecipitated (IP) using an anti-FLAG affinity gel. Cell lysates and precipitated proteins were analyzed by immunoblotting (IB) with anti-FLAG or anti-HA antibody. The blots are from one experiment representative of three independent experiments. C, immunocytochemistry analysis; all cells were co-stained with anti-FLAG and anti-HA antibodies, followed by appropriate Alexa Fluor–conjugated secondary antibodies, and analyzed by confocal microscopy. Green, FKBP–SMS1–FLAG; red, GCS–FRB–HA. Scale bar, 50 μm. The images shown are from one experiment representative of three independent experiments. D-H, SMS1–GCS DKO cells were transfected with indicated plasmids. At 24 h post-transfection, each cells were treated with or without 500 nm rapamycin for 3 h. D and E, assay for SMS (D) and GCS (E) activities in vitro; all cell lysates were incubated with C6–NBD–Cer in the absence (D) or presence of UDP–Glc (E). The values are expressed as percentages of enzyme activity to rapamycin-untreated control cells and represent the means ± S.D. from four independent experiments. F–H, assay for SMS (F and G) and GCS (F and H) activities in vivo by metabolic labeling; all cells were cultured in the culture medium containing 0.5 μCi of [14C]stearic acid (SA) and 500 nm rapamycin for 3 h. Lipids were extracted, saponified, and separated by TLC (F). The values of SM (G) and GlcCer (H) are expressed as percentages of radioactive SM or GlcCer to rapamycin-untreated control cells and represent the means ± S.D. from five independent experiments. *, p < 0.01; NS, not significant.
The rapamycin treatment did not affect in vitro SMS activity (Fig. 7D, column I versus column II) and GCS activity (Fig. 7E, column I versus column II), when FKBP–SMS1–FLAG or GCS–FRB–HA was expressed solely. Even when the two enzymes were expressed simultaneously, the rapamycin treatment had no effect on their activities (Fig. 7, D, column III versus column IV, and E, column III versus column IV).
We next examined SMS and GCS activities in vivo by metabolic labeling with [14C]stearic acid (Fig. 7F). SM synthesis was augmented by the rapamycin-induced heteromeric complex, showing a ∼30% increase as compared with cells in the absence of rapamycin (Fig. 7G, column III versus column IV), whereas GlcCer synthesis was not notably changed (Fig. 7H, column III versus column IV). These results suggested that the increased level and stability of SMS1–GCS heterodimerization indeed augmented SMS1 activity in vivo. Interestingly, SM synthesis in FKBP–SMS1–FLAG–expressing cells was decreased by ∼16% upon rapamycin treatment (Fig. 7, F and G, column I versus column II), whereas GlcCer synthesis in GCS–FRB–HA–expressing cells was not significantly affected (Fig. 7, F and H, column I versus column II). Given that rapamycin is an inhibitor of mammalian target of rapamycin (mTOR) (29), SM synthesis of SMS1 might be positively regulated through mTOR signaling. Thus, SMS1 activation by SMS1–GCS heterodimer formation overcomes the inhibitory effect of rapamycin, although rapamycin can also inhibit SMS1 activity through the inhibition of mTOR signaling.
SMS1 activity is increased, but GCS activity is decreased in engineered direct tandem fusion proteins of SMS1 and GCS
To further increase the rate of heterodimer formation of SMS1 and GCS, we engineered direct tandem fusion proteins of SMS1 and GCS, in which the C terminus of GCS was fused to the N terminus of SMS1 directly or through glycine-rich linkers of different lengths (Fig. 8A). When the combination of SMS1 and GCS, and the GCS–SMS1 fusion proteins were expressed in SMS1–GCS DKO cells, each protein was expressed at nearly the same level (Fig. 8B). Confocal microscopy showed that the all GCS–SMS1 fusion proteins co-localized with trans-Golgi network marker p230 (Fig. 8C, panels a–c), suggesting that the subcellular location does not change by the direct fusion. Given that GCS–SMS1 fusion proteins localized in their final destinations, it was unlikely that GCS–SMS1 fusion proteins were misfolded.
Figure 8.

Augmented heteromeric formation by direct fusion of SMS1 and GCS increases SM synthesis but decreases GlcCer synthesis. A, schematic representation of the fusion proteins. Constructs for expression of GCS–SMS1, GCS–(GGGGS)3–SMS1, and GCS–(GGGGS)6–SMS1 fusion proteins were engineered to connect the C terminus of GCS to the N terminus of SMS1 directly or with glycine-rich flexible linkers of different lengths. The locations of V5 tags are indicated. B-H, SMS1–GCS DKO cells were transfected with the indicated plasmids. The amount of plasmid was adjusted so that each protein was expressed at nearly the same level. B, all cells were lysed and analyzed by immunoblotting (IB) with anti-V5 antibody. Anti-GAPDH was used as a loading control. The blots are from one experiment representative of three independent experiments. C, immunocytochemistry analysis. All cells were stained with anti-V5 antibody, followed by appropriate Alexa Fluor–conjugated secondary antibodies, and analyzed by confocal microscopy. Localization was confirmed by co-staining with antibody against the trans-Golgi network marker p230. Green, GCS–SMS1, GCS–(GGGGS)3–SMS1 and GCS–(GGGGS)6–SMS1; red, p230. Scale bar, 50 μm. The images are from one experiment representative of three independent experiments. D and E, assay for SMS (D) and GCS (E) activities in vitro. Cell lysates were incubated with C6–NBD–Cer in the absence (D) or presence of UDP–Glc (E). The values represent the means ± S.D. from six independent experiments. F–H, assay for SMS (F and G) and GCS (F and H) activities in vivo by metabolic labeling. The cells were cultured in medium containing 0.5 μCi of [14C]stearic acid (SA) for 3 h. The lipids were extracted, saponified, and separated by TLC (F). The amount of SM (G) and GlcCer (H) were calculated from the standard curve of [14C]stearic acid. The values represent the means ± S.D. from five independent experiments. *, p < 0.01; **, p < 0.05; NS, not significant.
In SMS and GCS activities in vitro, SMS activity tended to increase for all GCS–SMS1 fusion proteins as compared with the combination of SMS1 and GCS; however, the increases were not statistically significant (Fig. 8D, column I versus columns II–IV). When SM synthesis in vivo was determined by metabolic labeling with [14C]stearic acid (Fig. 8F), SM synthesis of all GCS–SMS1 fusion proteins was markedly higher than that of simultaneously expressed SMS1 and GCS (Fig. 8G, column I versus columns II–IV). These results clearly indicated that heteromeric complex formation between SMS1 and GCS augmented SMS1 activity, because higher rates of complex formation resulted in higher SMS activities.
Interestingly, even in vitro, GCS activities of GCS–SMS1 and GCS–(GGGGS)3–SMS1 were decreased by ∼20% as compared with those of simultaneously expressed SMS1 and GCS (Fig. 8E, column I versus column II and III). These findings were confirmed by metabolic labeling; GlcCer synthesis was markedly lower for all GCS–SMS1 fusion proteins than for simultaneously expressed SMS1 and GCS (Fig. 8, H, column I versus column II–IV). It should be noted that GCS activity in vivo was affected by the distance between SMS1 and GCS in fusion proteins. GlcCer synthesis by GCS–SMS1 was lower than that by GCS–(GGGGS)6–SMS1, suggesting that the inhibition of GCS activity depended on the distance between GCS and SMS1 in the heterodimer. However, the stimulatory effect of the heteromeric complex formation on SMS1 activity in vivo was independent on the distance (Fig. 8G, columns II–IV). Thus, SM synthesis was up-regulated in a manner independent of the distance between both enzymes in the heterodimer, whereas GlcCer synthesis was down-regulated in a manner that depended on the proximity of both enzymes.
Complex formation of SMS1–GCS alters sensitivity to the CERT inhibitor HPA-12
Next, we examined whether Cer metabolism is regulated by the SMS1–GCS heteromeric complex under physiological conditions. We focused on CERT-transported Cer in the trans-Golgi, because SMS1 and GCS co-localize mainly in the medial/trans-Golgi (13). Therefore, we examined the effects of a CERT inhibitor (30), HPA-12, on the metabolism of CERT-transported Cer in SMS1 KO and GCS KO cells (Fig. 5, D and E). The inhibition of SM synthesis by HPA-12 in HEK293T cells was dose-dependent and reached a plateau at 2–4 μm, without cytotoxicity (Fig. 9, A and B). The results suggested that CERT-dependent and -independent SM synthesis contributed ∼70 and 30%, respectively. Similar inhibition of SM synthesis was observed in GCS KO cells, but the effective dose of HPA-12 was lower than that in the parent HEK293T cells (EC50: 1.1 μm in GCS KO#1 cells and 1.0 μm in GCS KO#2 cells versus 1.5 μm in HEK293T cells), although the contribution ratio of CERT-dependent or -independent mechanisms did not change by GCS KO. Cer synthesis was not dramatically altered by HPA-12 in HEK293T and GCS KO cells (Fig. 9C). The differences in the effective dose were most likely0 due to the lack of up-regulation of SMS1 by GCS KO.
Figure 9.
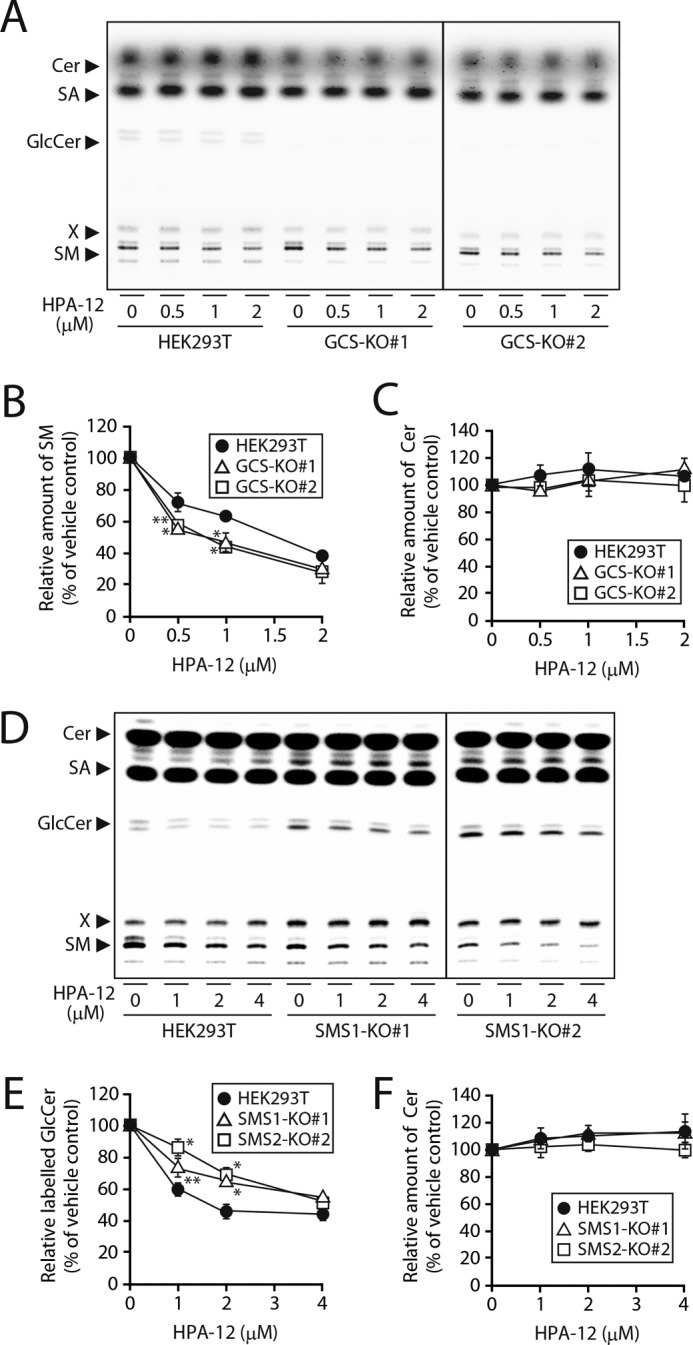
Complex formation of SMS1–GCS alters sensitivity to CERT inhibitor HPA-12. HEK293T, GCS KO, and SMS1 KO cells were pretreated with HPA-12 at the indicated concentrations in the growth medium for 1 h and further cultivated with 0.5 μCi of [14C]stearic acid (SA) for 3 h. Lipids were extracted, saponified, and separated by TLC (A and D). The values of SM (B), Cer (C and F), and GlcCer (E) are expressed as percentages of radioactive SM, Cer, or GlcCer to vehicle-treated control cells and represent the means ± S.D. from three independent experiments. *, p < 0.01; **, p < 0.05.
Interestingly, GlcCer synthesis was also inhibited by HPA-12 in a dose-dependent manner in HEK293T cells (Fig. 9, D and E), although no change in Cer synthesis was observed (Fig. 9, D and F). Similar inhibition of GCS synthesis was observed in SMS1 KO cells, but the effective dose of HPA-12 was higher than that in HEK293T cells (EC50: 4.2 μm in SMS1 KO#1 cells and 4.0 μm in SMS1 KO#2 cells versus 2.7 μm in HEK293T cells) (Fig. 9E). Similar results were obtained upon metabolic labeling with [14C]palmitic acid (Fig. S1, C–E). Again, the differences in the effective dose of HPA-12 may be due to the lack of down-regulation of GCS by SMS1 KO.
Contribution to GlcCer synthesis by GCS in cis-Golgi and to that by SMS1–GCS complex in trans-Golgi
HPA-12 does not affect the activity of serine palmitoyl transferase, sphingosine-N-acyltransferase, or GCS (30); however, when transport of Cer to trans-Golgi is inhibited, de novo synthesis of sphingolipid base seems to be suppressed (12, 30, 31). Thus, the decreases in [14C]stearic acid-derived GlcCer by HPA-12 treatment in HEK293T and SMS1 KO cells might be due to a secondary effect of CERT inhibition (Fig. 9E), although [14C]stearic acid-derived Cer was not affected by HPA-12 (Fig. 9F). To confirm their contribution to the SMS1–GCS activities, another substrate, [3H]sphingosine, was employed for metabolic labeling, which bypasses de novo synthesis of sphingolipid bases in the labeling of Cer (12, 30, 31). Inhibition of CERT by 4 μm HPA-12 decreased SM synthesis in HEK293T cells, in parallel with increased Cer, as well as GlcCer levels (Fig. 10, B–D), as expected on the basis of previous reports (12, 30). This can be explained by the fact that Cer accumulates in the ER through inhibition of CERT-mediated transport of Cer, which results in an overflow Cer to cis-Golgi—where GCS is located—by a CERT-independent process, leading to an increase in GlcCer (Fig. S2A). Similar results were obtained in SMS1 KO cells; the inhibition of CERT activity decreased SM synthesis and increased Cer levels (Fig. 10, B and C), indicating that SMS2 also uses CERT-transported Cer. However, in contrast to HEK293T cells, GlcCer synthesis in HPA-12–treated SMS1 KO cells was almost comparable with that in nontreated control cells (Fig. 10D). These results suggested that, in SMS1 KO cells, GlcCer synthesis in trans-Golgi might become CERT-dependent because the down-regulation of GlcCer synthesis by SMS1 is canceled in these cells. Therefore, HPA-12 treatment in SMS1 KO cells decreased CERT-dependent GlcCer synthesis in trans-Golgi, whereas there was an increase in GlcCer synthesis from the overflow of Cer in cis-Golgi, and as a result, overall GlcCer levels were not substantially affected (Fig. S2B). These results suggested that CERT-transported Cer in trans-Golgi is partly regulated by the SMS1–GCS heteromeric complex in HEK293T cells.
Figure 10.
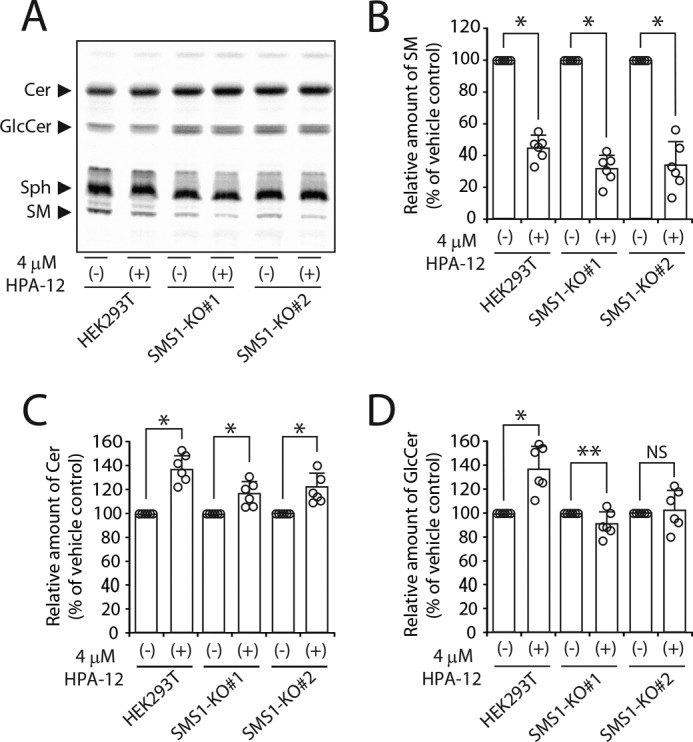
Contribution to GlcCer synthesis by GCS in cis-Golgi and to that by SMS1–GCS complex in trans-Golgi. A–D, HEK293T and SMS1 KO cells were pretreated with 4 μm HPA-12 in the growth medium for 1 h and further cultivated with 0.25 μCi of [3H]sphingosine for 3 h. A, lipids were extracted and separated by TLC. The locations of Cer, GlcCer, sphingosine (Sph), and SM are indicated. B–D, values of SM (B), Cer (C), and GlcCer (D) are expressed as percentages of radioactive SM, Cer, or GlcCer to vehicle-treated control cells and represent the means ± S.D. from six independent experiments. *, p < 0.01; **, p < 0.05; NS, not significant.
Discussion
SMS1 and GCS are known to partially co-localize in cisternae, especially in the medial/trans-Golgi. However, the mechanism by which CERT-transported Cer in the trans-Golgi is selectively used for SM, but not GlcCer synthesis, remains unclear. This study revealed that complex formation between SMS1 and GCS is part of a novel mechanism controlling the metabolic fate of Cer in the Golgi, which may be one of the reasons why CERT-transported Cer is specifically used for SM synthesis.
In the present study, we found that SMS1 directly interacts with GCS. The N terminus of SMS1 contains a predicted SAM domain (4). SAM domains are present in a wide variety of proteins involved in many biological processes, including cell signaling, calcium homeostasis, transcriptional repression, and ER-to-Golgi transport (23, 24, 32). Because SAM domains recognize a large variety of binding partners, such as protein, RNA, and lipids (33–35), they cannot easily be classified. Indeed, even close homologs can have different functions (16, 25, 36, 37). In mammals, SMS1 has two homologs: SMS2 and sphingomyelin synthase-related protein (SMSr); SMSr is an ER-resident protein and produces Cer phosphoethanolamine, but not SM (36). SMS1 and SMSr, not SMS2, contain a SAM domain; however, phylogenetic analysis has revealed that the SAM domain of SMS1 is not closely related to that of SMSr (37). The SAM domain of SMSr reportedly is required for homomeric oligomerization and efficient ER retention (36, 37). In contrast, the function of the SAM domain in SMS1 had not been established, although Yeang et al. (16, 25) and we (16) indicated that it is not involved in subcellular localization of SMS1, SMS activity in vitro, or homomeric oligomerization.
We postulate that the N-terminal SAM domain of SMS1 is important for the interaction affinity to form a stable SMS1–GCS heteromeric complex. The SAM domains can bind to other related SAM domains or to non-SAM domains (23, 24). It is likely that the SAM domain of SMS1 interacts with the unrelated structure of GCS, because no SAM domain is found in the amino acid sequence of GCS. Although the precise binding region on GCS has not been elucidated, the proximal C-terminal region of GCS is likely involved, because the N terminus of SMS1 and the C terminus of GCS are in close proximity in the heteromeric complex (Fig. 2C). Furthermore, SAM truncation did not completely abolish the interaction with GCS (Fig. 4, A and B), suggesting that other region(s) may be involved in complex formation. Lateral interactions between transmembrane helices are known to be important for oligomer formation of membrane proteins (38). Further research is needed to elucidate the intermolecular helix–helix interactions in the transmembrane domains of SMS1 and GCS, which will aid in establishing the functions of the SMS1–GCS heteromeric complex.
We considered the functions of the heteromeric complex and evaluated whether or not complex formation affects the activities of SMS1 and GCS. Our carefully designed in vivo experiments based on metabolic labeling of [14C]stearic acid demonstrated that SMS1 activity was augmented, whereas GSC activity was decreased by heteromeric complex formation. Based on these results, we propose a model in which SM synthesis is up-regulated, whereas GlcCer synthesis is down-regulated by SMS1–GCS heterodimer formation (Fig. 11).
Figure 11.
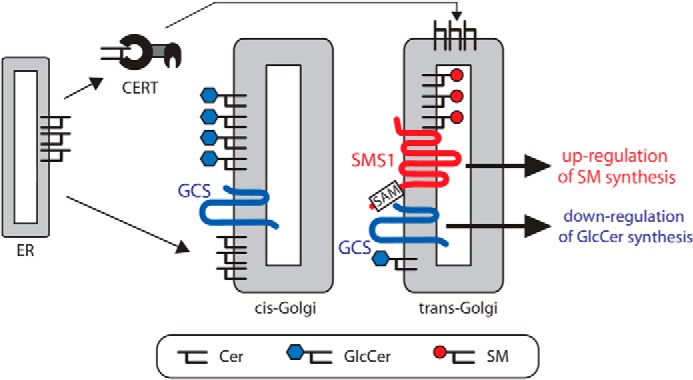
Heteromeric complex between SMS1 and GCS regulates Cer metabolism. A model for the role of the SMS1–GCS heteromeric complex in cellular Cer metabolisms is shown. The de novo synthesized Cer is transported from the ER to the trans-Golgi in a CERT-dependent manner or to the cis-Golgi in a CERT-independent manner. The Cer transported to cis-Golgi is mainly used for GlcCer synthesis, because SMS1 is localized mainly in the medial/trans-Golgi, whereas GCS has a broader distribution than SMS1 in the Golgi. We found that SMS1 forms a heteromeric complex with GCS through its N terminus, and the complex is involved in the regulation of the cellular Cer. The SAM domain in the N terminus of SMS1 is associated with the stability of the heteromeric complex. Based on these results, we propose a model in which SM synthesis is up-regulated by stable SMS1–GCS heterodimer formation, whereas GlcCer synthesis is down-regulated under conditions in which both proteins are in close proximity. Given that SMS1 and GCS partially co-localize in the cisternae, especially in medial/trans-Golgi, CERT-transported Cer in trans-Golgi might be partially regulated by the SMS1–GCS heteromeric complex.
We also considered the effects of the distance between SMS1 and GCS in the SMS1–GCS heterodimer on enzyme activities. The stimulatory effect on SMS1 activity was independent of spacer length (Fig. 8G). In contrast, the inhibitory effect on GSC activity depended on the proximity between SMS1 and GCS; the shorter the spacer in the direct fusion protein, the stronger the reduction in GCS activity (Fig. 8H). These results were consistent with the fact that GCS activity in vivo remained unchanged upon co-expression of SMS1–ΔSAM (Fig. 6F) and FKBP–FRB–based and rapamycin-induced heterodimer (Fig. 7H). GlcCer synthesis was not significantly different between WT SMS1 and SMS1–ΔSAM with GCS co-expression (Fig. 6F) because the SAM truncation hardly affected the proximity between both enzymes in the heteromeric complex (Fig. 4D). Furthermore, GlcCer synthesis did not show a notable change in the rapamycin-induced heteromeric complex (Fig. 7H), suggesting that the insertion of the relatively large molecular mass of FKBP (∼12 kDa) and FRB (∼11 kDa) extended the distance between SMS1 and GCS in the complex.
We found it difficult to examine the effects of complex formation by in vitro enzyme assay using C6–NBD–Cer (Figs. 6–8). This might be simply because we used a Cer analog bearing a short-chain fatty acid as a substrate, which dramatically differs from natural Cer in its physiological properties and its behavior in membranes (39, 40). Alternatively, the excess supply of C6–NBD–Cer as a substrate might have interfered with examining the influence of the heteromeric complex. In support of the latter possibility, in an in vivo assay based on metabolic labeling, SMS1 and GCS competed for de novo synthesized Cer, because SMS activity of SMS1 was decreased by the co-expression of GCS (Figs. 6D and 7F). GCS and SMS activities can be evaluated more specifically and with physiologic relevance in the metabolic labeling assay, which is limited in terms of Cer supply, than in vitro assays.
De novo synthesized Cer is transported from the ER to the trans-Golgi in a CERT-dependent manner (Fig. 11) or to the cis-Golgi via vesicle-dependent (11, 41) and/or CERT-independent nonvesicular transport (31). CERT-transported Cer is selectively destined for SM but not GlcCer synthesis, as suggested by experiments using KO/knockdown or pharmacological inhibition of CERT (12, 30, 42), despite the fact that SMS1 and GCS co-localize in trans-Golgi (13). However, the mechanism for the selective usage of Cer by SMS1 and GCS had not been yet elucidated. Several mechanisms regulating the alternate usage of Cer by SMS1 and GCS have been hypothesized. The presence of Cer on different sides of the Golgi membrane bilayer likely determines the usage of Cer by the two enzymes, because the active site of SMS1 is located on the luminal side of the membrane, whereas the active site of GCS is on the cytosolic side (10, 11). CERT seems to transport Cer on the cytosolic side of the Golgi, because ceramide kinase, a cytosolic enzyme, uses Cer provided by CERT (43). The CERT-transported Cer then translocates to the Golgi lumen via transbilayer movement for SM synthesis (11, 41); however, the mechanism of this transbilayer movement is not fully understood. As the asymmetric increase in Cer at one side of the lipid bilayer promotes transbilayer movement (44–46), CERT-transported Cer may translocate to the lumen spontaneously, without the need for membrane proteins. Alternatively, because ABCA12 was identified as the translocase for esterified omega-hydroxy-ultra-long-chain Cer and/or its metabolites (47), the related translocase(s) for Cer may be important for the delivery of Cer to SMS1. Very recently, Ishibashi et al. (48) found another possible mechanism, because they observed that phosphatidylinositol-4-phosphate (PI4P) inhibited GCS activity, but not SMS activity, by competing with UDP–Glc. PI4P-dependent inhibition of GCS activity may facilitate the delivery of Cer to SMS1, because CERT specifically delivers Cer to trans-Golgi, where PI4P is present.
Here, we propose another possible mechanism; the heteromeric complex formation of SMS1 and GCS leads to up-regulation of SMS1 activity and down-regulation of GCS activity. The exact mechanism by which the complex formation influences the metabolic fate of Cer is unclear from this study. Based on the results that SMS1 directly interacts with GCS, we speculate that the physical association of SMS1 with GCS induces the conformational change at the active site of SMS1. It may be difficult to determine the direct evidence for the conformational change in SMS1–GCS complex by examining the values of Km and Vmax, because the SMS1–GCS complex had negligible effect on their in vitro activities. We might be able to prove this hypothesis using the model for SMS1–GCS complex, although the structures of SMS1 and GCS have not been determined yet. On the other hand, because SMS1 seems to reside in a PI4P-rich environment in trans-Golgi, trapping of GCS at this location may reduce GlcCer synthesis.
We examined the SMS1–GCS heteromeric complex under overexpression conditions. Because overproduced Golgi proteins sometimes overflow to incorrect compartments or incorrectly associate with the proteins, this study may not be able to determine the behavior of the endogenous SMS1. To confirm the subcellular localization or immunoprecipitation of endogenous SMS1, immunocytochemical analysis and Western blotting were performed with SMS1 antibody (Fig. S3). However, unfortunately, the appropriate antibody to detect the endogenous SMS1 was unavailable, at least in HEK293T cells. The lack of information on endogenous SMS1 is one of the limitations of the present study. However, another approach employing KO cells of SMS1 or GCS combined with pharmacological inhibition of CERT revealed the evidence for association and regulation of SMS1 and GCS activities (Figs. 9 and 10). The sensitivity of CERT inhibitor was increased by GCS KO, whereas the sensitivity of the inhibitor was decreased by SMS1 KO. These results for the endogenous SMS1 and GCS were consistent with those found through the overexpression of SMS1–GCS. We believe that the physical association and their regulation of SMS1–GCS occur in endogenous or physiological conditions, at least in HEK293T cells.
In view of the relationship of homo- and heterodimerization of SMS1, we previously demonstrated that the C-terminal tail of SMS1 is involved in the stability of the homodimeric complex (16). However, the C-terminal tail of SMS1 is in close proximity to the N- and C-terminal tails of another SMS1 in the homodimer (16) (Fig. 2, B and C). Thus, one of the reasons why not all SMS1 forms a heteromeric complex with GCS may be the competition between GCS and SMS1 for the N-terminal tail of another SMS1 as a dimer partner. Further study of the alternative function of the N terminus of SMS1 and the switching of the binding to either GCS or another SMS1 is needed to uncover the regulation of homo- and heterodimer formation.
In summary, this study showed for the first time that SMS1 and GCS form a heteromeric complex. The N-terminal SAM domain of SMS1 is important for stable heteromeric interaction with GCS. SM synthesis is up-regulated by stable SMS1–GCS heterodimer formation, whereas GlcCer synthesis is down-regulated when the two proteins are in close proximity. Thus, this study suggests that complex formation between SMS1 and GCS is part of a novel mechanism controlling the metabolic fate of Cer in the Golgi.
Experimental procedures
Antibodies
Mouse IgG1 monoclonal anti-FLAG antibody (catalog no. F1804, lot no. 124K6106), rabbit polyclonal anti-V5 antibody (catalog no. V8137, lot no. 112M4850V), anti-FLAG affinity gel (catalog no. A2220, lot no. SLBG5784V), and anti-HA affinity gel (catalog no. A2095, lot no. 026M4810V) were obtained from Sigma. Mouse anti-p230 antibody (catalog no. 611280, lot no. 7208954) was from BD Biosciences. The rat anti-HA antibody (catalog no. 1867423, lot no. 15645900) was purchased from Roche, and the goat horseradish peroxidase (HRP)–conjugated anti-rat IgG antibody (catalog no. sc-2032, lot no. A1816) was purchased from Santa Cruz Biotechnology. Mouse IgG2a monoclonal anti-V5 antibody (catalog no. 46-0705, lot no. 1718556), goat anti-mouse IgG-HRP antibody (catalog no. 62-6520, lot no. QG215721), anti-rabbit IgG-Alexa Fluor 405 antibody (catalog no. A31556, lot no. 1145173), anti-rat IgG-Alexa Fluor 488 antibody (catalog no. A11006, lot no. 1301839), and anti-mouse IgG-Alexa Fluor 546 antibody (catalog no. A11030, lot no. 1829584) were obtained from Invitrogen. Rat PerCP/Cy5.5-conjugated anti-mouse IgG2a antibody (catalog no. 407111, lot no. B207066) was from BioLegend. Rabbit anti-GOSR1 antibody (catalog no. ab53288, lot no. GR3187187-2), rabbit anti-GCS antibody (catalog no. ab124296), and mouse APC-conjugated anti-FLAG antibody (catalog no. ab72569, lot no. GR193571–8) were obtained from Abcam.
Plasmids
We use the nomenclature for epitope-tagged proteins as follows: TagA–(protein)–TagB means that TagA and TagB are located in the N and C termini of the protein, respectively. The expression vectors for SMS1 (pcDNA4TO/SMS1–FLAG, pcDNA4TO/SMS1–V5, pQCXIP/SMS1–V5) were constructed by PCR using specific primers containing sequences corresponding to each epitope in front of the stop codon. PCR products were subcloned into pcDNA4TO (Invitrogen) or pQCXIP (Takara, Japan). The expression vector for the truncation mutant of N-terminal SAM in SMS1 (pcDNA4TO/SMS1–ΔSAM–V5 and pQCXIP/SMS1–ΔSAM–V5) was constructed by PCR with primers designed to truncate the indicated region. The expression vectors for GCS (pcDNA4TO/GCS-FLAG, pcDNA4TO/GCS-HA, pcDNA4TO/FLAG–GCS–Myc, pcDNA4TO/nontagged GCS, and pQCXIH/GCS-V5) were constructed using the same methods as described above. The mutated ORF of GCS (W276A) that lacked GCS activity was prepared by QuikChange site-directed mutagenesis (Stratagene). The chimeric expression vectors for use in the BiFC assay, VN (N-terminal residues 1–173 of Venus)–SMS1–V5, VC (C-terminal residues 155–238 of Venus)–SMS1–FLAG, SMS1–V5–VN, SMS1–FLAG–VC, VC–GCS–FLAG, and FLAG–GCS–VC, were constructed by PCR with overlapping primers as described previously (49). The chimeric expression vectors (pQCXIP/FKBP–SMS1—FLAG–P2A–GCS–(GGGGS)3—FRB–HA, pQCXIP/FKBP–SMS1–FLAG, pQCXIP/GCS–(GGGGS)3–FRB–HA, pQCXIP/GCS–SMS1–V5, pQCXIP/GCS–(GGGGS)3–SMS1–V5, and pQCXIP/GCS–(GGGGS)6–SMS1–V5) were constructed using the same methods as described above. Because the C-terminal tail of GCS is short, the spacer (GGGGS)3 was inserted between GCS and FRB to facilitate the proper folding of FRB. pJ-INPP5E was a gift from Robin Irvine (Addgene plasmid no. 38001) (50). pEGFP-FRB was gift from Klaus Hahn (Addgene plasmid no. 25919) (51). pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid no. 48138) (52).
Generation of CRISPR/Cas9–based SMS1–GCS DKO, SMS1 KO, and GCS KO cells
To establish SMS1–GCS DKO, SMS1 KO, and GCS KO cells, we designed guide RNAs (pSpCas9-SMS1gRNA#1-For, CACCGATGCTGAAGCTGCCGTCGG; pSpCas9-SMS1gRNA#1-Rev, AAACCCGACGGCAGCTTCAGCATC; pSpCas9-SMS1g RNA#2-For, CACCGTACTGAGAGCGCTCCAGTTC; pSpCas9-SMS1gRNA#2-Rev, AAACGAACTGGAGCGCTCTCAGTAC; pSpCas9-GCSgRNA#1-For, CACCGTGGAGGGAATGGCCGTCTTC; pSpCas9-GCSgRNA#1-Rev, AAACGAAGACGGCCATTCCCTCCAC; pSpCas9-GCSgRNA#2-For, CACCGAAGAGGACGAACCCGAAGA; and pSpCas9-GCSgRNA#2-Rev, AAACTCTTCGGGTTCGTCCTCTTC) using the target design software developed by Dr. Feng Zhang's lab at the Massachusetts Institute of Technology (http://crispr.mit.edu).3 The corresponding sequences to these sgRNAs were cloned into the pSpCas9(BB)–2A–GFP (PX458) vector. The constructs were transfected into HEK293T cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The transfected cells were cultured for 72 h and selected by the GFP marker using FACS Aria II (BD Bioscience). Using limiting dilution, we isolated clonal populations of SMS1–GCS DKO, SMS1 KO, and GCS KO cells. Disruption of SMS1–GCS, SMS1, and GCS was confirmed by DNA sequencing and enzyme activities in vitro.
Cell culture and cDNA transfection
COS7, HEK293T, SMS1–GCS DKO, SMS1 KO, and GCS KO cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, 100 μg/ml streptomycin, and 100 units/ml penicillin at 37 °C in a humidified incubator containing 5% CO2. HEK293T cells were authenticated by the National Institute of Biomedical Innovation (Osaka, Japan) using a short tandem repeat method. For cDNA transfection, 2 × 105 cells were plated in 6-well plates for all experiments, except for the confocal microscopy analysis, in which 4 × 104 cells were seeded in 35-mm-diameter glass-bottomed dishes (MatTek). After 24 h, the cells were transfected using Lipofectamine 2000 with 1 μg of plasmid for 6-well plates or 100 ng of plasmid for 35-mm-diameter glass-bottomed dishes, unless stated otherwise. After transfection, the cells were cultured for an additional 24 h and used for the experiments described below.
Co-immunoprecipitation analysis
COS7 cells transfected with plasmids were washed three times in PBS and lysed in buffer containing 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, complete protease inhibitors (Roche), and 1% CHAPS. The insoluble nuclear materials were removed by centrifugation at 900 × g for 10 min. Co-immunoprecipitation was performed by incubating cell extracts with an anti-FLAG or anti-HA affinity gel at 4 °C for 4 h. The gels were then washed four times with lysis buffer and eluted in SDS sample buffer by incubation at 65 °C for 4 min. Co-immunoprecipitated proteins were subjected to SDS–PAGE with WIDE-VIEW prestained protein size marker III (Fujifilm Wako Pure Chemical Corporation). After the proteins were transferred to nitrocellulose membranes, the membranes were incubated with the appropriate primary antibody and subsequently with the appropriate HRP-conjugated secondary antibody. Signals were detected with Western blot Quant HRP substrate (Takara, Japan) and were analyzed and quantified using the Amersham Biosciences Imager 600 with ImageQuant TL software (GE Healthcare).
Immunocytochemistry and fluorescence microscopy
COS7 cells transfected with plasmids in 35 mm-diameter glass-bottomed dishes (MatTek) were fixed with 3% paraformaldehyde in PBS for 10 min. After being rinsed with 50 mm NH4Cl in PBS, the cells were permeabilized with 0.1% Triton X-100 in PBS at 25 °C for 10 min or 0.002% digitonin in PBS containing 10 mm Hepes (pH 7.5), 300 mm sucrose, 100 mm KCl, 2.5 mm MgCl2, and 0.5% BSA at 4 °C for 15 min. After treatment with ImmunoBlock (DS Pharma, Japan) for 30 min, the samples were incubated with specific antibodies at 4 °C overnight followed by incubation with Alexa Fluor–conjugated secondary antibodies at 25 °C for 2 h. For microscopy, an Olympus FV10i confocal microscope equipped with a 60× water objective was used, with Olympus Fluoview version 3.1 viewer.
BiFC assay by flow cytometry
The details of this procedure, a modification of the original procedure described by Ozalp et al. (19), have been described previously (16).
Assays of SMS and GCS activities in vitro
SMS and GCS activities were assayed according to the method of Ichikawa et al. (9), with slight modifications. SMS1–GCS DKO cells transfected with plasmids were washed three times in PBS and lysed by sonication in buffer containing 50 mm Tris-HCl (pH 7.5), 1 mm EDTA, and complete protease inhibitors (Roche). C6–NBD–Cer (400 pmol) and lecithin (6.5 nmol) were mixed in 100 μl of ethanol, and the solvent was evaporated. Aqueous solution (20 μl) was added, and the mixture was sonicated to form liposomes. For the assay of SMS activity, a reaction mixture (40 μl) composed of 50 mm Tris-HCl (pH 7.5), 1 mm EDTA, 10 μm C6–NBD–Cer, and 20 μg of cell protein was incubated at 37 °C for 30 min. For the assay of GCS activity, a reaction mixture (40 μl) composed of 100 μm UDP–Glc, 50 mm Tris-HCl (pH 7.5), 1 mm EDTA, 10 μm C6–NBD–Cer, and 5 μg of cell protein was incubated at 37 °C for 10 min. The reaction was stopped by adding 200 μl of chloroform/methanol (2/1, v/v) and mixing well. Lipids were extracted by the Bligh and Dyer method (53), dried, dissolved in 10 μl of chloroform/methanol (2/1, v/v), and then applied to a TLC plate that was developed with chloroform/methanol/H2O (65:25:4, v/v/v). Each band of C6–NBD–Cer, C6-NBD-SM, and C6-NBD-GlcCer was quantified using a Typhoon FLA9000 fluorescence imaging analyzer (GE Healthcare) with ImageQuant TL software.
Metabolic labeling and lipid extraction
SMS1–GCS DKO, SMS1 KO, and GCS KO cells were incubated with 0.5 μCi of [14C]stearic acid or 0.25 μCi of [3H]sphingosine (American Radiolabeled Chemicals) for 3 h at 37 °C in DMEM supplemented with 10% FBS in a humidified incubator containing 5% CO2. The cells were washed three times with 0.25% BSA in PBS. The lipids were extracted by the Bligh and Dyer method, and the solutions were evaporated to dryness. The lipids labeled with [14C]stearic acid were dissolved in methanolic 0.2 n NaOH at 37 °C for 1 h to eliminate ester-containing glycerolipids and then neutralized by the addition of 1 n HCl. Then alkali-stable lipids were re-extracted by the Bligh and Dyer method, and the solutions were evaporated to dryness. The dried lipids were dissolved in 10 μl of chloroform/methanol (2/1, v/v), and applied to a TLC plate, which was developed with chloroform/methanol/H2O (65:25:4, v/v/v). Radioactive SM and GlcCer on the TLC plate was detected with a Typhoon FLA 9500 (GE Healthcare) and quantified using ImageQuant TL software.
Rapamycin-induced heterodimerization
SMS1–GCS DKO cells transfected with plasmids were incubated in DMEM supplemented with 10% FBS containing 0.1% DMSO or 500 nm rapamycin for 3 h at 37 °C in a humidified incubator containing 5% CO2. Then the cells were harvested for co-immunoprecipitation analysis or assay of enzyme activities in vitro, or fixed for immunocytochemistry analysis as described above. In the metabolic labeling experiment, the cells were further cultured in medium containing 0.5 μCi of [14C]stearic acid and 0.1% DMSO or 500 nm rapamycin for 3 h, and then the lipids were extracted as described above.
Statistical analysis
All analyses were performed with GraphPad PRISM 6 (GraphPad Software). For comparisons, the Student's t test or one-way analysis of variance followed by the Tukey–Kramer multiple comparison test were used. The results are shown as the means ± S.D. The statistical significance is indicated as follows: *, p < 0.01; **, p < 0.05.
Author contributions
Y. H., Y. N.-S., N. M., K. H., T. T., S. O., T. Saeki, T. Kumasaka, T. Koizumi, S. A., and I. W. conceptualization; Y. H. investigation; Y. H. writing-original draft; K. Y., T. Sugiura, and A. Y. funding acquisition; A. Y. project administration.
Supplementary Material
Acknowledgments
We are grateful to Prof. J. Aoki and Dr. A. Inoue (Tohoku University, Sendai, Japan) for valuable technical advice regarding the CRISPR-Cas9 system. We acknowledge the assistance of A. Harima, A. Takuma, H. Taka, S. Kawakami, and A. Nomizu (Teikyo University, Tokyo, Japan) in plasmid construction. We thank Editage for English language editing.
This work was supported in part by Grant-in-Aid for Young Scientists (B) 15K18868 (to Y. H.) and Grants-in-Aid for Scientific Research (C) 18K06635 (to Y. H.) and 15K07946 (to A. Y.) from the Japan Society for the Promotion of Science. This work was also supported in part by funds from the Japan Foundation for Applied Enzymology (to Y. H.) and by the Science Research Promotion Fund from the Japan Private School Promotion Foundation (to Y. H., K. H., K. Y., and A. Y.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- SM
- sphingomyelin
- BiFC
- bimolecular fluorescence complementation
- Cer
- ceramide
- CERT
- ceramide transport protein
- FKBP
- FK506-binding protein
- FRB
- FKBP-rapamycin-binding domain
- GCS
- glucosylceramide synthase
- GlcCer
- glucosylceramide
- GSL
- glycosphingolipid
- PI4P
- phosphatidylinositol-4-phosphate
- SAM
- sterile α-motif
- SMS
- sphingomyelin synthase
- VN
- N-terminal residues 1–173 of Venus protein
- VC
- C-terminal residues 155–238 of Venus protein
- KO
- knockout
- DKO
- double knockout
- mTOR
- mammalian target of rapamycin
- ER
- endoplasmic reticulum
- SMSr
- sphingomyelin synthase-related protein
- HA
- hemagglutinin
- HRP
- horseradish peroxidase
- DMEM
- Dulbecco's modified Eagle's medium
- APC
- allophycocyanin.
References
- 1. Rietveld A., and Simons K. (1998) The differential miscibility of lipids as the basis for the formation of functional membrane rafts. Biochim. Biophys. Acta 1376, 467–479 10.1016/S0304-4157(98)00019-7 [DOI] [PubMed] [Google Scholar]
- 2. Kiyokawa E., Baba T., Otsuka N., Makino A., Ohno S., and Kobayashi T. (2005) Spatial and functional heterogeneity of sphingolipid-rich membrane domains. J. Biol. Chem. 280, 24072–24084 10.1074/jbc.M502244200 [DOI] [PubMed] [Google Scholar]
- 3. Huitema K., van den Dikkenberg J., Brouwers J. F., and Holthuis J. C. (2004) Identification of a family of animal sphingomyelin synthases. EMBO J. 23, 33–44 10.1038/sj.emboj.7600034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yamaoka S., Miyaji M., Kitano T., Umehara H., and Okazaki T. (2004) Expression cloning of a human cDNA restoring sphingomyelin synthesis and cell growth in sphingomyelin synthase-defective lymphoid cells. J. Biol. Chem. 279, 18688–18693 10.1074/jbc.M401205200 [DOI] [PubMed] [Google Scholar]
- 5. Tafesse F. G., Huitema K., Hermansson M., van der Poel S., van den Dikkenberg J., Uphoff A., Somerharju P., and Holthuis J. C. (2007) Both sphingomyelin synthases SMS1 and SMS2 are required for sphingomyelin homeostasis and growth in human HeLa cells. J. Biol. Chem. 282, 17537–17547 10.1074/jbc.M702423200 [DOI] [PubMed] [Google Scholar]
- 6. Mitsutake S., and Igarashi Y. (2013) Sphingolipids in lipid microdomains and obesity. Vitam. Horm. 91, 271–284 10.1016/B978-0-12-407766-9.00012-2 [DOI] [PubMed] [Google Scholar]
- 7. Mitsutake S., Zama K., Yokota H., Yoshida T., Tanaka M., Mitsui M., Ikawa M., Okabe M., Tanaka Y., Yamashita T., Takemoto H., Okazaki T., Watanabe K., and Igarashi Y. (2011) Dynamic modification of sphingomyelin in lipid microdomains controls development of obesity, fatty liver, and type 2 diabetes. J. Biol. Chem. 286, 28544–28555 10.1074/jbc.M111.255646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hayashi Y., Nemoto-Sasaki Y., Tanikawa T., Oka S., Tsuchiya K., Zama K., Mitsutake S., Sugiura T., and Yamashita A. (2014) Sphingomyelin synthase 2, but not sphingomyelin synthase 1, is involved in HIV-1 envelope-mediated membrane fusion. J. Biol. Chem. 289, 30842–30856 10.1074/jbc.M114.574285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ichikawa S., Sakiyama H., Suzuki G., Hidari K. I., and Hirabayashi Y. (1996) Expression cloning of a cDNA for human ceramide glucosyltransferase that catalyzes the first glycosylation step of glycosphingolipid synthesis. Proc. Natl. Acad. Sci. U.S.A. 93, 4638–4643 10.1073/pnas.93.10.4638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Futerman A. H., and Pagano R. E. (1991) Determination of the intracellular sites and topology of glucosylceramide synthesis in rat liver. Biochem. J. 280, 295–302 10.1042/bj2800295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Futerman A. H., and Riezman H. (2005) The ins and outs of sphingolipid synthesis. Trends Cell Biol. 15, 312–318 10.1016/j.tcb.2005.04.006 [DOI] [PubMed] [Google Scholar]
- 12. Hanada K., Kumagai K., Yasuda S., Miura Y., Kawano M., Fukasawa M., and Nishijima M. (2003) Molecular machinery for non-vesicular trafficking of ceramide. Nature 426, 803–809 10.1038/nature02188 [DOI] [PubMed] [Google Scholar]
- 13. Halter D., Neumann S., van Dijk S. M., Wolthoorn J., de Mazière A. M., Vieira O. V., Mattjus P., Klumperman J., van Meer G., and Sprong H. (2007) Pre- and post-Golgi translocation of glucosylceramide in glycosphingolipid synthesis. J. Cell Biol. 179, 101–115 10.1083/jcb.200704091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sprong H., Degroote S., Claessens T., van Drunen J., Oorschot V., Westerink B. H., Hirabayashi Y., Klumperman J., van der Sluijs P., and van Meer G. (2001) Glycosphingolipids are required for sorting melanosomal proteins in the Golgi complex. J. Cell Biol. 155, 369–380 10.1083/jcb.200106104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jeckel D., Karrenbauer A., Burger K. N., van Meer G., and Wieland F. (1992) Glucosylceramide is synthesized at the cytosolic surface of various Golgi subfractions. J. Cell Biol. 117, 259–367 10.1083/jcb.117.2.259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hayashi Y., Nemoto-Sasaki Y., Matsumoto N., Tanikawa T., Oka S., Tanaka Y., Arai S., Wada I., Sugiura T., and Yamashita A. (2017) Carboxyl-terminal tail-mediated homodimerizations of sphingomyelin synthases are responsible for efficient export from the endoplasmic reticulum. J. Biol. Chem. 292, 1122–1141 10.1074/jbc.M116.746602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu C.-D., Chinenov Y., and Kerppola T. K. (2002) Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9, 789–798 10.1016/S1097-2765(02)00496-3 [DOI] [PubMed] [Google Scholar]
- 18. Yang W., Qiu C., Biswas N., Jin J., Watkins S. C., Montelaro R. C., Coyne C. B., and Wang T. (2008) Correlation of the tight junction-like distribution of Claudin-1 to the cellular tropism of hepatitis C virus. J. Biol. Chem. 283, 8643–8653 10.1074/jbc.M709824200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ozalp C., Szczesna-Skorupa E., and Kemper B. (2005) Bimolecular fluorescence complementation analysis of cytochrome p450 2c2, 2e1, and NADPH-cytochrome p450 reductase molecular interactions in living cells. Drug Metab. Dispos. 33, 1382–1390 10.1124/dmd.105.005538 [DOI] [PubMed] [Google Scholar]
- 20. Marks D. L., Wu K., Paul P., Kamisaka Y., Watanabe R., and Pagano R. E. (1999) Identification of active site residues in glucosylceramide synthase. A nucleotide-binding catalytic motif conserved with processive β-glycosyltransferases. J. Biol. Chem. 274, 451–456 10.1074/jbc.274.1.451 [DOI] [PubMed] [Google Scholar]
- 21. Marks D. L., Dominguez M., Wu K., and Pagano R. E. (2001) Oligomerization and topology of the Golgi membrane protein glucosylceramide synthase. J. Biol. Chem. 276, 26492–26498 10.1074/jbc.M102612200 [DOI] [PubMed] [Google Scholar]
- 22. Yasuda S., Nishijima M., and Hanada K. (2003) Localization, topology, and function of the LCB1 subunit of serine palmitoyltransferase in mammalian cells. J. Biol. Chem. 278, 4176–4183 10.1074/jbc.M209602200 [DOI] [PubMed] [Google Scholar]
- 23. Kim C. A., and Bowie J. U. (2003) SAM domains: uniform structure, diversity of function. Trends Biochem. Sci. 28, 625–628 10.1016/j.tibs.2003.11.001 [DOI] [PubMed] [Google Scholar]
- 24. Qiao F., and Bowie J. U. (2005) The many faces of SAM. Sci. STKE 2005, re7 [DOI] [PubMed] [Google Scholar]
- 25. Yeang C., Varshney S., Wang R., Zhang Y., Ye D., and Jiang X. C. (2008) The domain responsible for sphingomyelin synthase (SMS) activity. Biochim. Biophys. Acta 1781, 610–617 10.1016/j.bbalip.2008.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chu B. B., Liao Y. C., Qi W., Xie C., Du X., Wang J., Yang H., Miao H. H., Li B. L., and Song B. L. (2015) Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 161, 291–306 10.1016/j.cell.2015.02.019 [DOI] [PubMed] [Google Scholar]
- 27. Klayman L. M., and Wedegaertner P. B. (2017) Inducible inhibition of Gβγ reveals localization-dependent functions at the plasma membrane and Golgi. J. Biol. Chem. 292, 1773–1784 10.1074/jbc.M116.750430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim J. H., Lee S. R., Li L. H., Park H. J., Park J. H., Lee K. Y., Kim M. K., Shin B. A., and Choi S. Y. (2011) High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One 6, e18556 10.1371/journal.pone.0018556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ballou L. M., and Lin R. Z. (2008) Rapamycin and mTOR kinase inhibitors. J. Chem. Biol. 1, 27–36 10.1007/s12154-008-0003-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yasuda S., Kitagawa H., Ueno M., Ishitani H., Fukasawa M., Nishijima M., Kobayashi S., and Hanada K. (2001) A novel inhibitor of ceramide trafficking from the endoplasmic reticulum to the site of sphingomyelin synthesis. J. Biol. Chem. 276, 43994–44002 10.1074/jbc.M104884200 [DOI] [PubMed] [Google Scholar]
- 31. Fukasawa M., Nishijima M., and Hanada K. (1999) Genetic evidence for ATP-dependent endoplasmic reticulum-to-Golgi apparatus trafficking of ceramide for sphingomyelin synthesis in Chinese hamster ovary cells. J. Cell Biol. 144, 673–685 10.1083/jcb.144.4.673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nagaya H., Wada I., Jia Y. J., and Kanoh H. (2002) Diacylglycerol kinase delta suppresses ER-to-Golgi traffic via its SAM and PH domains. Mol. Biol. Cell 13, 302–316 10.1091/mbc.01-05-0255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qiao F., Song H., Kim C. A., Sawaya M. R., Hunter J. B., Gingery M., Rebay I., Courey A. J., and Bowie J. U. (2004) Derepression by depolymerization; structural insights into the regulation of Yan by Mae. Cell 118, 163–173 10.1016/j.cell.2004.07.010 [DOI] [PubMed] [Google Scholar]
- 34. Aviv T., Lin Z., Lau S., Rendl L. M., Sicheri F., and Smibert C. A. (2003) The RNA-binding SAM domain of Smaug defines a new family of post-transcriptional regulators. Nat. Struct. Biol. 10, 614–621 10.1038/nsb956 [DOI] [PubMed] [Google Scholar]
- 35. Barrera F. N., Poveda J. A., González-Ros J. M., and Neira J. L. (2003) Binding of the C-terminal sterile α motif (SAM) domain of human p73 to lipid membranes. J. Biol. Chem. 278, 46878–46885 10.1074/jbc.M307846200 [DOI] [PubMed] [Google Scholar]
- 36. Vacaru A. M., Tafesse F. G., Ternes P., Kondylis V., Hermansson M., Brouwers J. F., Somerharju P., Rabouille C., and Holthuis J. C. (2009) Sphingomyelin synthase-related protein SMSr controls ceramide homeostasis in the ER. J. Cell Biol. 185, 1013–1027 10.1083/jcb.200903152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cabukusta B., Kol M., Kneller L., Hilderink A., Bickert A., Mina J. G., Korneev S., and Holthuis J. C. (2017) ER residency of the ceramide phosphoethanolamine synthase SMSr relies on homotypic oligomerization mediated by its SAM domain. Sci. Rep. 7, 41290 10.1038/srep41290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li E., Wimley W. C., and Hristova K. (2012) Transmembrane helix dimerization: beyond the search for sequence motifs. Biochim. Biophys. Acta 1818, 183–193 10.1016/j.bbamem.2011.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van Blitterswijk W. J., van der Luit A. H., Veldman R. J., Verheij M., and Borst J. (2003) Ceramide: second messenger or modulator of membrane structure and dynamics? Biochem. J. 369, 199–211 10.1042/bj20021528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goñi F. M., and Alonso A. (2006) Biophysics of sphingolipids: I. membrane properties of sphingosine, ceramides and other simple sphingolipids. Biochim. Biophys. Acta 1758, 1902–1921 10.1016/j.bbamem.2006.09.011 [DOI] [PubMed] [Google Scholar]
- 41. Ishibashi Y., Kohyama-Koganeya A., and Hirabayashi Y. (2013) New insights on glucosylated lipids: metabolism and functions. Biochim. Biophys. Acta 1831, 1475–1485 10.1016/j.bbalip.2013.06.001 [DOI] [PubMed] [Google Scholar]
- 42. D'Angelo G., Polishchuk E., Di Tullio G., Santoro M., Di Campli A., Godi A., West G., Bielawski J., Chuang C. C., van der Spoel A. C., Platt F. M., Hannun Y. A., Polishchuk R., Mattjus P., and De Matteis M. A. (2007) Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature 449, 62–67 10.1038/nature06097 [DOI] [PubMed] [Google Scholar]
- 43. Lamour N. F., Stahelin R. V., Wijesinghe D. S., Maceyka M., Wang E., Allegood J. C., Merrill A. H. Jr, Cho W., and Chalfant C. E. (2007) Ceramide kinase uses ceramide provided by ceramide transport protein: localization to organelles of eicosanoid synthesis. J. Lipid Res. 48, 1293–1304 10.1194/jlr.M700083-JLR200 [DOI] [PubMed] [Google Scholar]
- 44. Contreras F. X., Villar A. V., Alonso A., Kolesnick R. N., and Goñi F. M. (2003) Sphingomyelinase activity causes transbilayer lipid translocation in model and cell membranes. J. Biol. Chem. 278, 37169–37174 10.1074/jbc.M303206200 [DOI] [PubMed] [Google Scholar]
- 45. Contreras F. X., Basañez G., Alonso A., Herrmann A., and Goñi F. M. (2005) Asymmetric addition of ceramides but not dihydroceramides promotes transbilayer (flip-flop) lipid motion in membranes. Biophys. J. 88, 348–359 10.1529/biophysj.104.050690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mitsutake S., and Igarashi Y. (2007) Transbilayer movement of ceramide in the plasma membrane of live cells. Biochem. Biophys. Res. Commun. 359, 622–627 10.1016/j.bbrc.2007.05.160 [DOI] [PubMed] [Google Scholar]
- 47. Akiyama M. (2017) Corneocyte lipid envelope (CLE), the key structure for skin barrier function and ichthyosis pathogenesis. J. Dermatol. Sci. 88, 3–9 10.1016/j.jdermsci.2017.06.002 [DOI] [PubMed] [Google Scholar]
- 48. Ishibashi Y., Ito M., and Hirabayashi Y. (2018) Regulation of glucosylceramide synthesis by Golgi-localized phosphoinositide. Biochem. Biophys. Res. Commun. 499, 1011–1018 10.1016/j.bbrc.2018.04.039 [DOI] [PubMed] [Google Scholar]
- 49. Wurch T., Lestienne F., and Pauwels P. J. (1998) A modified overlap extension PCR method to create chimeric genes in the absence of restriction enzymes. Biotechnol. Techniques 12, 653–657 10.1023/A:1008848517221 [DOI] [Google Scholar]
- 50. Hammond G. R., Fischer M. J., Anderson K. E., Holdich J., Koteci A., Balla T., and Irvine R. F. (2012) PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 337, 727–730 10.1126/science.1222483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Karginov A. V., Ding F., Kota P., Dokholyan N. V., and Hahn K. M. (2010) Engineered allosteric activation of kinases in living cells. Nat. Biotechnol. 28, 743–747 10.1038/nbt.1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Blight E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 10.1139/y59-099,10.1139/o59-099 [DOI] [PubMed] [Google Scholar]
- 54. Hirokawa T., Boon-Chieng S., and Mitaku S. (1998) SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics 14, 378–379 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.