

Abstract
Regulation of chromatin compaction is an important process that governs gene expression in higher eukaryotes. Although chromatin compaction and gene expression regulation are commonly disrupted in many diseases, a locus-specific, endogenous, and reversible method to study and control these mechanisms of action has been lacking. To address this issue, we have developed and characterized novel gene-regulating bifunctional molecules. One component of the bifunctional molecule binds to a DNA-protein anchor so that it will be recruited to an allele-specific locus. The other component engages endogenous cellular chromatin-modifying machinery, recruiting these proteins to a gene of interest. These small molecules, called chemical epigenetic modifiers (CEMs), are capable of controlling gene expression and the chromatin environment in a dose-dependent and reversible manner. Here, we detail a CEM approach and its application to decrease gene expression and histone tail acetylation at a Green Fluorescent Protein (GFP) reporter located at the Oct4 locus in mouse embryonic stem cells (mESCs). We characterize the lead CEM (CEM23) using fluorescent microscopy, flow cytometry, and chromatin immunoprecipitation (ChIP), followed by a quantitative polymerase chain reaction (qPCR). While the power of this system is demonstrated at the Oct4 locus, conceptually, the CEM technology is modular and can be applied in other cell types and at other genomic loci.
Keywords: Bioengineering, Issue 139, Chemical biology, bifunctional molecules, chemical induced proximity, chromatin regulation, gene repression, histone deacetylase, epigenetics, chemical epigenetic modifiers, CEMs
Introduction
Chromatin consists of DNA wrapped around histone octamer proteins that form the core nucleosome particle. Regulation of chromatin compaction is an essential mechanism for proper DNA repair, replication, and expression1,2,3. One way in which cells control the level of compaction is through the addition or removal of various post-translational histone tail modifications. Two such modifications include (1) lysine acetylation, which is most commonly associated with gene activation, and (2) lysine methylation, which can be associated with either gene activation or repression, depending on the amino acid context. The addition of the acetylation and methylation marks is catalyzed by histone acetyltransferases (HATs) and histone methyltransferases (HMTs), respectively, whereas the removal of the mark is done by histone deacetylases (HDACs) and histone demethylases (HDMs), respectively4,5. Although the existence of these proteins has been known for decades, many mechanisms of how chromatin-modifying machinery works to properly regulate gene expression remain to be defined. Since chromatin regulatory processes are dysregulated in many human diseases, new mechanistic insights could lead to future therapeutic applications.
The chromatin in vivo assay (CiA) is a recently described technique that uses a chemical inducer of proximity (CIP) to control chromatin-modifying machinery recruitment to a specific locus6. This technology has been used to study an expanding list of chromatin dynamics, including histone-modifying proteins, chromatin remodelers, and transcription factors6,7,8,9. CIP-based chromatin tethering of exogenously expressed proteins has also been extended past CiA to modulate non-modified genetic loci by use with a deactivated Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated protein (dCas9)10,11. In the CiA system, mESCs have been modified to express a GFP reporter gene at the Oct4 locus, a highly expressed and strictly regulated area of the mESC genome. Previously, this system has been applied to describe the dose-dependent and reversible recruitment of exogenously expressed heterochromatin protein 1 (HP1), an enzyme that binds H3K9me3 and propagates repressive marks (e.g., H3K9me3) and DNA methylation6. To accomplish this type of recruitment strategy, the mESCs are infected with a plasmid that expresses an FK506-binding protein (FKBP) linked to a Gal4, which is a DNA-protein anchor that binds to a Gal4-binding array upstream of the GFP reporter. The cells are also infected with an FKBP-rapamycin-binding domain (FRB) connected to HP1. When the mESCs are exposed to low nanomolar concentrations of the CIP, rapamycin, both FRB and FKBP, are brought together at the target locus. Within days of rapamycin treatment, expression of the target gene is repressed, as evidenced by fluorescence microscopy and flow cytometry, and histone acetylation is decreased, shown by ChIP and bisulfite sequencing. While the development and validation of this approach have been significant advances in the field of chromatin research and regulation, one drawback is the required exogenous expression of chromatin-modifying machinery.
To make the technology based on recruitment of only endogenous physiologically-relevant enzymes and make a more modular system, we designed and characterized novel bifunctional molecules, termed CEMs (Figure 1)12. One component of the CEMs includes FK506 which, similar to rapamycin, binds tightly and specifically to FKBP. Thus, the CEMs will still be recruited to the Oct4 locus in the CiA mESCs. The other component of the CEMs is a moiety that binds endogenous chromatin-modifying machinery. In a pilot study, we tested CEMs that contain HDAC inhibitors. While the concept of using an inhibitor to recruit HDACs seems counter-intuitive, the inhibitor is nonetheless able to recruit HDAC activity to the gene of interest. This is accomplished by (1) redirecting the HDAC to the locus, releasing the enzyme, and increasing the density of un-inhibited HDACs in the area and (2) maintaining HDAC inhibition at the locus, but recruiting repressive complexes that bind to the inhibited HDACs, or (3) a combination of both. In a previous study, we showed that the CEMs were able to successfully repress the GFP reporter in a dose- and time-dependent manner, as well as in a manner that was rapidly reversible (i.e., within 24 hours)12. We characterized the ability of the CEM technology presented here to control gene expression using fluorescence microscopy, flow cytometry, and the ability to control the chromatin environment using ChIP-qPCR6. Here, we describe a method for using and characterizing the CEMs, which will facilitate the adaptation of this system to answer additional questions related to chromatin biology.
Protocol
1) Cell Line Culture for Producing Lentivirus
Grow fresh, low-passage (less than passage 30) 293T human embryonic kidney (HEK) cells in high-glucose Dulbecco's modified Eagle's medium (DMEM) base media supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, 1x non-essential amino acids (NEAA), Pen/Strep, 2-mercaptoenthanol in a 37 °C incubator with 5% CO2. Split the cells every 3 - 5 d and before they become > 95% confluent.
- Passage 293T HEK cells with 18 x 106 per 15-cm plate (one 15-cm plate for each virus produced).
- For a 15-cm format, aspirate the old media, add 10 mL of 1x phosphate-buffered saline (PBS), aspirate the PBS, and add 3 mL of 0.05% trypsin. Incubate the cells in trypsin for 8 min at 37 °C, rocking and tapping the cells halfway through the incubation. NOTE: The goal of this step is to get the cells into a single-cell suspension.
- When the cells have reached 60% - 80% confluency the following day, perform a polyethylenimine (PEI) transfection with 13.5 µg of the psPAX2 plasmid, 4.5 µg of the pMD2.G plasmid, 18 µg of lentiviral-compatible delivery vector with the gene of interest (i.e., FKBP-Gal4), 108 µL of PEI, and 1.8 mL of transfection media.
- Gently mix the DNA, PEI, and transfection media in a 15-mL conical tube, incubate the sample at room temperature for 15 min, and add it dropwise to the 15-cm plate. NOTE: Please use appropriate safety measures and follow all institutional and laboratory safety procedures after this step, as all reagents will contain a live virus.
After 16 h of transfection and incubation in a 37 °C incubator with 5% CO2, aspirate the media and gently add fresh 293 HEK media (prewarmed in a 37 °C water bath) to the 15-cm plates. Incubate the cells in a 37 °C incubator with 5% CO2 for 48 h after the media change. The virus is then ready to be harvested.
2) Infection of Mouse Embryonic Stem Cells (mESC) with Lentivirus
- Grow low-passage (less than passage 35), feeder-free adapted CiA mESCs in high-glucose DMEM base media supplemented with 20% ESC-grade FBS, 10 mM HEPES, 1x NEAA, Pen/Strep, 2-mercaptoenthanol, and 1:500 leukemia inhibitor factor (LIF) in a 37 °C incubator with 5% CO2. NOTE: The LIF is obtained from the supernatant of COS LIF-producing cells, which are collected in batch and frozen at -80 °C.
- Grow cells on plates that have been preincubated for 1 - 3 h with 0.1% gelatin in 1x PBS, and subsequently washed with 1x PBS. Feed the cells daily and split them every 2 - 3 d, depending on their density. NOTE: Morphologically, embryonic stem cell (ES) colonies should be small, round, and distinct from each other.
- Passage mESC into a 12-well plate format (100,000 cells/well) 1 d prior to infection.
- Count the cells with a hemocytometer. For cells grown in a 10-cm format, aspirate the old media, add 5 mL of 1x PBS, aspirate the PBS, and add 1 mL of 0.25% trypsin. Incubate the cells in trypsin for 8 min at 37 °C, rocking and tapping the cells halfway through the incubation. The goal of this step is to get the cells into a single-cell suspension.
48 h after changing the media from the 293T15-cm viral packaging cells from step 1.4, remove and transfer the supernatant to a 50-mL conical tube.
Centrifuge the supernatant at 300 x g for 5 min to pellet cell debris.
Filter the supernatant through a 0.45-µm membrane. NOTE: Make sure to use surfactant-free cellulose acetate (SFCA) membranes, as some other materials can retain virus and significantly reduce yields.
Concentrate the virus with ultracentrifugation, using a centrifuge with an SW32 rotor, and spin it at 20,000 rpm (~72,000 x g) for 2.5 h at 4 °C.
While the virus concentrates under ultracentrifugation, treat the CiA mESCs in the 12-well plate with fresh ES media containing 5 µg/mL of polybrene.
When the virus concentration has finished, carefully aspirate the supernatant and suspend the virus pellet in 100 µL of 1x PBS (avoid excess bubbles).
Add the virus and 1x PBS to a 1.5-mL microfuge tube. Vortex/shake the tube at 300 x g (or at the lowest setting) to fully suspend the virus. Spin down the tube in a mini-tabletop centrifuge for 5 - 10 s to remove bubbles.
- Add 30 µL of the virus to each well of a 12-well plate. Swirl the plate and then centrifuge the plate at 1,000 x g for 20 min. NOTE: The amount of virus added can be varied depending on the viral titer, which is inversely related to delivery construct size.
- Alternatively, flash freeze the virus with liquid nitrogen and store it at -80 °C. However, freezing the virus will lower the viral titer.
Place the 12-well plate back in the 37 °C incubator and change the media the following morning (~16 h later) with fresh ES media (as described in step 2.1).
After 48 h of infection, select for cells that integrated the viral plasmid by adding the appropriate antibiotic (i.e., 1.5 µg/mL of puromycin, 10 µg/mL of blasticidin, etc.). Change the media daily and wash the cells with 1x PBS if a majority of the cells are floating/dead. Keep the selection media on the cells for 72 - 96 h in a 37 °C incubator with 5% CO2.
3) Chemical Epigenetic Modifiers (CEM)s Preparation and Treatment
Suspend the powdered CEM into dimethyl sulfoxide (DMSO) to a stock concentration of 1 mM and a working concentration of 100 µM.
- After the cells have undergone the selection for 72 - 96 h, split the mESCs into a 12-well format with 100,000 cells/well. Incubate the cells in a 37 °C incubator with 5% CO2 for 24 h. Afterward, prepare a media solution of 100 nM CEM with ES media. Use this media to feed the CiA mESC with 1 mL/well. To serve as a control, keep a well of cells without any added CEMs.
- Change the media by aspirating old media and adding 1 mL/well of freshly prepared CEM-containing or CEM-free ES media every 24 h for the duration of an experiment.
4) Analysis of the Expression by Microscopy and Flow Cytometry
After 48 hours of CEM treatment, image the cells without CEM treatment and the cells treated with 100 nM CEM using a fluorescence microscope. Take representative images using phase or brightfield to record the mESC morphology at 20X magnification; the cells should have formed round colonies, with a few differentiated cells. Under the FITC fluorescence channel, image both cell conditions.
Isolate the control and CEM-treated cells for flow cytometry by aspirating the media, washing the cells with 1 mL of 1x PBS, aspirating the PBS, and adding 0.25 mL of 0.25% trypsin with EDTA for 8 - 10 min.
Confirm that the cells are no longer in large clumps by looking in the microscope. Use a 20X magnification. If the cells do not appear to be in a single-cell suspension, gently pipette up and down with a P1000 pipette to fully trypsinize the cells.
Quench the trypsin with 1 mL of fresh media and resuspend the cells at high speed with a serological pipette (or using a P1000 pipette and tip) until the cells are in a single-cell suspension.
Spin down the cells at 300 x g for 5 min. Aspirate the supernatant. Wash with 1x PBS and recentrifuge the cells. Aspirate the PBS supernatant.
- Resuspend the cell pellet in 200 mL of FACS buffer (1 mM ethylenediaminetetraacetic acid [EDTA], 1x PBS, and 0.2% bovine serum albumin [BSA]).
- The total volume of FACS buffer will be dependent on how many cells are present, but the concentration should be 1,000 - 3,000 cells/µL. Count the number of cells in 3 samples and average the concentration of cells. Add more FACS buffer if needed.
Perform flow cytometry on > 50,000 cells with the suspended cells, keeping the cells on ice when they are not being analyzed. Use a 530/40 filter (FITC, AF488, GFP, etc.) for recording the changes in expression. Determine the appropriate fluorescent voltage using an untreated control cell sample and set the voltage to have the fluorescent peak be in the middle of the recorded intensity spectrum. Run the samples at a sheath speed of around 5,000 cells/s.
Export the data and analyze the FCS files on a flow cytometry program (i.e., FlowJo) by gating first on forward scatter vs. side scatter for live cells and then on forward scatter height vs. area for singlets. Then, visualize GFP channel data on a histogram to determine the relative GFP expression in the targeted and control populations.
5) Analysis of Chromatin by Chromatin Immunoprecipitation (ChIP) Followed by qPCR
Split the mESCs into a 10-cm plate with 3 million cells and 10 mL of ES media (one 10-cm plate for each experimental or control condition). The following day, add 10 mL of CEM-containing or CEM-free media. After 48 hours of CEM treatment, aspirate the old media. Wash and aspirate the cells with 5 mL of 1x PBS. Next, dissociate the cells with 0.25% trypsin, and quench the trypsin with 10 mL of ES media. Count and isolate a sample of 10 million cells per condition.
Spin down each of the 10 million cell samples at 300 x g for 5 min.
Resuspend each pellet in 10 mL of 1x PBS and recentrifuge the cells at 300 x g for 5 min.
Resuspend each pellet in 10 mL of CiA Fix buffer (50 mM pH 8, 1 mM EDTA pH 8, 0.5 mM ethylene glycol-bis[ß-aminoethyl ether]-N,N,N',N'-tetraacetic acid [EGTA] pH 8, and 100 mM sodium chloride [NaCl]). Add 1 mL of 11% formaldehyde solution and immediately invert the samples 5x - 10x. Incubate the sample at room temperature for 10 min.
Add 0.5 mL of 2.5 M glycine. Invert the samples immediately and incubate the sample on ice for 5 min.
Centrifuge the sample at 1,200 x g for 5 min at 4 °C. Aspirate the supernatant.
Resuspend the pellet in 10 mL of CiA Rinse 1 (50 mM HEPES pH 8, 140 mM NaCl, 1 mM EDTA pH 8, 10% glycerol, 0.5% Nonidet P-40 [NP40], and 0.25% Triton X100). Incubate the sample on ice for 10 min. Centrifuge it at 1,200 x g for 5 min at 4 °C.
Resuspend the pellet in 10 mL of CiA Rinse 2 (10 mM tris[hydroxymethyl]aminomethane [Tris] pH 8, 1 mM EDTA pH 8, 0.5 mM EGTA pH 8, and 200 mM NaCl). Centrifuge the sample at 1,200 x g for 5 min at 4 °C.
Gently add 5 mL of CiA shearing buffer (0.1% SDS, 1 mM EDTA, and 10 mM Tris pH 8) along the sides of the conical tube to wash off any salt without disrupting the pellet. Centrifuge the sample at 1,200 x g for 3 min at 4 °C. Repeat the wash step.
Resuspend the pellet in 90 µL of CiA shearing buffer and fresh protease inhibitors (use a mixture of leupeptin, chymostatin, and pepstatin A dissolved together at 10mg/mL each in DMSO to make a 1,000x stock solution, making a final concentration of 10 µg/mL). Add 10 µL of sonication-enhancing nanodroplets while keeping everything on ice13,14.
Transfer 100 µL of the solution to a glass tube and cap the tube.
Sonicate the samples for 3.5 min (determined based on the cell line and number of cells). To avoid overheating the samples, process the samples in 2-min-maximum cycle times if the total sonication time exceeds 2 min. Note: The focused ultrasonicator used here functions at a high frequency (500 kHz). Run the machine at an acoustic power of 55 W. The sonication duration should be predetermined by each individual lab.
Transfer each sonicated chromatin sample to a new 1.5-mL microcentrifuge tube. Spin the tubes at 8,000 x g for 2 min at 4 °C.
To analyze the shearing efficiency and to quantify the relative amount of chromatin, follow the ChIP kit manufacturer's protocol. NOTE: Determine the DNA concentration spectrophotometrically (e.g., using a DNA nanodrop instrument), and run ~200 ng of DNA on a 1% agarose gel to verify that the chromatin is sonicated to roughly 200 - 500 basepairs (bp) in size.
To perform immunoprecipitation and isolate the chromatin, follow the ChIP kit manufacturer's protocol. NOTE: For the immunoprecipitation of samples with higher DNA concentrations, warm the elution buffer at 37 °C and elute the samples 2x with 30 µL of elution buffer (spinning for 1 min each time).
- To perform qPCR, load the reagents into a 384-well plate. Add 5 µL of 2x asymmetrical cyanine dye master mix, 2.5 µM of each primer, water, and 0.1 - 10 ng of DNA for each reaction.
- Design primers to amplify 50 - 100 bp amplicons, and CT values are normalized to a house-keeping gene, intracisternal A-type particles (IAP). NOTE: For further qPCR methods, see the kit manufacturer's protocol. The primer set used to target the Oct4 locus was: (F) CACATGAAGCAGCACGACTT; (R) CCTTGAAGAAGATGGTGCGC. The control primer set used to target IAP was: (F) ATTTCGCCTAGGACGTGTCA; (R) ACTCCATGTGCTCTGCCTTC.
- Based on the primers and the desired product, run the qPCR with 40 cycles of an annealing temperature of 60 °C for 1 min. NOTE: The resulting data was quantitated using the delta-delta Ct comparisons between samples, with CiA:Oct4 normalized over IAP. The final analysis results are displayed as averages of triplicate experimental samples, with the error represented as the standard deviation from the mean.
Representative Results
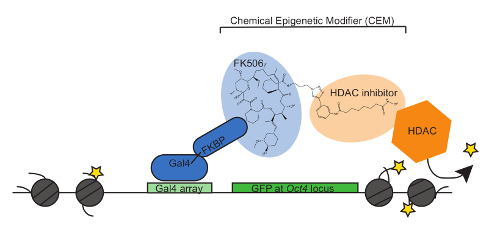
We recently developed CEMs and demonstrated that this technology can be applied to regulate gene expression and the chromatin environment at a reporter locus in a dose-dependent and reversible manner. In Figure 1, a model of the lead CEM, CEM23, is shown. HDAC machinery is recruited to the reporter locus by the HDAC inhibitor which, in this case, is the GFP reporter inserted at the Oct4 locus.
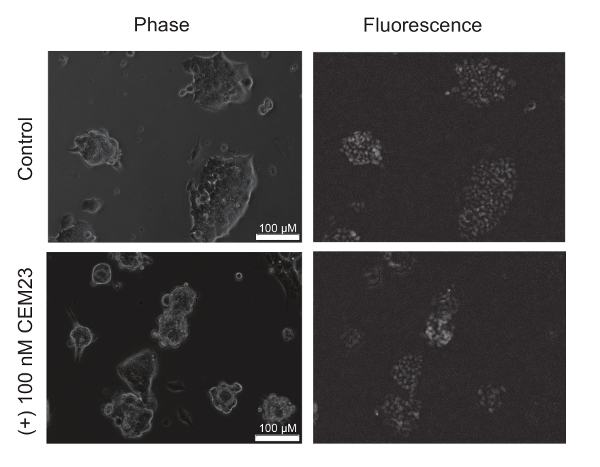
We sought to characterize the CEM system at the Oct4 locus in CiA mESCs. Because the cells express GFP, it was possible to quickly visualize the expression of the reporter with fluorescence microscopy. The cells were imaged after 48 hours of treatment with 100 nM of CEM23, along with untreated cells. The phase images show healthy mESCs, which is important because unhealthy or differentiated mESCs would indicate a non-specific GFP repression. The fluorescence images show a bright GFP expression in the control cells and a reduced GFP expression in mESCs treated with CEM23. Representative images are displayed in Figure 2.
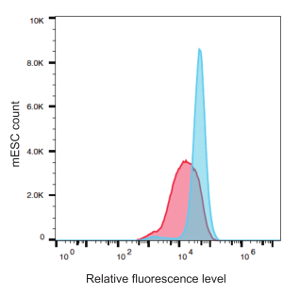
To quantify the changes in the GFP expression, flow cytometry was used. Again, the CiA mESCs were treated with 100 nM of CEM23 for 48 hours. Experimental cells treated with CEM23 and control cells were prepared for flow cytometry, using > 100,000 cells per sample. Consistently, we observed a > 30% decrease in GFP-expressing cells among those treated with CEMs. A representative histogram is shown in Figure 3.
Once evidence of CEM-induced GFP gene expression decrease was shown, we tested for changes in the chromatin environment using ChIP. One important factor for preparing samples for ChIP is the extent of chromatin sonication. To have the samples as consistent and properly sheared as possible, 10 million cells were sonicated for 3.5 minutes to obtain chromatin between 200 and 500 bp in size (Figure 4). Because we hypothesized that the repressive CEMs were binding to and recruiting HDAC proteins and repressive complexes to the reporter, we tested for changes in histone tail acetylation. Histone 3 Lysine 27 acetylation (H3K27ac) is commonly found along the transcriptional start site (TSS) of genes. We performed ChIP with an antibody for H3K27ac and then performed a qPCR with primer sets upstream and downstream of the TSS. The results show that a 48-hour treatment with 100 nM of CEM23 decreased the level of H3K27ac at the target locus when compared to control cells not treated with CEMs (p < 0.01, two-tailed Student's t-test with three biological replicates, Figure 5).
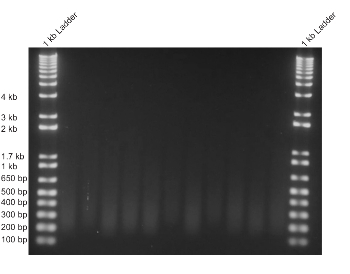
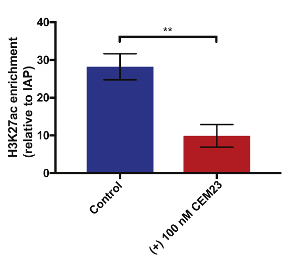
Figure 1: CEMs bind to the CiA:Oct4 locus through recruitment to the FKBP and tether endogenous epigenetic machinery. The model of the CEM system shows Gal4-FKBP as the protein-to-DNA anchor. The FK506 portion of the CEM binds to FKBP and the HDAC inhibitor recruits endogenous HDAC proteins to repress GFP. Please click here to view a larger version of this figure.
Figure 2: Fluorescence images of mouse embryonic stem cells (mESCs) with a CiA:Oct4-GFP reporter. Fluorescence microscopy images show a decrease in GFP expression upon CEM treatment. The top panel shows phase and fluorescent images of mESCs grown with untreated media. The bottom panel shows phase and fluorescent of the mESCs treated with 100 nM of CEM23 for 48 hours. The images adapted with permission from ACS Synthetic Biology12. Copyright (2018) American Chemical Society. Please click here to view a larger version of this figure.
Figure 3: Flow cytometry analysis quantitates a decreased expression of GFP in mESCs upon CEM23 treatment. mESCs without CEM23 (blue) were compared with mESCs treated with 100 nM of CEM23 for 48 hours (red). Please click here to view a larger version of this figure.
Figure 4: Sonication of chromatin is uniform, and a smear is visible between 200 and 500 bp. To perform ChIP-qPCR, the chromatin from mESCs was sonicated. Each sample consisted of approximately 10 million cells, which were sonicated for 3.5 minutes, to produce similarly-sheared chromatin samples between 200 and 500 bp in length. Please click here to view a larger version of this figure.
Figure 5: CEM23 treatment causes a decrease in H3K27ac at the CiA locus. ChIP-qPCR was performed to test for changes in the chromatin environment. After a 48-hour treatment with 100 nM of CEM23, a decrease in H3K27ac was observed at CiA:Oct4 (**p < 0.01, two-tailed Student's t-test with three biological replicates. The error bars represent the standard deviation). Please click here to view a larger version of this figure.
Discussion
Here, we described the recently developed CEM system being applied to regulate gene expression and chromatin environment at a specific gene in a dose-dependent manner. We provide an accurate method to study the dynamics involved in regulating gene expression through the selective recruitment of specific endogenous chromatin regulatory proteins. This is a highly modular technology that can be applied to investigate how different protein- and chromatin-modifying complexes work in concert to properly regulate the chromatin environment, as well as to study how these processes are dysregulated, with the benefit of achieving gene specificity.
Here, three ways were demonstrated to visualize changes in the chromatin structure and gene expression with new technology. After perturbation of specific targeted loci with CEMs, real-time changes to gene expression could be analyzed by fluorescence microscopy. Then, changes in gene expression levels could be measured more quantitatively with flow cytometry at defined endpoints. Finally, changes to posttranslational epigenetic marks on chromatin were examined by ChIP; specifically, in this case, H3K27ac. We did not test HDAC recruitment with ChIP-qPCR because of the diversity of HDACs that act in concert at a single locus. It is likely that a heterogenous population of HDAC enzymes was recruited and it would, therefore, be technically difficult to test them all by ChIP. In a previously published work, we have demonstrated that the direct recruitment of HDAC3 by a GAL4 fusion caused similar activity on gene expression and chromatin modification by ChIP12. If a non-fluorescent reporter is being modulated, changes in gene expression can also be measured by (1) an RNA expression analysis with reverse transcriptase qPCR or (2) protein expression with western blotting, or imaging and conducting flow cytometry with fluorescent secondary antibodies.
In relation to current CIP-based techniques like the CiA system12, this CEM technology has the advantage of recruiting endogenous epigenetic modulators to the target gene. Redirecting the cell's own machinery creates a more physiologically relevant means of modulating expression. Exogenously expressing master enzymes, like HATs and transcription factors, might result in side effects from their increased expression, especially while not being actively recruited to the gene locus.
While this article shows the functionality of this novel system with CEMs composed of HDAC inhibitors, several other inhibitors or protein recruiters can be synthesized in place of the HDAC inhibitor. To achieve gene activation, HAT inhibitor- or HDMT inhibitor-based CEMs can be synthesized. To repress and then overexpress the same gene, it is possible to treat the cells with a repressive CEM, wash them with regular media, and then add an activating CEM. This would allow for a clean system to study the dynamics of recruiting repressive and activating complexes to the same genomic region. When designing future CEMs, several characteristics need to be considered, including cell permeability, inhibitor potency, structure, and inhibitory kinetics. The linker length between FK506 and the recruiter moiety will influence the permeability of the CEMs, as well as the position of the recruited protein. When comparing two CEMs that differed only in linker length, the CEM with the shorter linker was more effective12. While it has not been tested directly here, the higher effectiveness is a result of greater cell permeability. Choosing inhibitors with chemical structures amenable to modification without disrupting the portion that binds the active site of the target is also important. The potency and specificity were also considered by selecting inhibitors with a high potency and specificity for a given class of proteins. The kinetics of inhibition may influence the effectiveness of the CEMs. CEMs comprised of inhibitors with slow on-off rates tend to be less effective.
One constraint of the current CEM technology is the requirement of a Gal4-binding array at the target gene locus. To recruit CEMs to a region other than the Oct4 CiA locus, any of the hundreds of engineered Gal4 upstream activation sequence (UAS) lines available to the scientific community can be used. One way the system will be made more modular is by incorporating a deactivated Cas9 (dCas9) with chemical epigenetic enzyme recruitment10,11,15. This nuclease-dead protein from the CRISPR-Cas9 system will still be recruited to any region of the genome to which a guide RNA (gRNA) is designed, but it will not cut the DNA. Using a dCas9-FKBP fusion, the FKBP component will recruit the CEMs where the dCas9 is recruited, greatly increasing the ease and flexibility of potential target genes. Imagine using this technology to target disease-relevant genes as a potential therapeutic or a means by which to reversibly and temporally study disease mechanisms at the chromatin level.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to thank the members of the Hathaway and Jin laboratories for their helpful discussions. The authors also thank Dan Crona and Ian MacDonald for their critical reading of the manuscript. This work was supported in part by Grant R01GM118653 from the U.S. National Institutes of Health (to N.A.H.); and by Grants R01GM122749, R01CA218600, and R01HD088626 from the U.S. National Institutes of Health (to J.J.). This work was also supported by a tier 3 and a student grant from the UNC Eshelman Institute for Innovation (to N.A.H and A.M.C, respectively). Additional funding from a T-32 GM007092 (to A.M.C) supported this work. Flow cytometry data was obtained at the UNC Flow Cytometry Core Facility funded by a P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center.
References
- Alabert C, Groth A. Chromatin replication and epigenome maintenance. Nature Reviews Molecular Cell Biology. 2012;13(3):153–167. doi: 10.1038/nrm3288. [DOI] [PubMed] [Google Scholar]
- Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nature Reviews Molecular Cell Biology. 2015;16(3):178–189. doi: 10.1038/nrm3941. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Research. 2011;21(3):381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräff J, Tsai L-H. Histone acetylation: molecular mnemonics on the chromatin. Nature Reviews Neuroscience. 2013;14(2):97–111. doi: 10.1038/nrn3427. [DOI] [PubMed] [Google Scholar]
- Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nature Reviews Molecular Cell Biology. 2015;16(4):258–264. doi: 10.1038/nrm3931. [DOI] [PubMed] [Google Scholar]
- Hathaway NA, et al. Dynamics and memory of heterochromatin in living cells. Cell. 2012;149(7):1447–1460. doi: 10.1016/j.cell.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Hathaway NA, Crabtree GR, Muegge K. Tethering of Lsh at the Oct4 locus promotes gene repression associated with epigenetic changes. Epigenetics. 2018;13(2):173–181. doi: 10.1080/15592294.2017.1338234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton BZ, Hodges C, et al. Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nature Genetics. 2017;49(2):282–288. doi: 10.1038/ng.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh AS, Miller EL, et al. Rapid chromatin repression by Aire provides precise control of immune tolerance. Nature Immunology. 2018;19(2):162–172. doi: 10.1038/s41590-017-0032-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Gao D, et al. Chemically Controlled Epigenome Editing through an Inducible dCas9 System. Journal of the American Chemical Society. 2017;139(33):11337–11340. doi: 10.1021/jacs.7b06555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun SMG, et al. Rapid and reversible epigenome editing by endogenous chromatin regulators. Nature Communications. 2017;8(1):560. doi: 10.1038/s41467-017-00644-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler KV, Chiarella AM, Jin J, Hathaway NA. Targeted Gene Repression Using Novel Bifunctional Molecules to Harness Endogenous Histone Deacetylation Activity. ACS Synthetic Biology. 2018;7(1) doi: 10.1021/acssynbio.7b00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasoji SK, et al. Cavitation Enhancing Nanodroplets Mediate Efficient DNA Fragmentation in a Bench Top Ultrasonic Water Bath. PLoS ONE. 2015;10(7):0133014. doi: 10.1371/journal.pone.0133014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarella AM, et al. Cavitation enhancement increases the efficiency and consistency of chromatin fragmentation from fixed cells for downstream quantitative applications. Biochemistry. 2018;57(19):2756–2761. doi: 10.1021/acs.biochem.8b00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, et al. Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nature Methods. 2016;13(12):1043–1049. doi: 10.1038/nmeth.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]