

Abstract
The human mitochondria possess a dedicated set of ribosomes (mitoribosomes) that translate 13 essential protein components of the oxidative phosphorylation complexes encoded by the mitochondrial genome. Since all proteins synthesized by human mitoribosomes are integral membrane proteins, human mitoribosomes are tethered to the mitochondrial inner membrane during translation. Compared to the cytosolic ribosome the mitoribosome has a sedimentation coefficient of 55S, half the rRNA content, no 5S rRNA and 36 additional proteins. Therefore, a higher protein-to-RNA ratio and an atypical structure make the human mitoribosome substantially distinct from its cytosolic counterpart.
Despite the central importance of the mitoribosome to life, no protocols were available to purify the intact complex from human cell lines. Traditionally, mitoribosomes were isolated from mitochondria-rich animal tissues that required kilograms of starting material. We reasoned that mitochondria in dividing HEK293-derived human cells grown in nutrient-rich expression medium would have an active mitochondrial translation, and, therefore, could be a suitable source of material for the structural and biochemical studies of the mitoribosome. To investigate its structure, we developed a protocol for large-scale purification of intact mitoribosomes from HEK cells. Herein, we introduce nitrogen cavitation method as a faster, less labor-intensive and more efficient alternative to traditional mechanical shear-based methods for cell lysis. This resulted in preparations of the mitoribosome that allowed for its structural determination to high resolution, revealing the composition of the intact human mitoribosome and its assembly intermediates. Here, we follow up on this work and present an optimized and more cost-effective method requiring only ~1010 cultured HEK cells. The method can be employed to purify human mitoribosomal translating complexes, mutants, quality control assemblies and mitoribosomal subunits intermediates. The purification can be linearly scaled up tenfold if needed, and also applied to other types of cells.
Keywords: Biochemistry, Issue 140, Mitochondria, translation, ribosome, cryo-EM, protein synthesis, biochemistry
Introduction
The process of mitochondrial protein synthesis is based on 13 essential mt-mRNAs that are translated by a specialized membrane-attached mitoribosome to form the catalytic core of the respiratory chain. The altered mitochondrial genomes and the need for the proteins to insert co-translationally into the membrane have substantially shaped the architecture of the mitochondrial ribosomes1. Recent high-resolution structures of the mammalian mitoribosome showed a strikingly different overall appearance to the bacterial counterpart2,3. Particularly, at least 36 mitochondria-specific proteins are added, contributing ~1 MDa extra molecular mass, while mt-rRNA is reduced by twofold and highly diverged. The structural rearrangements alter almost all critical functional features that were previously generally accepted to be universally conserved4.
New principal elements have been acquired by each one of the mitoribosomal subunits, for example, the small subunit has incorporated an intrinsic GTPase protein mS29 into its 'head' region. GTPase activity has not been found in other translation systems, andthe structure indicates that the GTPase might have a role in subunit assembly2. The 5S rRNA that was believed to be a landmark of all known ribosomal large subunits, forming the core of the central protuberance, is missing in the mammalian mitoribosome and has adopted mt-tRNA-Val as an integral building block instead2. Ban and colleagues showed that the porcine mitoribosome has mt-tRNA-Phe and not –Val5. Chrzanowska-Lightowlers, Minczuk, and colleagues followed up on these data and found that mitoribosomes from patients with compromised mt-tRNA-Val stability can in principle accommodate mt-tRNA-Phe6,7. Why the mitoribosomes have incorporated these specific elements and what pathways and trans-factors are required for these unique assemblies remain unknown.
Overall, the high complexity of the human mitoribosome, new protein components, and the unique association of the mt-tRNA as a structural element imply the involvement of as-yet-unknown mitochondria-specific trans-factors. However, because many of the features of this system are unique to mitochondria, which have traditionally been difficult to investigate8, little is known about the molecular and quality control mechanism. With the development of high-resolution single particle analysis by electron cryo-microscopy (cryo-EM)20, opportunities now arise to comprehensively study the molecular mechanisms underlying the assembly, action and quality control of the human mitoribosome. Our report of the first structure of the human mitoribosome assembly intermediate provides the recognition that it is possible to visualize how the human mitoribosome is formed and shows that cryo-EM is instrumental in the identification of new trans-acting assembly factors9.
To expand on this initial effort, we describe a rapid protocol for human mitoribosome purification in detail. In the first part of the protocol, a large-scale isolation of highly pure intact mitochondria from suspension cells is described. This procedure requires 9 h and can be easily modified and adapted to different cell types and scales. A significant step in this protocol is the use of nitrogen cavitation to break open the cells. The second part of the protocol was developed to purify mitoribosomes. This procedure requires 7 h and yields a sufficient amount of mitoribosomes for biochemical and structural studies. Use of pure mitochondria as a starting material offers high-quality final preparations and can be extrapolated to other mitochondrial macromolecules.
Protocol
All mammalian cell culture work must be performed inside a biological safety cabinet. Use sterile equipment if contact with cells. Wear nitrile gloves and a lab coat and follow good tissue culture practice.
1. Cell Culture
Maintain HEK293S suspension cells in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% tetracycline-free fetal bovine serum (FBS), 5 μg/mL blasticidin and 200 μg/mL Azithromycin, at 37 °C and 5% CO2.
Scale up to 9 x T175 flasks. At 90% confluency, harvest cells and spin them down at 500 x g for 5 min. Resuspend the pelleted cells in Freestyl supplemented with 5% tetracycline-free FBS. Count cells using an automated cell counter and adjust cell concentration to 1.5 x 106 cells/mL in a vented shaking flask. Note: This will typically correspond to a starting volume of 300 mL in a 1,000 mL vented flask.
Incubate cells in a shaking incubator at 37 °C and 5% CO2 at 120 rpm (50 mm shaking diameter).
After 2 days count the cells and proceed to split the cells if the cell density is above 3.0 x 106 cells/mL. Split the cells by spinning down 2 x 150 mL cell culture in 2 x 250 mL conical bottles at 500 g for 5 min. Resuspend cells in 2 x 10 mL fresh media and transfer to a 2 x 1, 000 mL vented flasks containing the appropriate volume for a final cell density of 1.5 x 106 cell/mL, e.g. 2 x 300 mL from a starting cell density of 3.0 x 106 cells/mL.
Incubate cells in a shaking incubator at 37 °C and 5% CO2 at 120 rpm (50 mm shaking diameter).
After two days count the cells and proceed to split the cells if the cell density is above 3.0 x 106 cells/mL. Split the cells by spinning down 4 x 150 mL cell culture in 4 x 250 mL conical bottles at 500 g for 5 min. Resuspend cells in 2 x 10 mL fresh media and transfer to 2 x 2,000 mL vented flask containing the appropriate volume for a final cell density of 1.5 x 106 cell/mL, e.g. 2 x 700 mL from a starting cell density of 3.0 x 106 cells/mL.
Incubate cells in a shaking incubator at 37 °C and 5% CO2 at 120 rpm (50 mm shaking diameter).
After two days count the cells and proceed to split the cells if the cell density is above 3.0 x 106 cells/mL. Split the cells by pouring the 2 x 700 mL cell culture into 2 x 2800 mL vented flasks and topping up with 2 x 300 mL fresh media to reach a volume of 2 x 1 L.
Incubate cells in a shaking incubator at 37°C and 5% CO2 at 120 rpm (50 mm shaking diameter).
After 24 h count the cells and harvest the cells if the cell density is between 3.0 - 4.0 x 106 cells/mL.
2. Mitochondria Isolation
- Buffers required Note: The quantities are given for a preparation from 2 L cells. Prepare the following stock solutions ahead of time for mitochondrial isolation buffer (MIB), sucrose/mannitol buffer (SM4), experimental buffer (MIBSM), resuspension buffer (SEM).
- Make 0.5L MIB buffer: 50mM HEPES-KOH, pH 7.5, 10 mM KCl, 1.5mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1mM DTT, protease inhibitors.
- Make 100 mL SM4 buffer: 280 mM sucrose, 840 mM mannitol, 50 mM HEPES-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1m M DTT, protease inhibitors.
- Make 160 mL MIBSM buffer: 120 mL MIB buffer + 40 mL SM4 buffer.
- Store 200 mL PBS at 4 °C.
- Make 5 mL SEM buffer: 250 mM sucrose, 20 mM HEPES-KOH, pH 7.5, 1 mM EDTA.
- Make 4 x 10 mL stock solutions for the stepwise sucrose gradient containing 20 mM HEPES-KOH, pH 7.5, 1 mM EDTA and 60% / 32% /23% and 15% sucrose, respectively.
Mitochondria isolation Note: It is important to work quickly and keep everything on ice throughout the procedure.
- Precool the nitrogen cavitation chamber prior to use.
- Harvest 2 x 1 L cells (3-4 x 109 cells per centrifuge tube) by centrifugation at 1,000 x g for 7 min, 4 °C.
- Decant the supernatant carefully and resuspend the pelleted cells quickly in 2 x 100 mL PBS. Pool the cells.
- Centrifuge the resuspended cells at 1,200 x g for 10 min, 4 °C.
- Decant the supernatant carefully and weigh the pellet (~20 g).
- Resuspend the pellet in 120 mL MIB buffer.
- Allow cells to swell by gently stirring in a cold room for 10 min.
- Place the nitrogen cavitation chamber on the ice and transfer the swelled cells to the nitrogen cavitation chamber. Add 45 mL SM4 buffer (1/3 of the final volume of resuspended cells, about 140 mL, yielding a final concentration of 70 mM sucrose and 210 mM mannitol).
- Fasten the nitrogen cavitation chamber and fill with nitrogen until the pressure reaches 500 psi. Close taps and keep isolated on ice for 20 min.
- Slowly release the pressure in the nitrogen cavitation chamber and collect the lysate (approximately 185 mL).
- Centrifuge the lysed material to remove the cell debris and nuclei at 800 x g for 15 min, 4 °C.
- Collect the supernatant by pouring it through the cheesecloth into a beaker kept on ice. Do not discard the pellet.
- Resuspend the pellet in 90 mL MIBSM buffer (1/2 the previous volume). Homogenize using Teflon/glass Dounce homogenizer and repeat the centrifugation at 800 x g for 15 min, 4 °C.
- Collect the supernatant by pouring it through the cheesecloth into a beaker kept on ice. Combine this supernatant with the supernatant from step 2.2.11.
- Centrifuge the combined supernatants at 1,000 x g for 15 min, 4 °C.
- Collect the supernatant by pouring it through the cheesecloth into a beaker kept on ice.
- Discard the pellet and centrifuge the supernatant containing crude mitochondria at 10,000 x g for 15 min, 4 °C. Note: The pellet will typically consist of two parts: loose and tight.
- Carefully wash out the loose pellet without disturbing the tight portion. Resuspend the tight pellet in 10 mL MIBSM buffer.
- Perform a protein concentration assay using a commercial Protein Assay Kit or similar method. Typical concentration yields from 2 L starting culture are ~2 mg/mL.
- Add 200 Units of RNase free DNase and leave to rotate on a roller in the cold room for 20 min to evenly mix the DNase for removal of the genomic DNA.
- Centrifuge at 10,000 x g for 15 min, 4 °C and resuspend the pellet in 2 mL SEM buffer. Homogenize gently using a small Teflon/glass Dounce homogenizer to resuspend any remaining aggregates. Perform not more than five up-and-down passes to avoid breakage. Keep on ice.
- Prepare the sucrose gradient in 14 mL SW40 tubes by carefully pipetting 1.5 mL of 60% sucrose stock buffer into the bottom of the tube. Carefully add 4.5 mL of the 32% sucrose stock buffer on top of the 60% band without disturbing it. Repeat with 1.5 mL of 23% sucrose stock buffer and again with 1.5 mL of 15% sucrose stock buffer.
- Load the entire mitochondrial suspension (approximately 3 mL) on top of the sucrose gradient.
- Centrifuge in SW40 rotor at 139, 065 x g for 60 min.
- Carefully collect the brown band migrating to the interface of 32% and 60% sucrose using a transfer pipette, typically 2-3 mL.
- Snap-freeze the purified mitochondria in liquid nitrogen and store at -80 °C.
3. Mitoribosome Preparation
- Buffers required Note: Prepare the following stock solutions ahead of time for the lysis buffer, sucrose cushion/gradient, and resuspension buffer.
- Make 10 mL lysis buffer: 25 mM HEPES-KOH, pH 7.5, 150 mM KCl, 50mM MgOAc, 2% Polyethylene glycol octylphenyl ether, 2 mM DTT, protease inhibitors.
- Make 10 mL sucrose cushion buffer: 1 M sucrose (34% w/v), 20 mM HEPES-KOH, pH 7.5, 100 mM KCl, 20 mM MgOAc, 1% Polyethylene glycol octylphenyl ether, 2 mM DTT.
- Make 5 mL resuspension buffer: 20 mM HEPES-KOH, pH 7.5, 100 mM KCl, 20mM MgOAc, 2 mM DTT
- Make 15%-30% linear sucrose gradients with resuspension buffer in TLS-55 polycarbonate tubes.
- Mitoribosome purification
- Defrost the frozen mitochondria on ice.
- Add 2 volumes of the lysis buffer,e.g. add 6 mL lysis buffer to 3 mL mitochondria. Mix immediately by inverting the tube several times.
- Homogenize with a small Teflon/glass dounce homogenizer to assist the lysis and incubate for 5-10 min on ice to complete the lysis.
- Centrifuge the lysed material (approximately 9 mL) at 30,000 x g for 20 min, 4 °C to remove the insoluble material. Decant the supernatant carefully from the pellet and discard the pellet.
- Repeat the centrifugation at 30,000 x g for 20 min at 4 °C to ensure clarification of the supernatant. Decant the supernatant carefully from the pellet and discard the pellet.
- Prepare the sucrose cushion in TLA 120.2 tubes (ultra-clear): 0.4 mL sucrose cushion per tube. Prepare one tube per mL of lysed material.
- Layer lysed mitochondria on the sucrose cushions: approximately 1 mL per tube, resulting in a lysate:cushion ratio of 2.5:1.
- Centrifuge the sample at 231, 550 x g for 45 min in TLA120.2 rotor at 4 °C.
- Discard the supernatant and rinse the tubes sequentially with 100 µl of the resuspension buffer to remove residual sucrose.
- Resuspend the pellets in total 100 µl resuspension buffer.
- Vortex on slow speed for 30 s to dissolve the remaining aggregates and centrifuge at 17,949 x g for 10 min in microtube centrifuge at 4 °C.
- Carefully collect the supernatant and repeat the centrifugation.
- Measure mitoribosome absorption at A260. Note: The typical yield is 7 A260, with A260: A280 ratio of 1.3.
- Load the entire sample onto a single linear sucrose gradient tube. Centrifuge in TLS-55 rotor at 213, 626 x g for 120 min at 4 °C.
- Fractionate the gradient, determine the optical density at A260 and pool the fraction corresponding to the nucleic acid peak together. Note: The typical A260: A280 ratio of the peak is >1.6.
- Exchange the buffer if necessary, using a method of choice. Calculate the final concentration by using the conversion 1 A260 = 0.1 mg/mL.
- Snap freeze the purified mitoribosome sample in the resuspension buffer and store at -80 °C.
Representative Results
Dividing and highly viable cells is an essential starting point for purification of active mitoribosomes. This protocol is applicable to any HEK293 suspension cells. We are using in-house cell line T501, which is stably expressing a transporter under tetracycline-inducible control. The parental cell line is HEK293S-GnTI- cells (Table of Materials)10. During the cell growth and expansion in the FreeStyle 293 Expression Medium the minimal density should be kept at 1.5 x 106 cell/mL, in order to ensure a doubling rate of every two days, whereas the maximal cell density should not exceed ~5 x 106 cell/mL. Upon reaching a cell density above 3 x 106 cell/mL the cells are pelleted and resuspended in a fresh, preheated media in an expanded volume to achieve the minimum cell density of 1.5 x 106 cell/mL. This splitting is performed repeatedly every 2-3 days until a desired cell mass for the procedure is achieved. The final cell density can vary in the range of 3-4.5 x 106 cell/mL, and at least 2 L is required as a starting point for the isolation of mitochondria. The cell viability should be generally maintained >90%, and for the final culture that is harvested >95%. Treatment of the large-scale cell culture with antibiotics is not recommended.
After the series of differential centrifugations, the volume of the mitochondrial suspension after step 2.2.20 is typically in the range of 3-5 mL. The mitochondria are then separated on the sucrose gradient (Figure 1) from other organelles. The stepwise sucrose gradient is prepared such that the volume of the gradient and the volume of the mitochondrial suspension together fill up the centrifugation tube to its maximal volume. Here special care is needed to collect the brown band migrating to the 60% / 32% interface with minimal contamination from the surrounding buffer. This is important in order to keep constant protein:detergent ratio in the following step of mitochondria solubilization. It is advised to assess the mitochondrial protein concentration at this stage, and a typical yield of 15-20 mg total mitochondrial protein is expected from ~1010 cultured HEK cells.

Upon successful mitochondria isolation, they are lysed by the addition of polyethylene glycol octylphenyl ether, and mitoribosomes are separated through a sucrose cushion (Figure 2). To separate mitoribosomes from the hydrophobic membranous substance, pellets are resuspended in the buffer not containing detergent, and hydrophobic complexes are pelleted by centrifugation. This procedure is repeated, and the purification degree of mitoribosomes is quantified by A260/A280 ratio, which is expected to be ~1.3. Typical yield is 7 A260 from a 2 L starting culture. This mitoribosomal fraction also contains additional large soluble mitochondrial complexes, such as pyruvate dehydrogenase and glutamate dehydrogenase. To separate mitoribosomes from the soluble mitochondrial complexes, the supernatant is applied to a sucrose density gradient. Fractions containing the mitoribosome are generally located at the bottom third of the tube. The fractionation of the sucrose gradient can then be done either with an automated piston or manually by carefully taking 50 µL fractions with a pipette or by punching the tube bottom with a 21 G needle and collecting the drops.

Two major mitoribosomal populations are identified in the gradient: monosome 55S and large subunit 39S, as illustrated in Figure 3 and Figure 4. The presence of the large subunit fraction suggests that cells are harvested in a highly active dividing state11. The ratio between the monosome and large subunit peaks may change. An additional peak located closer to the bottom may appear in preparations with contaminating cytoplasmic ribosomes 80S. Please note that the small sucrose gradient using the swinging bucket rotor TLS-55 allows for the rapid purification of ~1 mL of mitoribosomes at an optical density of 0.4-1 A260. The separation of the monosome and large subunit peaks may vary slightly depending on the fractionation, but there is usually some overlap of the two peaks. Hence, which fractions to collect, and pool should be taken into consideration in order to ensure the highest proportion of monosome, or alternatively large subunit, in the sample. For high-resolution cryo-EM studies, the separation between the two mitoribosomal populations is not absolutely required (due to additional in silico operations). However, if better separation is necessary, it is recommended to use larger tubes and corresponding running times.
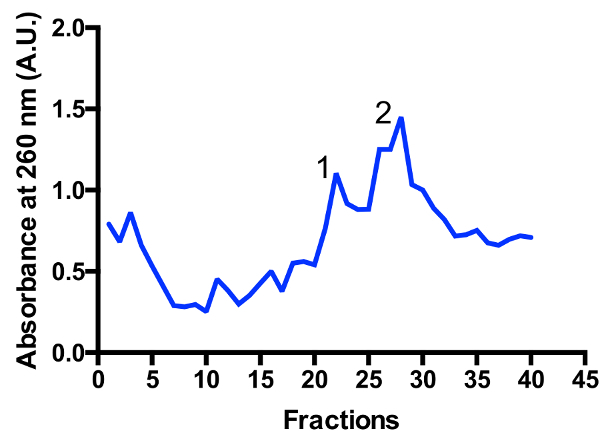

Figure 1: Purification of mitochondria on a sucrose gradient. Subcellular organelles from a 2 L starting culture were fractionated through a series of differential centrifugations as described in the protocol, and mitochondria were separated on a discontinuous sucrose gradient. The purified mitochondria are found in the lower band at the 32%/60% interface.
Figure 2: Purification of mitoribosomes on a sucrose cushion. Crude mitoribosomes from a 2 L starting culture are sedimented through 1 M sucrose cushion. The pellets are resuspended in a detergent-free buffer, and mitoribosomes are clarified by two centrifugations as described in the protocol. The absorbance is recorded to assess the quality of the preparation and typical A260/A280 ratio of ~1.3 (right panel) certifies a mitoribosome rich fraction. Please click here to view a larger version of this figure.
Figure 3: Fine purification of mitoribosomes on a sucrose gradient. Absorbance trace from a 2 L starting culture. Fractions are numbered from the top to the bottom of the gradient. Two major mitoribosomal populations are identified: large subunit (peak 1) and monosome 55S (peak 2). The ratio between the populations may change. Please click here to view a larger version of this figure.
Figure 4: Electron micrograph, 2D classes, and 3D reconstruction.Left panel: a micrograph with the sample from the monosome peak 2 at a calibrated magnification of 1.23 A/pixel. Middle panel: A representative post-processing data (2D classes) revealing intact monosomes. Right panel: 3D reconstruction. Please click here to view a larger version of this figure.
Discussion
Regarding the source of starting material, although the relatively easy availability of animal tissues that are known to be a rich source of mitochondria, makes it a popular choice for mitoribosomes3,5,12,13, they cannot be genetically modified and reproduced in the lab easily. Hence, there is a clear practical need for developing protocols involving homogeneously cultured human cell lines. The major difference between the protocols dealing with tissues and cultured cells is the mode of lysis and homogenization. For cultured cells grown as a monolayer, a typical method is Teflon/glass dounce homogenization14. The original preparation protocols while efficient were developed for small scales15. A direct scaling up employing a homogenizer with the capacity of 500 mL is possible11, however, it requires ~2 h of manual labor to achieve 80% lysis. This introduced at least three issues: aggregation of organelles due to long waiting times, non-homogeneous lysis due to heavy material sinking to the bottom of a large vessel, heating of the sample due to the necessity of multiple strokes. Therefore, a preferable method of lysis utilizes nitrogen cavitation, which is based on decompression from a pressurized vessel16,17. In this method, cells are first swelled in the cold room in order to soften the cells and make them more susceptible to lysis. As they are placed in the nitrogen cavitation device a buffer containing sucrose and mannitol is added in order to maintain an osmotic pressure that will help keep mitochondria intact. The nitrogen cavitation device is then pressurized with a large volume of oxygen-free nitrogen, which dissolves into the cells. As the pressure is released nitrogen bubbles out of solution resulting in rupturing of the cell membranes. This method offers several advantages over mechanical homogenizing methods involving shear stresses and friction as follows: 1) any external physical stress on the cells is avoided; 2) an adiabatic expansion that cools the sample, ensures no heat damage to organelles; 3) cell components are protected from oxidation by the inert nitrogen gas; 4) no pH alteration of the suspending medium; 5) the process is uniform and reproducible because the same disruptive forces are applied within each cell and throughout the sample; 6) the process is fast and can be completed within 20-30 min.
Isolation of mitochondria from cultured cells and tissues has been described in the literature extensively, and it is based on a gentle cell disruption followed by a series of differential centrifugations. Most of the currently used protocols follow the original procedures developed in the middle of the previous century18. While the basic biochemical approaches are correct, there are several misconceptions that are highlighted in the literature and remained undetected. To optimize the protocol for mitoribosomes, we systematically investigated the reported general principles and conclude that: 1) Inclusion of Mg2+ and K+ in the buffer is not crucial. It has been argued that the KCl helps to prevent cytoplasmic proteins from forming a gel19, however, provided a sufficient dilution as described in our protocol, this phenomenon does not occur. Furthermore, exclusion of Mg2+ is useful to reduce contaminations by cytoplasmic ribosomes20; 2) There is no need to keep maximal possible volume ratio of cells to medium to protect the released organelles from the hypo-osmotic environment. The osmotic support in our protocol is sufficiently provided by sucrose-containing buffers, and the dilution of cells with mitochondria isolation buffer (step 2.II.5) is critical for the efficient separation of organelles in a large-scale preparation.
The pH of mitochondria isolation buffer is close to physiological values, i.e. 7.5, which is also the optimal pH for the following mitoribosome preparation. Binding agents EDTA and EGTA are added to the isolation media to chelate contaminating ions, and they chelate free magnesium and calcium, respectively. It has been discussed that inclusion of EDTA can lead to the damage of the inner mitochondrial membrane14, therefore the concentration is limited to 1 mM as a precaution. We have not observed any difference when using 5 mM EDTA in the final purification step of sucrose density gradient.
The protocol described herein uses HEK293S-derived human cells for purification of the mitoribosome. The quality of the preparation allows the obtained sample to be investigated biochemically and structurally to atomic resolution. This allows one to apply the method to human mitoribosomal translating complexes, quality control assemblies, and mitoribosomal subunits intermediates. Moreover, since cancer cells have amplified OXPHOS capacity and elevated mitochondrial protein translation compared with adjacent stromal tissue21, mitoribosomes are established drug targets for cancer22. Therefore, using this protocol for specific purification of mitoribosomes in presence of inhibitors will have medical applications. Also, mitoribosomal mutations have been linked to hereditary mitochondrial diseases23. Since these mutations have direct effects on the structure, the approach presented here will be useful for their structural characterization. The protocol can be experimentally expanded and applied to a variety of scientific questions tackling the fundamental understanding of translation in human mitochondria and its medical importance.
Disclosures
None
Acknowledgments
This work was supported by the Swedish Foundation for Strategic Research (Future Leaders Grant FFL15:0325), Ragnar Söderberg Foundation (Fellowship in Medicine M44/16), Swedish Research Council (Starting Grant NT×2015-04107), FEBS Long-Term Fellowship (SA), H2020-MSCA-ITN-2016 project 721757 (VS).
References
- Ott M, Amunts A, Brown A. Organization and regulation of mitochondrial protein synthesis. Annual review of biochemistry. 2016;85:77–101. doi: 10.1146/annurev-biochem-060815-014334. [DOI] [PubMed] [Google Scholar]
- Amunts A, Brown A, Toots J, Scheres SH, Ramakrishnan V. The structure of the human mitochondrial ribosome. Science. 2015;348(6230):95–98. doi: 10.1126/science.aaa1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber BJ, Bieri P, Leibundgut M, Leitner A, Aebersold R, Boehringer D, Ban N. The complete structure of the 55S mammalian mitochondrial ribosome. Science. 2015;348(6232):303–308. doi: 10.1126/science.aaa3872. [DOI] [PubMed] [Google Scholar]
- Greber BJ, Ban N. Structure and function of the mitochondrial ribosome. Annual review of biochemistry. 2016;85:103–132. doi: 10.1146/annurev-biochem-060815-014343. [DOI] [PubMed] [Google Scholar]
- Greber BJ, Boehringer D, Leitner A, Bieri P, Voigts-Hoffmann F, Erzberger JP, Ban N. Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature. 2014;505(7484):515–519. doi: 10.1038/nature12890. [DOI] [PubMed] [Google Scholar]
- Rorbach J, Gao F, Powell CA, D'Souza A, Lightowlers RN, Minczuk M, Chrzanowska-Lightowlers ZM. Human mitochondrial ribosomes can switch their structural RNA composition. Proceedings of the National Academy of Sciences. 2016. p. 201609338. [DOI] [PMC free article] [PubMed]
- Chrzanowska-Lightowlers Z, Rorbach J, Minczuk M. Human mitochondrial ribosomes can switch structural tRNAs-but when and why? RNA biology. 2017;14(12):1668–1671. doi: 10.1080/15476286.2017.1356551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammage PA, Moraes CT, Minczuk M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends in Genetics. 2017. [DOI] [PMC free article] [PubMed]
- Brown A, Rathore S, Kimanius D, Aibara S, Bai XC, Rorbach J, Ramakrishnan V. Structures of the human mitochondrial ribosome in native states of assembly. Nature Structural and Molecular Biology. 2017;24(10):866. doi: 10.1038/nsmb.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves PJ, Callewaert N, Contreras R, Khorana HG. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogenous N-glycosylation by tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proceedings of National Academy of Sciences USA. 2002;99:13419–13424. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Amunts A, Bai XC, Sugimoto Y, Edwards PC, Murshudov G, Ramakrishnan V. Structure of the large ribosomal subunit from human mitochondria. Science. 2014;346(6210):718–722. doi: 10.1126/science.1258026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien TW, Kalf GF. Ribosomes from rat liver mitochondria I. Isolation procedure and contamination studies. Journal of Biological Chemistry. 1967;242(9):2172–2179. [PubMed] [Google Scholar]
- Spremulli LL. Large-scale isolation of mitochondrial ribosomes from mammalian tissues. Mitochondria: Practical Protocols. 2007. pp. 265–275. [DOI] [PubMed]
- Rice JE, Lindsay JG. Subcellular fractionation of mitochondria. In: Grahamand JM, Rickwood D, editors. Subcellular Fractionation: A Practical Approach. Oxford: IRL Press; 1997. pp. 107–142. [Google Scholar]
- Attardi G, Ching E. Biogenesis of mitochondrial proteins in HeLa cells. Methods in enzymology. 1979;56:66–79. doi: 10.1016/0076-6879(79)56010-8. [DOI] [PubMed] [Google Scholar]
- Gottlieb RA, Adachi S. Nitrogen cavitation for cell disruption to obtain mitochondria from cultured cells. Methods in enzymology. 2000;322:213–221. doi: 10.1016/s0076-6879(00)22022-3. [DOI] [PubMed] [Google Scholar]
- Simpson RJ. Disruption of cultured cells by nitrogen cavitation. Cold Spring Harbor Protocols. 2010;2010(11) doi: 10.1101/pdb.prot5513. [DOI] [PubMed] [Google Scholar]
- Kennedy EP, Lehninger AL. Oxidation of fatty acids and tricarboxylic acid cycle intermediates by isolated rat liver mitochondria. Journal of Biological Chemistry. 1949;179(2):957–972. [PubMed] [Google Scholar]
- Graham JM. Isolation of mitochondria from tissues and cells by differential centrifugation. Curr Protoc Cell Biol. 2001. Chapter 3, Unit 3.3, [DOI] [PubMed]
- Amunts A, Brown A, Bai XC, Llácer JL, Hussain T, Emsley P, Ramakrishnan V. Structure of the yeast mitochondrial large ribosomal subunit. Science. 2014;343(6178):1485–1489. doi: 10.1126/science.1249410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annual Review of Pathology: Mechanisms of Disease. 2012;7:423–467. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- Škrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Lai CK. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer cell. 2011;20(5):674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boczonadi V, Horvath R. Mitochondria: impaired mitochondrial translation in human disease. The international journal of biochemistry & cell biology. 2014;48:77–84. doi: 10.1016/j.biocel.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]