
Figure 2.
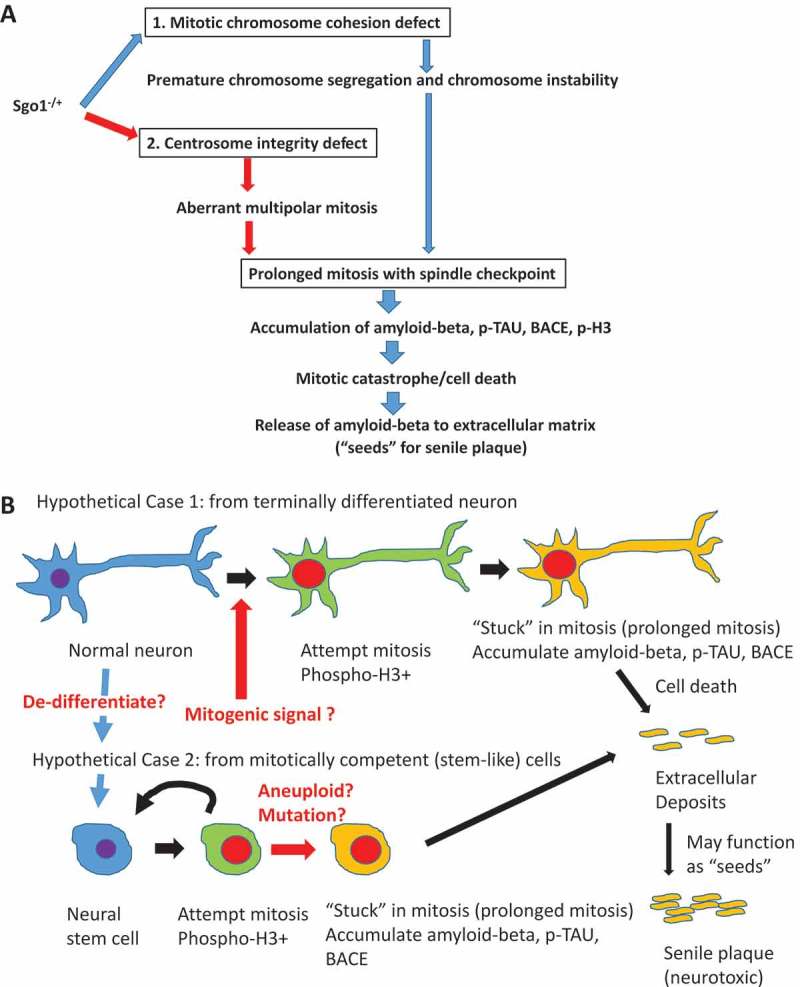
Critical role of prolonged mitosis in amyloid-beta accumulation, suggested by Sgo1−/+ model. (a) Two consequences of Sgo1−/+ with common destinationSgo1−/+ induces two defects in the cell cycle; 1) Mitotic chromosome cohesion defect followed by premature chromosome segregation and chromosome instability, and 2) Centrosome integrity defect followed by aberrant multipolar mitosis. Both defects provoke the mitotic spindle checkpoint that leads to prolonged mitosis. Amyloid-beta (and p-TAU and BACE) accumulation occurred in mitotic marker p-H3-positive, live cells. We propose that the amyloid-beta accumulating cells in prolonged mitosis state eventually die of mitotic catastrophe, releasing amyloid-beta to extracellular matrix. As amyloid-beta possesses prion-like self-aggregating property, the released amyloid-beta may serve as “seeds” for further aggregation and eventual development of neurotoxic senile plaques. (b) “Mitotic origin of deposits” model. Whether amyloid-beta accumulating cells are originated from (case1) terminally differentiated neuron or (case 2) mitotically competent (stem-like) cells has not been determined. In “Hypothetical Case 1: from terminally differentiated neuron”, terminally differentiated neuronal cells de-differentiate to stem-like cells, or receive mitogenic signal and re-enter mitotic cycle. In “Hypothetical Case 2: from mitotically competent (stem-like) cells”, mitotically-competent stem (-like) cells “got stuck in mitosis” possibly due to aneuploidy or other mutation/stimulus that provokes spindle checkpoint leading to mitotic prolongation. In either case, cells in the state of prolonged mitosis are the source of amyloid-beta accumulation. When these cells die (of mitotic catastrophe), the accumulated amyloid-beta is released to extracellular matrix, followed by senile plaque development.