

Summary
At early stages of organismal development, endothelial cells self-organize into complex networks subsequently giving rise to mature blood vessels. The compromised collective behavior of endothelial cells leads to the development of a number of vascular diseases, many of which can be life-threatening. Cerebral cavernous malformation is an example of vascular diseases caused by abnormal development of blood vessels in the brain. Despite numerous efforts to date, enlarged blood vessels (cavernomas) can be effectively treated only by risky and complex brain surgery. In this work, we use a comprehensive simulation model to dissect the mechanisms contributing to an emergent behavior of the multicellular system. By tightly integrating computational and experimental approaches we gain a systems-level understanding of the basic mechanisms of vascular tubule formation, its destabilization, and pharmacological rescue, which may facilitate the development of new strategies for manipulating collective endothelial cell behavior in the disease context.
Subject Areas: Biological Sciences, Biomechanics, Biophysics, Cell Biology
Graphical Abstract
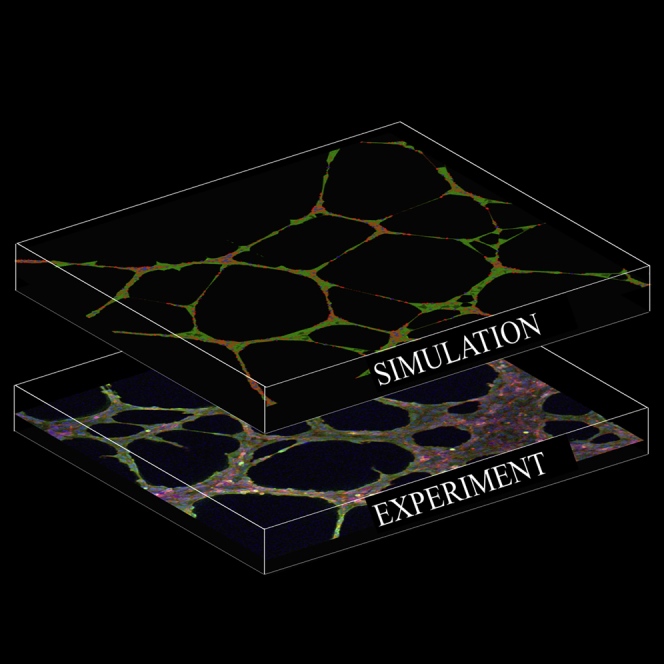
Highlights
-
•
A biophysical model reveals the differential effects of CCM proteins on cell behavior
-
•
CCM proteins are critical for the balance of cell-cell and cell-matrix interactions
-
•
Altered cell biomechanics explains the limited phenotype rescue by ROCK inhibition
-
•
Knockdown of CCM3 expression leads to unique defects in the actomyosin organization
Biological Sciences; Biomechanics; Biophysics; Cell Biology
Introduction
Collective cell behavior is a hallmark of organismal development, tissue regeneration, and disease progression. Among the most illustrious examples of this phenomenon are collective cell migration, which underlies embryonic development (Scarpa and Mayor, 2016) and cancer spreading (Mayor and Etienne-Manneville, 2016), and multicellular self-organization at the early stages of embryogenesis, when groups of vascular precursor cells give rise to the vascular tree (Betz et al., 2016). Coordinated behavior in multicellular clusters is dependent on the characteristics of individual cells, their active communication with neighbors, and interactions with the extracellular matrix (ECM).
When studying collective cell behavior, understanding the complex interplay of multiple parameters that change simultaneously across different spatial and temporal scales often requires computational modeling. Indeed, conceptual models and verbal reasoning cannot predict the behavior of complex systems with many interacting components. In contrast, holistic simulations models informed and validated by experimental data enable systems-level analysis through the predict-test-refine cycle. Here we designed a simulation model that explicitly incorporates cytoskeletal stiffness and protrusive activity of individual cells, as well as forces developed through cell-cell and cell-matrix interactions, into the morphodynamic context. Using this model, we gain insights into the emergence of multicellular structures at the initial stages of vasculogenesis and establish the effects of CCM protein deficiency on the integrity of the endothelial tubule networks.
Cerebral cavernous malformations (CCMs) are common vascular lesions that predispose affected individuals to brain hemorrhage. CCM disease is characterized by the presence of aberrantly enlarged “cavernous” endothelial channels found exclusively in the central nervous system. Genetic studies identified that CCMs result from homozygous inactivating mutations in one of three genes, krit1 (ccm1), ccm2, or pdcd10 (ccm3) (Pagenstecher et al., 2009). Products of these genes, CCM proteins, form a complex involved in the regulation of cytoskeletal dynamics through controlling RhoA function (Fischer et al., 2013). An increase in RhoA activity is a signature feature of CCM lesions at the molecular level. It was shown that pharmacological inhibition of RhoA decreases vascular permeability, improves vascular stability in vitro and in vivo, and decreases the number of lesions in KRIT1- and CCM2-deficient animals (Borikova et al., 2010, Stockton et al., 2010, McDonald et al., 2012).
Several studies identified additional mechanisms contributing to the development of CCM, such as endothelial-to-mesenchymal transition (Maddaluno et al., 2013), down-regulation of Notch signaling (Schulz et al., 2015), and KLF4 signaling (Zhou et al., 2016). Although dysregulation of RhoA activity appears central for disease development, an increasing volume of evidence indicates that, in addition to forming a complex that controls RhoA, CCM proteins have non-overlapping functions (Fisher and Boggon, 2014). These independent characteristics of KRTI1, CCM2, and PDCD10 may explain documented differences in clinical phenotypes caused by mutations in either of the genes (Shenkar et al., 2015). Recently it has been proposed that loss of KRIT1, CCM2, or PDCD10 gives rise to tubular structures with unique phenotypes, which are attributed not only to increased cellular stiffness but also to an imbalance of cell-cell (on PDCD10 loss) or cell-ECM adhesion (on KRIT1 loss) (Beltran et al., 2018).
Here, by using an integrative approach that couples computational modeling and experimental characterization, we report the biomechanical factors that contribute to the observed CCM phenotypes and its limited rescue by ROCK inhibition, which further expands our understanding of non-overlapping functions of ccm genes and advances the general knowledge of vascular tubule formation.
Results
Inhibition of ROCK Does Not Fully Restore Endothelial Tubule Formation in Cells with CCM Expression Knockdown
Knockdown of either of CCM protein expression disrupts endothelial tubule formation on Matrigel (Borikova et al., 2010). In addition, previous studies indicated that inhibiting ROCK function effectively increases mean tubule length thus restoring vascular networks in endothelial cell cultures with knockdown of CCM protein expression (Borikova et al., 2010). However, the visual appearance of cellular structures on pharmacological inhibition of ROCK activity by H1152 does not closely resemble the wild-type (WT) patterns. Here, we aimed to quantitatively evaluate this difference in the patterns of treated and untreated endothelial cells with and without CCM knockdown. To this end, we transduced HUVEC cells with lentiviral particles carrying shRNAs or transfected them with siRNA against krit1, ccm2, and pdcd10 genes (see Figure S1) before plating on an 800-μm-thick layer of Matrigel.
Consistent with previously published work, tubule patterns generated by either of the CCM protein KD cells were distinct from those in WT cultures and could be easily distinguished from each other (Figure 1A, top panels). As expected, treatment with H1152 resulted in a partial rescue of vascular networks of KD cells (Figure 1A, lower panels).
Figure 1.

Inhibition of Rho Kinase Leads to Incomplete Rescue of Vascular Tubule Defects in Endothelial Cell Cultures with Loss of KRIT1, CCM2, and PDCD10
(A) Representative images of vascular tubules generated by HUVEC cells infected with control PLKO.1 (WT) or shRNA carrying lentiviral particles (see also Figure S1). Cells with knockdown of KRIT1, CCM2, or PDCD10 expression were plated on growth factor-reduced Matrigel in the presence or absence of 1 μM of H1152, fixed at 18 hr after plating, and stained with rhodamine-phalloidin to visualize actin cytoskeleton.
(B) Results of morphometric analysis of the tubule network integrity. Void area (in pixels) measures the area of the plate not covered by cells. Mean loop count measures the number of background regions enclosed by interconnected cells. When considering both measures, all knockdown cultures and all treatment cultures are significantly different from WT cultures (p < 0.05). Data are represented as mean ±2·SEM.
To obtain quantitative measures of the differences between the CCM knockdown tubule patterns, and a degree of “rescue” in ROCK inhibitor treated cultures, we performed morphometric analysis of tubules formed in the absence or presence of H1152 in the media at the time of cell plating. The measurements included the void area, the number of background pixels corresponding to the area of the substrate not covered by cells, and the mean loop count, the number of isolated regions in the dish enclosed by cells (Figure 1B).
Quantitative analysis showed that all CCM knockdown cultures had a significantly increased void area, as compared with WT endothelial cell (EC) tubules (Table S1). For KRIT1 and PDCD10 knockdown cultures, void areas were approximately 10% larger and CCM2 approximately 5% larger than in WT cultures. The mean loop count was significantly decreased for KRIT1 (p < 0.01) and PDCD10 (p < 0.01) but not for CCM2 (p = 0.21). KRIT1 cultures had 75% fewer loops on average compared with WT cultures, whereas PDCD10 failed to form tubule networks.
On treatment with H1152, all CCM knockdowns showed significant differences with respect to their untreated cultures, with a trend toward WT tubule pattern. This includes a lower void area and a higher loop count. However, even though the void area in KRIT1 KD and PDCD10 KD cultures treated by the ROCK inhibitor is “rescued” (no significant difference from WT), the loop count in these KDs remains significantly different from that of untreated WT (Figure 1B).
These results might be explained by two factors: (1) the increased ROCK activity in CCM KD cells contributes to but does not fully explain the phenotypes of vascular tubule patterns generated by CCM KD cells and (2) pharmacological inhibition of ROCK activity imposes additional biomechanical perturbations on cell behavior, resulting in a significant change of both WT and CCM KD pattern, such as, for example, a suboptimal level of cytoskeletal tension required for efficient vascular network development. To investigate which of the potential factors could plausibly explain our findings, we turned to computational modeling.
Computational Modeling Links EC Biomechanics to Disrupted Tubule Patterns in CCM KD Cells and Incomplete Rescue upon ROCK Inhibition
Our model is designed to explicitly account for protrusive activity, cytoskeletal stiffness, and forces developed through cell interactions. For this work, we developed a three-dimensional simulation model that differs from our previously reported two-dimensional cell model design in several critically important aspects. First, we model cell-ECM interaction explicitly through the extension of cell protrusions and establishment of elastic contacts within the 3D substrate, as opposed to the viscous drag interaction between a cell and the substrate surface in the old model. Second, even with the fixed total volume, the ellipsoidal cell body allows the cell to stretch and spread on the substrate due to lateral cell-cell interactions. Previously, the fixed area of the elliptical cell body would allow cells to stretch but not spread. Finally, in contrast to the old model, here we introduce a (presumably substrate-mediated) long-distance sensing between plated cells during their directed protrusion extension toward each other. This change was necessary for achieving close correspondence between the simulated and the experimentally observed dynamics at the cellular level (see Figures S2–S4). Indeed, human umbilical vein endothelial cells (HUVECs) with an average diameter of 17.21 ± 2.13 μm are surprisingly efficient at reaching each other by extending protrusions from distances as long as 120 μm (Video S1).
Optical z-stack images were acquired every 3 min starting at 20 min after cell plating on Matrigel, over 7 hr. Scale bar, 100 μm.
We choose to represent the body of each endothelial cell as an extendable ellipsoid (Figure 2A) with viscoelastic axes to account for cell stiffness while maintaining high efficiency of simulations with thousands of interacting cells. Each cell interacts with the other cells by mechanosensitive lateral protrusions, initiated radially from the edge of the cell body in the xy-plane and forming attachments (cell-cell contacts) on reaching cell bodies of other cells (see Figures S2 and S3). The protrusions function in two distinct modes. In the “extending” mode, protrusions extend with a rate uprot (0.017 μm/s) toward other cells. If the distance between the centers of two cells, R, is such that Rshort < R < Rlong, the protrusions extended by the cell are labeled as long range. Each cell has a limit on the maximum number of long-range protrusions at any given time, Nlong ≤ 5, whereas short-range protrusions extend toward the cells at close proximity R < Rshort (see Figure S4). On reaching the body of another cell, both types of protrusions switch to the “pulling” mode and begin to retract with a rate uretr (0.003 μm/s). If the distance between the centers of two cells connected by a retracting long-range protrusion becomes less than Rshort, this protrusion is automatically relabeled as short range, so that new long-range protrusions can be initiated and the total number of protrusions per cell, Ntotal = Nlong + Nshort, can increase. However, new short-range protrusions are not initiated if Ntotal > Nlim = 15. This setup ensures that there is a limit on how many protrusions a given cell can initiate at any given time while allowing for initiating new long-range interactions after the retraction of previously initiated protrusions is completed. Because cell-cell contacts in the model are defined as an attachment of a protrusion to another cell's body, we do not take into consideration interactions (or collisions) between different protrusions. In addition to lateral interactions between each other, cells also interact with the ECM that they are platted on. In the model, vertically directed protrusions establish cell-ECM contacts on reaching a length Rbott. Up to Nbott contacts per cell can be formed. Each of the above-mentioned parameters (see Table S2) has been adjusted through simulation scans to closely reproduce WT cell dynamics observed in our live imaging experiments.
Figure 2.

Simulations of Endothelial Tube Formation by WT and CCM KD Cells Untreated and Treated with the ROCK Inhibitor H1152
(A) An illustration of the cell model with an ellipsoidal cell body, mechanosensitive lateral protrusion responsible for cell-cell interactions, and downward-directed protrusions responsible for cell-ECM interactions (see also Figures S2–S4).
(B) Simulated cell formations that reproduce experimental patterns of untreated cells in the top row of Figure 1A (see also Figure S5).
(C) Comparison of experimental images (top row) and simulated multicellular formations (bottom row) of H1152-treated cells (see also Figures S6–S8).
(D) Simulated patterns resulted from the same klat and kbott as in B, but with the decreased cytoskeletal stiffness, pulling force, and the range of cell-cell sensing.
(E) Simulated patterns resulting from an adjustment (partial rescue) of only the phenotype-defining parameters klat and kbott.
The cell-cell and cell-ECM contacts were modeled as elastic springs with spring constant kpull. By extending these springs, cells exert contractile forces on each other, which ultimately leads to the emergence of a multicellular pattern (a network of connected cells). To model contact breakage events, we assumed that each contact spring has a finite extension-dependent probability, , of being removed, so that the corresponding protrusion can switch back to the extending mode. The parameter regulating the breakage probability is effectively an indicator of the cell-cell adhesion strength. Similarly, the breakage of the contacts between endothelial cells and the ECM, , defines the resistance of the cell to lateral displacement under external forces and, thus, represents the cell-ECM adhesion strength. On the timescale of the tube formation, we can assume that ECs are not motile by themselves and move only through the interactions with other cells.
The state of the system is defined by (1) the location {x,y}, orientation {φ}, and deformation of each cell body with a constant volume V; (2) the status (attached or detached) and the length L of each protrusion; (3) the length of each contact spring; and (4) an additional variable {ξ}, which defines the orientation of uniformly distributed lateral protrusion bases on the cell body with respect to the orientation of the semi-principle axes in the horizontal plane. The latter is needed for the proper reorientation of the cell body with respect to the direction of forces acting on it. This ensures very realistic deformation of the simplified geometry of the cell under mechanical stresses. At each time step, the set of coordinates i = {x,y,φ,a,b,ξ} is updated according to the force balance along each of the coordinates: Fi = −ηiνi−∇iH = 0, where ηi and νi are the viscous drag and the rate of change of the coordinate . For example, the change in cell body orientation is found as , where is the total Hamiltonian of the system, which includes all elastic components from the cell bodies and contact springs. The variables and are updated according to the status of each protrusion at each time step. Collectively these steps lead to the evolution of the system and the emergence of the multicellular patterns.
First, we used this model to establish the parameters that are responsible for the CCM phenotype. KRIT1 and PDCD10 KD cells form characteristic, easily distinguishable patterns, whereas the CCM2 phenotype appears to be less pronounced, being closer to the WT in terms of both the void area and the hole count measures (Figure 1A). Thus, for modeling purposes, we focused on the analysis of KRIT1 and PDCD10 patterns. Consistent with the previous work (Beltran et al., 2018), the 3D cell model readily reproduces a highly interconnected mesh in WT, elongated and branching but disconnected fragments in KRIT1, and round isolated cell clusters in PDCD10 by properly adjusting the stability/strength of cell-ECM and cell-cell contacts (Figures 2B and S5). Because our live cell imaging never showed a single event of cell-cell contact breakage in WT, we set the probability of breakage for WT cells to zero (klat = ∞). For the KRIT1 cells, the breakage probability is small but finite (klat = 6000 μm), whereas for the PDCD10 cells, the breakage probability is high (klat = 300 μm). In contrast, the resistance to the lateral forces due to cell-ECM attachments is low in KRIT1 (kbott = 5 μm) and high in PDCD10 (kbott = 20 μm).
Next, we sought to reproduce the patterns of WT, KRIT1, and PDCD10 cells treated by the ROCK inhibitor H1152. An extensive set of simulation scans systematically exploring multiple model parameters (Figures S6–S8) allowed us to successfully reproduce the experimentally observed cell formations (Figure 2C). This close correspondence was achieved by adjusting the parameters in the following way: (1) the stability of cell-cell contacts was increased 2-fold (klat = ∞ for WT, klat = 12,000 μm for KRIT1, klat = 600 μm for PDCD10); (2) the stability of cell-ECM contacts was also increased 2-fold (kbott = 40 for WT, kbott = 10 μm for KRIT1, kbott = 40 μm for PDCD10); (3) the cytoskeletal stiffness was reduced from kxy = 5 and kz = 100 to kxy = 1 and kz = 1 before and after treatment, respectively; (4) the pulling force resulting from the extension of the contact spring was decreased, changing the spring constant from kpull = 0.5 to kpull = 0.1; (5) the cell-sensing distance reduced from Rlong = 120 μm and Rshort = 40 μm to Rlong = 95 μm and Rshort = 30 μm. The convergence toward these parameter values suggests that not only the phenotype-defining parameters klat and kbott but also the parameters associated with the cytoskeletal stiffness, protrusion contractility, and the range of cell-cell sensing (presumably due to a reduced contraction of the Matrigel) are responsible for the specific arrangement of WT and CCM KD cells in tubular structures on the treatment with the ROCK inhibitor.
For the “control,” we ran simulations with the same parameters as earlier except for the phenotype-defining parameters klat and kbott, which we kept as in Figure 2B. The resulting patterns (Figure 2D) are not consistent with the experimental images, indicating that the change in cytoskeletal stiffness, protrusion contractility, and the range of cell-cell sensing is not exclusively responsible for the observed effects of ROCK inhibition. Therefore, our simulation results argue that the rescue still takes place, although to a limited degree.
In another important “control simulation,” we set the phenotype-defining parameters klat and kbott to the partially rescued values as in Figure 2B and left the other parameters unchanged. Again, the resulting patterns (Figure 2E) clearly deviate from the experimental images. This discrepancy justifies the need for careful considerations of the secondary, off-target effects of the perturbation of RhoA signaling in the CCM-deficient cells. This result emphasizes the critical importance of the holistic modeling approaches, which accounts for multiple factors that could be potentially affected by a pharmacological treatment targeting a key cell-signaling molecule, such as ROCK.
To confirm the model predictions regarding the modulation of cell-cell and cell-ECM interaction in conjunction with the alterations of the contractile characteristics of the CCM KD cells, we turn back to the experimental characterization, now focusing on the cell stiffness and the gene expression profiles.
Knockdown of CCM Protein Expression Leads to Changes in Actomyosin Architecture and Increases the Cellular Stiffness of KRIT1 and CCM2 but Not PDCD10 KD Cells
Despite growing evidence of unique features of KRIT1, CCM2, and PDCD10 knockdown phenotypes in human disease (Fischer et al., 2013, Fisher and Boggon, 2014) and in vitro (Beltran et al., 2018), and multiple pathways involved in CCM disease progression, RhoA-ROCK activity remains the most extensively characterized signaling pathway responsible for CCM lesions development (Beltran et al., 2018). To date, several pharmacological compounds targeting RhoA signaling are being tested to prevent or reduce the development of CCM lesions in pre-clinical trials (Bond et al., 2015).
According to predictions of the model described earlier, changes in cytoskeletal tension and cellular responses to external forces have a dramatic effect on tubule pattern formation. In the context of CCM KD, increased cellular stiffness is expected as a consequence of dysregulated RhoA function (Sreenivasappa et al., 2014). Thus, we aimed to directly measure cellular stiffness in CCM KD and compare it with WT cultures.
Atomic force microscopy (AFM) measurements were taken from 30–50 cells in each of the experimental groups at 24 hr after plating. The results of three independent experiments showed significantly higher stiffness of KRIT1 KD (p = 0.00053) and CCM2 KD (p = 0.00014) cells as compared with WT cells. However, the stiffness of PDCD10 KD cell was not different from that of WT cultures, p = 0.23 (Figures 3A and 3B). This was unexpected, because similarly to previously published data (Borikova et al., 2010, Beltran et al., 2018), we did observe statistically significant increase in myosin light chain (MLC) and cofilin phosphorylation in all CCM KD cells (Figures 3C and 3D), thus excluding a possibility that ROCK activity in PDCD10 KD cells was lower than in KRIT1 or CCM2 KD cultures.
Figure 3.

Changes in Actomyosin Cytoskeleton Associated with Loss of CCM Protein Expression Result in Increased Stiffness of Cells with Knockdown of KRIT1 and CCM2 but Not PDCD10
(A) AFM stiffness measurement results for single PLKO.1 (WT) and cells with knockdown of KRIT1, CCM2, and PDCD10. The graph shows mean values and standard deviations of measurements from up to 30 cells in each group. Note higher stiffness of KRIT1 and CCM2 KD but not PDCD10 KD cells when compared with WT. The data shown are from one representative of three independent experiments.
(B) The graph shows representative force curves of WT cells and cells with loss of CCM protein expression recorded by AFM, with shaded area representing ±2·SEM.
(C) Representative immunoblot showing increased phosphorylation of myosin light chain (MLC-P) and cofilin in HUVEC cells with loss of KRIT1, CCM2, and PDCD10 expression. Total actin used as a loading control.
(D) Quantification of fold change of MLC-P and phospho-cofilin levels. Optical densities of the corresponding bands were normalized to the loading control; the results represent an increase over PLKO.1 (WT) expression levels (mean ± SEM, n = 3 experiments. In the bar graphs p values are indicated for each of the KD cultures compared with WT).
(E) Upper row: representative images of cells immunolabeled with an antibody against phosphorylated myosin light chain (MLC-P) (green), and incubated with rhodamine-phalloidin to visualize F-actin (red). Lower row: results of subtraction of thresholded signals from the F-actin and MLC-P channels from the images of cells above.
(F) Percentage of the active area defined here as a fraction of the pixels, which have an over-threshold intensity of the F-actin signal but a sub-threshold intensity of the MLC-P signal, among all pixels within a near-edge strip. Contrast and homogeneity are statistical measures of the image derived from the gray-level co-occurrence matrix (see Transparent Methods). Data are represented as mean ±2·SEM.
Taken together, AFM measurement data demonstrated that, despite activation of a common signaling pathway and comparable increase in MLC and cofilin phosphorylation levels, biomechanical characteristics of the CCM KD cells are far from identical. Thus, a decrease in cell-cell or cell-ECM adhesion predicted by the model cannot be attributed solely to an increased cytoskeletal tension. This is clearly the case for cells with PDCD10 gene knockdown.
We reasoned that unexpectedly low stiffness of PDCD10 cells might be due to specifics of the architecture of actin cytoskeleton in these cells. Indeed, as demonstrated in previous studies, cells with highly aligned stress fibers had lower overall stiffness when compared with cells with a more intertwined organization of the cytoskeleton (Zhou et al., 2017). To address this possibility, we visualized filamentous actin with fluorescently labeled phalloidin and performed immunocytochemical staining of cultures using antibodies against MLC-P (Figure 3E, top row) to study the distribution of actomyosin fibers in WT and KD cells. Consistent with western blotting data (Figures 3C and 3D), the overall intensity of staining of EC cultures with anti-MLC-P antibodies was not different among all types of KD cells. To quantify the distribution of total filamentous actin as well as myosin II-containing contractile fibers in the cortical layer and throughout cytoplasm, we performed two types of analysis: (1) quantification of percent presence of non-bundled F-actin near the cell edge and (2) assessment of the internal structuring of actin bundles using the standard texture measures, contrast and homogeneity, derived from the gray-level signal distribution in cell images (see Transparent Methods). As shown in Figures 3E and 3F, the width of lamellipodia, the cortical layer characterized by high F-actin staining, was higher in WT than in all CCM KD cells (Figure 3E). This area of high F-actin activity was dramatically reduced in PDCD10 KD cells. Analysis of F-actin distribution showed that KRIT1 and CCM2 KD cell cytoskeleton had a higher contrast of actin bundles and was characterized by less homogeneity of staining as compared with WT and PDCD10 KD cells (Figure 3F).
Although elevated RhoA activity is a common feature of CCM KD cells, the overall stiffness and the underlying architecture of actomyosin cytoskeleton are drastically different among CCM KD cultures. The characteristics of actin cytoskeleton organization may contribute to differences in biomechanical properties of KD cells, the patterns of vascular networks, and the response of KD cultures to ROCK inhibition.
Gene Expression Analysis and Functional Assays Confirm Differences in Cell-Cell and Cell-ECM Adhesion in CCM KD Cultures
To test predictions of computational modeling, we studied gene expression profiles of WT and CCM KD cells cultured under 2D and 3D (Matrigel) conditions. We reasoned that expression profiles would change depending on the length of exposure to Matrigel and performed deep sequencing of RNA collected from cells at the initial (2 hr) as well as late (6 hr) stages of EC tubule formation (see Table S3 for all studied conditions).
Functional enrichment analysis using the DAVID v6.8 bio-informatics resources identified several significantly enriched gene ontology (GO) terms and KEGG pathways genes differentially expressed under specified conditions. Consistent with computational model predictions, enrichment of terms associated with cell-cell adhesion was characteristic of PDCD10 cultures, whereas terms associated with ECM organization and adhesion to ECM were over-represented in KRIT1 KD cultures. The highest enrichment scores were observed for EC tubules at 6 hr after plating on Matrigel (Figures 4 and S9 for full dataset), emphasizing the importance of genes involved in the formation of vascular networks under these conditions.
Figure 4.

Gene Expression Analysis and Adhesion Assay Reveal Differences in Cell-Cell Adhesion and Cell-ECM Adhesion in CCM KD Cultures
(A) GO term enrichment analysis of selected cellular component gene groups, representing cell-cell and cell-ECM interactions. The graphs show negative log(p values) for KRIT1 KD cells (blue bars) and PDCD10 cells (red bars) compared with WT cells. Genes associated with ECM interaction (extracellular matrix organization and ECM-receptor interaction) were significantly enriched in cultures lacking KRIT1 expression, whereas genes associated with cell-cell adhesion were over-represented in PDCD10 KD cells (see also Figure S9).
(B) Heatmap of a subset of differentially expressed genes involved in cell-cell adhesion. For a selected set of genes, PDCD10 KD cells showed over-expression in comparison with KRIT1 and CCM2 KD. Text annotation in the heatmap represents fold change of RPKM values in comparison with WT cells (see also Figure S10).
(C) Heatmap of selected differentially expressed genes involved in cell-ECM adhesion.
(D) Adhesion assay demonstrates decreased attachment of KRIT1 KD cells to extracellular matrix. The percentage of adherent cells was quantified as a ratio of absorbance at 590 nm of wells from which non-adherent cells were removed at 30 min after plating.
Detailed analysis of differentially expressed genes identified several sets of genes potentially involved in destabilizing cell-cell adhesion in PDCD10 KD and cell-ECM adhesion in KRIT1 KD cultures (Figure 4, showing the data for 3D culture, 6 hr, and Figure S10 for other studied conditions).
For example, α-adducin, involved in regulating the assembly of actin filaments at the areas of cell-cell contacts, thus stabilizing endothelial barrier (Kugelmann et al., 2015), was significantly decreased in PDCD10 KD cultures across studied culture conditions.
Enrichment of GO terms related to matrix composition and cell-matrix adhesion was higher for KRIT1 KD cultures than for PDCD10 or CCM2 KD cells (Figures 4A and 4C). Although some of the integrin receptor genes and secretory matrix components were down-regulated in KRIT1 KD, expression of other isoforms was increased (Figures 4A and 4C). To study whether KRIT1 protein KD affects adhesion of cells to the ECM, as suggested by the modeling, we plated cells on the Matrigel-coated surface, allowed them to attach for 30 min, and evaluated the number of adherent cells in WT and CCM protein KD cultures. Among all, KRIT1 KD cells showed the lowest adhesion to the substrate (Figure 4D). This result confirms that loss of KRIT1 expression reduces the strength of cell-ECM interactions.
Consistent with predictions of the computational model, RNA sequencing analysis revealed the differential impairment of cell-cell versus cell-substrate adhesion in KRIT1 and PDCD10 KD endothelial cells.
Discussion
Although RhoA activity has been identified as the key player in the development of CCM (Stockton et al., 2010, Borikova et al., 2010), CCM proteins are involved in other signaling pathways (Maddaluno et al., 2013, Schulz et al., 2015, Zhou et al., 2016, Beltran et al., 2018), creating clear phenotypic differences in the collective behavior of endothelial cells with the knockdown of the different members of the CCM complex: KRIT1, CCM2, and PDCD10. Indeed, in this work, we used direct measurements of cell stiffness and showed that despite significantly elevated phosphorylation of cofilin and MLC in all three knockdowns, only KRIT1 and CCM2 have significantly increased stiffness as compared with the control cells. Subsequent analysis of F-actin distribution offers an explanation of this surprising observation. Our quantitative texture-based image analysis confirmed the visually apparent difference in the spatial arrangement of actin filaments: all three knockdowns show an increased density of actin bundles throughout the cell, but in contrast to KRIT1 and CCM2 KDs, PDCD10 KD cells have shorter and less organized bundles, indicating the lack of the cell-level connectivity of the stress fibers.
This result is important from the pharmacological treatment perspective because targeting the components of the RhoA signaling pathway might rescue the phosphorylation levels of cofilin and MLC but has limited effect on the collective behavior of PDCD10 cells since the overall stiffness of these cells appears not to be the major factor defining their deficient biomechanics. Then, what are the major factors that give rise to the distinct patterns of the collective behavior of CCM KD cells? To address this question, we developed a detailed cell model that explicitly accounts for cell body stiffness and deformation, protrusion-based interactions with the ECM, and the mechanosensitive interaction between the cells initially plated at sub-confluent densities. Our strategy was to relate the biophysical properties of individual cells to the emergent patterns of their collective behavior through the systematic parameter scanning until a close correspondence between the simulated and the experimental images was achieved. Such integrated in silico/in vitro experimentation led us to the conclusion that central to the loss of multicellular integrity in KRIT1 and PDCD10 KD cells is the decrease in cell-ECM and cell-cell contact stability, respectively. Our RNA-seq analysis of gene expression is consistent with this result. Although the changes in expression of many genes overlap between KRIT1 and PDCD10 KD cells, GO analysis clearly shows that more genes associated with the ECM re-organization and ECM-receptor interactions are affected by KRIT1 KD, whereas more genes associated with cell-cell adhesion are affected by PDCD10 KD. Furthermore, a more directed assessment of the strength of cell-ECM interaction—the non-adherent cell washout assay—also confirmed the deficiency of the KRIT1 KD cell attachment to Matrigel-coated substrate.
Collectively, these results raise another question: to what degree does normalizing cell signaling downstream of RhoA rescue the interaction of CCM KD cells during the formation of tubular structures? Our ROCK inhibition experiments showed that the patterns of H1152-treated cells are noticeably different from those of the untreated ones. For example, the WTH1152 and KRIT1H1152 cells form very similar (visually indistinguishable) patterns, but these patterns are very different from the patterns of untreated control cells. Could it be that, although the rescue of cell interactions takes place on H1152 treatment, additional perturbations of cell biomechanics unrelated to the deficiency of CCM proteins continue to affect the resulting endothelial network? Our simulations suggest that both factors play a role. We achieve close correspondence between the simulated and experimental images only if we partially “rescue” the parameters directly affecting the stability of cell-cell and cell-ECM contacts and adjust (perturb) the parameters that affect collective cell behavior through the modulation of stretching and spreading, contractility of protrusions, and the efficiency of long-range cell-cell sensing.
Although this work only scratches the surface of the systems-level understanding of the complex interplay between the regulatory and biomechanical processes during the collective behavior of endothelial cells, our simulation model provides a platform for further investigation that could take advantage of the integration of genetic and biochemical characterization, quantitative imaging, and systematic predict-test-refine modeling. Many parameters can be involved in the setup of a comprehensive cell model, but focusing on the characterization and experimental verification of the emergent behavior allows us to constrain the range of parameters and gain mechanistic insight as we did in this paper.
Limitations of the Study
The two main measures used for quantitative comparison of experimental and simulated cellular patterns in this study, the void area and the mean loop count, capture the significant variations in the structure of cell formations. However, accounting for subtle but still important geometric features, such as the curvature of the bridges or the variation in their thickness, requires a more comprehensive characterization of complex and diverse cellular patterns. Such a methodology is currently under development.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Adriana Beltran for the gift of lentiviral vectors carrying shRNAs against krit1, ccm2, and pdcd10. We also thank Drs. Timothy C. Elston and Gary L. Johnson for their immense support, stimulating discussions, and the ideas regarding the design of this project. We wish to acknowledge the core facilities at the Parker H. Petit Institute for Bioengineering and Bioscience at the Georgia Institute of Technology for the use of their shared equipment, services, and expertise. This work was supported by the U.S. Army Research Office (ARO) grant W911NF-17-1-0395 to D.T. and by funds from the Marcus Foundation, The Georgia Research Alliance, and the Georgia Tech Foundation through their support of the Marcus Center for Therapeutic Cell Characterization and Manufacturing (MC3M) at Georgia Tech.
Author Contributions
D.T. designed the computational and experimental studies, analyzed imaging data, and wrote the paper. O.C. performed the majority of experiments and wrote the paper. A.Z. developed the model, performed simulations, and edited the paper; S.H. analyzed RNA-seq data; W.P. analyzed images of cell patterns and edited the paper; K.Y. performed AFM experiments and edited the paper; J.O. and V.A. assisted in AFM data acquisition; T.A.S. provided resources and supervision of AFM experiments.
Declaration of Interests
The authors declare no competing interests.
Published: November 30, 2018
Footnotes
Supplemental Information includes Transparent Methods, 10 figures, 3 tables, and 1 video and can be found with this article online at https://doi.org/10.1016/j.isci.2018.11.001.
Data and Software Availability
The accession number for the sequencing data reported in this paper is GEO: GSE116323.
Supplemental Information
References
- Beltran A.S., Olivares-Quintero J.F., Tsygankov D., Richardson B.T., Dibble C.F., Borikova A.L., Miller C.R., Zawistowski J.S., Sciaky N., Licea A.F. Cerebral cavernous malformation proteins and mekk3 coordinately modulate SMURF1 abundance to control RhoA-dependent endothelial cell phenotypic state. Dev. Cell. 2018 [submitted] [Google Scholar]
- Betz C., Lenard A., Belting H.G., Affolter M. Cell behaviors and dynamics during angiogenesis. Development. 2016;143:2249–2260. doi: 10.1242/dev.135616. [DOI] [PubMed] [Google Scholar]
- Bond L.M., Sellers J.R., Mckerracher L. Rho kinase as a target for cerebral vascular disorders. Future Med. Chem. 2015;7:1039–1053. doi: 10.4155/fmc.15.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borikova A.L., Dibble C.F., Sciaky N., Welch C.M., Abell A.N., Bencharit S., Johnson G.L. Rho kinase inhibition rescues the endothelial cell cerebral cavernous malformation phenotype. J. Biol. Chem. 2010;285:11760–11764. doi: 10.1074/jbc.C109.097220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A., Zalvide J., Faurobert E., Albiges-Rizo C., Tournier-Lasserve E. Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol. Med. 2013;19:302–308. doi: 10.1016/j.molmed.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Fisher O.S., Boggon T.J. Signaling pathways and the cerebral cavernous malformations proteins: lessons from structural biology. Cell. Mol. Life Sci. 2014;71:1881–1892. doi: 10.1007/s00018-013-1532-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugelmann D., Waschke J., Radeva M.Y. Adducin is involved in endothelial barrier stabilization. PLoS One. 2015;10:e0126213. doi: 10.1371/journal.pone.0126213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddaluno L., Rudini N., Cuttano R., Bravi L., Giampietro C., Corada M., Ferrarini L., Orsenigo F., Papa E., Boulday G. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature. 2013;498:492–496. doi: 10.1038/nature12207. [DOI] [PubMed] [Google Scholar]
- Mayor R., Etienne-Manneville S. The front and rear of collective cell migration. Nat. Rev. Mol. Cell Biol. 2016;17:97–109. doi: 10.1038/nrm.2015.14. [DOI] [PubMed] [Google Scholar]
- McDonald D.A., Shi C., Shenkar R., Stockton R.A., Liu F., Ginsberg M.H., Marchuk D.A., Awad I.A. Fasudil decreases lesion burden in a murine model of cerebral cavernous malformation disease. Stroke. 2012;43:571–574. doi: 10.1161/STROKEAHA.111.625467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagenstecher A., Stahl S., Sure U., Felbor U. A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum. Mol. Genet. 2009;18:911–918. doi: 10.1093/hmg/ddn420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa E., Mayor R. Collective cell migration in development. J. Cell Biol. 2016;212:143–155. doi: 10.1083/jcb.201508047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz G.B., Wieland E., Wustehube-Lausch J., Boulday G., Moll I., Tournier-Lasserve E., Fischer A. Cerebral cavernous malformation-1 protein controls DLL4-Notch3 signaling between the endothelium and pericytes. Stroke. 2015;46:1337–1343. doi: 10.1161/STROKEAHA.114.007512. [DOI] [PubMed] [Google Scholar]
- Shenkar R., Shi C., Rebeiz T., Stockton R.A., Mcdonald D.A., Mikati A.G., Zhang L., Austin C., Akers A.L., Gallione C.J. Exceptional aggressiveness of cerebral cavernous malformation disease associated with PDCD10 mutations. Genet. Med. 2015;17:188–196. doi: 10.1038/gim.2014.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreenivasappa H., Chaki S.P., Lim S.M., Trzeciakowski J.P., Davidson M.W., Rivera G.M., Trache A. Selective regulation of cytoskeletal tension and cell-matrix adhesion by RhoA and Src. Integr. Biol. (Camb) 2014;6:743–754. doi: 10.1039/c4ib00019f. [DOI] [PubMed] [Google Scholar]
- Stockton R.A., Shenkar R., Awad I.A., Ginsberg M.H. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J. Exp. Med. 2010;207:881–896. doi: 10.1084/jem.20091258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Tang A.T., Wong W.Y., Bamezai S., Goddard L.M., Shenkar R., Zhou S., Yang J., Wright A.C., Foley M. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature. 2016;532:122–126. doi: 10.1038/nature17178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.L., Sun X.X., Ma J., Tong M.H., To S.K.Y., Wong A.S.T., Ngan A.H.W. Actin cytoskeleton stiffness grades metastatic potential of ovarian carcinoma Hey A8 cells via nanoindentation mapping. J. Biomech. 2017;60:219–226. doi: 10.1016/j.jbiomech.2017.06.040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Optical z-stack images were acquired every 3 min starting at 20 min after cell plating on Matrigel, over 7 hr. Scale bar, 100 μm.