

Abstract
Aims:
Chloramphenicol (CAP), a broad spectrum antibiotic, was shown to protect the heart against ischemia/reperfusion (I/R) injury. CAP also induces autophagy, however, it is not known whether CAP-induced cardioprotection is mediated by autophagy. Therefore, here we aimed to assess whether activation of autophagy is required for the infarct size limiting effect of CAP and to identify which component of CAP-induced autophagy contributes to cardioprotection against I/R injury.
Main methods:
Hearts of Sprague-Dawley rats were perfused in Langendorff mode with Krebs-Henseleit solution containing either vehicle (CON), 300µM CAP (CAP), CAP and an inhibitor of autophagosome-lysosome fusion chloroquine (CAP+CQ), or an inhibitor of autophagosome formation, the functional null mutant TAT-HA-Atg5K130R protein (CAP+K130R), and K130R or CQ alone, respectively. After 35 min of aerobic perfusion, hearts were subjected to 30 min global ischemia and 2 h reperfusion. Autophagy was determined by immunoblot against LC3 from left atrial tissue. Infarct size was measured by TTC staining, coronary flow was measured, and the release of creatine kinase (CK) was assessed from the coronary effluent.
Key findings:
CAP treatment induced autophagy, increased phosphorylation of Erk1/2 in the myocardium and significantly reduced infarct size and CK release. Autophagy inhibitor TAT-HA-Atg5K130R abolished cardioprotection by CAP, while in CAP+CQ hearts infarct size and CK release were reduced similarly to as seen in the CAP-treated group.
Conclusion:
This is the first demonstration that autophagosome formation but not autophagosomal clearance is required for CAP-induced cardioprotection.
Significance:
Inducing autophagy sequestration might yield novel therapeutic options against acute ischemia/reperfusion injury.
Keywords: (3-5) autophagy, cardioprotection, chloramphenicol
1. Introduction
Autophagy is an intracellular degradation process which eliminates dysfunctional organelles and long-lived proteins through lysosomal breakdown [1]. During starvation degradation of intracellular components promotes cell survival by maintaining cellular energy levels. Autophagy consists of sequential steps: the early stage, which includes initiation of the autophagosomal membrane and autophagosome formation, and the late stage, where autophagosome-lysosome fusion and lysosomal degradation happen [2, 3]. Autophagy has a constitutive, low activity under normal conditions in most cells, including cardiac myocytes. A growing amount of evidence suggests that increased autophagy plays a role in cardioprotective interventions [4, 5].
Previous studies reported that several therapeutic agents which are already in clinical use, for example hydrophobic statins [6], sevoflurane [7], sulfaphenazole [8] and certain antibiotics, such as chloramphenicol (CAP) [9], may also induce autophagy in addition to their primary effects. Since previous studies reported that CAP protects the heart against ischemia/reperfusion injury and upregulates autophagy markers [10–12], we hypothesized that CAP induces cardioprotection via induction of autophagy.
It has been shown that the induction of autophagy is required for cardioprotective mechanisms but no detailed investigation has been performed on which stage of autophagy is necessary for cardioprotection. Since autophagy is a multi-step process, it can be inhibited at different steps. Previous studies have shown that inhibition of early-stage autophagy can be achieved by 3-methyladenine [13] or TAT-HA-Atg5K130R, a dominant negative mutant fusion protein of a key mediator of autophagy, Atg5 [14–16]. The late phase of autophagy, lysosomal fusion and degradation, can be arrested by elevating lysosomal pH, e.g., by the use of an antimalarial drug, chloroquine (CQ, see for review: [17]). Although a few studies utilized these substances to investigate the mechanism of cardioprotective stimuli, it is unknown which phase of CAP-induced autophagy is necessary for cardioprotection.
Therefore, in this study we aimed to investigate whether CAP-induced autophagy is necessary for cardioprotection. Furthermore, we assessed whether sequestration and/or degradation phases of autophagy are necessary for the cardioprotective effect of CAP.
2. Materials and methods
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85-23, revised 1996) and was approved by the animal ethics committee of the San Diego State University, San Diego, California and Semmelweis University, Budapest, Hungary. Chemicals were purchased from Sigma-Aldrich (St. Louis, MO), unless indicated otherwise.
2.1. Study design
To identify the most suitable model system, in a pilot study we examined the effect of CAP on autophagy in neonatal rat cardiomyocytes (NRCMs), in H9c2 cardiac myoblast cells and in isolated hearts. We found that CAP induced autophagy in isolated hearts but not in NRCMs or in H9c2 cells (see Fig. 1A-C). The efficacy of TAT-HA-Atg5K130R was also assessed in a pilot experiment where we observed that in the left atrium LC3-II/I ratio was decreased after 15 min of administration of 200nM TAT-HA-Atg5K130R to isolated hearts as compared to vehicle controls (Fig. 1D).
Fig. 1.
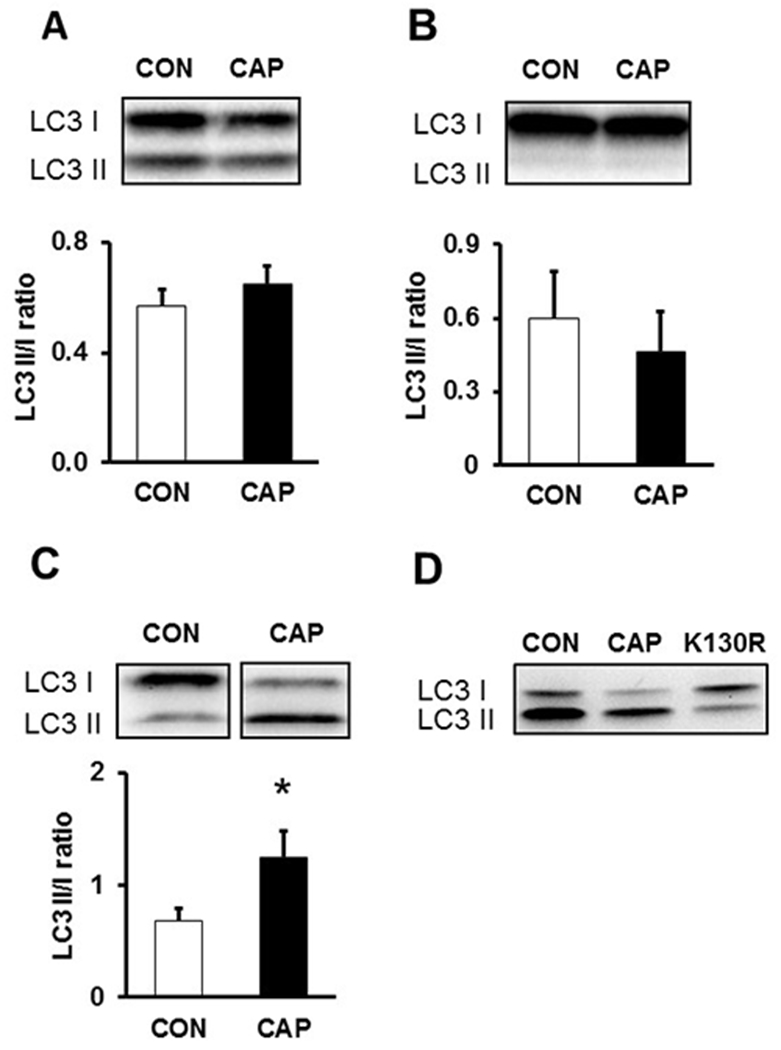
Pilot study: The effect of CAP on autophagy-related protein LC3 levels in (A) NRCMs (B) in H9c2 cells and (C) ex vivo heart lysates *p <0.05 vs. CON n=3-8 (D) Pilot study: effect of CAP and TAT-HA-Atg5K130R on autophagy-related protein LC3 levels in whole heart. n=2 Data are presented as means ± SEM. Chloramphenicol - CAP, Control - CON, Microtubule-associated protein 1A/1B-light chain 3 - LC3, Neonatal rat cardiac myocytes – NRCMs
Therefore, in the main series of experiments, we used an ex vivo model of acute cardiac ischemia/reperfusion injury to assess the effects of autophagy inhibitors (TAT-HA-Atg5K130R and CQ) on CAP-induced cardioprotection. Since the availability of TAT-HA-Atg5K130R was limited due to technical limitations in production and purification of the protein in quantities necessary for ex vivo heart perfusion experiments, we had to reduce the number of isolated hearts treated with TAT-HA-Atg5K130R.
2.2. Isolation and treatment of NRCMs
Preparation of NRCM culture used in the pilot study was described before in details [18]. Briefly, neonatal rats were sacrificed by cervical dislocation and hearts were removed and placed into ice-cold phosphate buffered saline (PBS) solution. Ventricles were minced and resuspended in 0.25% trypsin (Thermo Fisher Scientific, USA) solution. Digested tissue was centrifuged at 400×g, for 15 min at 4°C. Cell pellet was resuspended in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with L-glutamine, Antibiotic-Antimycotic solution and 10% fetal bovine serum (FBS, Thermo Fisher Scientific, USA). Cells were counted in a hemocytometer, and seeded into 75 cm2 flasks at a density of 4×106 cell/flask. After 24 hours, the growth medium was replaced with differentiation medium containing 1% FBS. Cardiomyocytes were kept under normoxic conditions (37°C, in 95% air and 5% CO2 gas mixture) for three days prior to treatment and cell collection.
On the 3rd day of culturing, cell medium was supplemented with vehicle (saline, 5% v/v) or 300μM CAP for 1 hour. Then cell cultures were washed with ice cold PBS and lysed for 5 minutes in 500µL homogenization buffer (1x Radio-Immunoprecipitation Assay Buffer, RIPA, supplemented with a protease and phosphatase inhibitor cocktail). Lysed cells were scraped, collected and sonicated with an ultrasonic homogenizer (Ultrasonic Processor UP200H, Hielscher, USA) for 10 seconds on ice. The homogenate was centrifuged at 10,000×g, 4°C for 10 minutes; and the supernatant was collected and stored at −80°C. Protein concentration was assessed by Bicinchoninic Acid Protein Assay Kit (BCA Protein Assay kit, Pierce, USA).
2.3. Treatment of H9c2 cells
H9c2 cells were seeded in T25 flasks (106 cells/flask) in 6mL DMEM supplemented with 10% FBS, Antibiotic-Antimycotic solution, glucose, MEM Non-Essential Amino Acid Solution and L-glutamine. Then incubated at 37°C in 5% CO2. After 24 hours cells were treated with 300μM CAP or vehicle (CON). Cells were incubated for 1 h, then scraped in 200μL RIPA lysis buffer and sonicated (Ultrasonic Processor UP200H, Hielscher, USA) for 3×15 sec at 50% power. Homogenates were centrifuged for 10 min at 10,000×g, 4°C. Supernatants were aliquoted and stored at −80°C. Protein concentration was assessed with BCA kit.
2.4. Preparation of K130R
TAT-HA-Atg5K130R protein was purified from BL21(DE3)pLysS E. coli bacteria transformed with pTAT-HA-Atg5K130R plasmid as described elsewhere [8]. Briefly, crude cellular extract was purified on a Ni-NTA column (Thermo Fisher Scientific, USA) followed by desalting on a PD-10 column (GE Healthcare, United Kingdom) into PBS. Purified protein was used immediately after assessing its concentration by the BCA method.
2.5. Ex vivo heart perfusion
Sprague-Dawley rats (250-300g) were anesthetized with i.p. pentobarbital (30 mg/kg) and anticoagulated with i.v. heparin (100U/kg). Hearts were excised and perfused in Langendorff mode with Krebs-Henseleit solution (KH) for 15 min [19]. From the 15th min, a group of heart received KH containing 300µM CAP. To inhibit CAP-induced autophagosome formation a group of hearts were perfused with KH containing 300µM CAP and 200nM cell-permeable recombinant TAT-HA-Atg5K130R (CAP+K130R) for 15 min at the beginning of the protocol, then received CAP alone.
Another group of hearts received 300µM CAP and 10µM CQ (CAP+CQ) throughout the protocol to inhibit lysosomal degradation of CAP-induced autophagosomes. Further groups of hearts were perfused with 10µM CQ or 200nM TAT-HA-Atg5K130R alone (CQ and K130R, respectively; see Fig. 2).
Fig. 2.
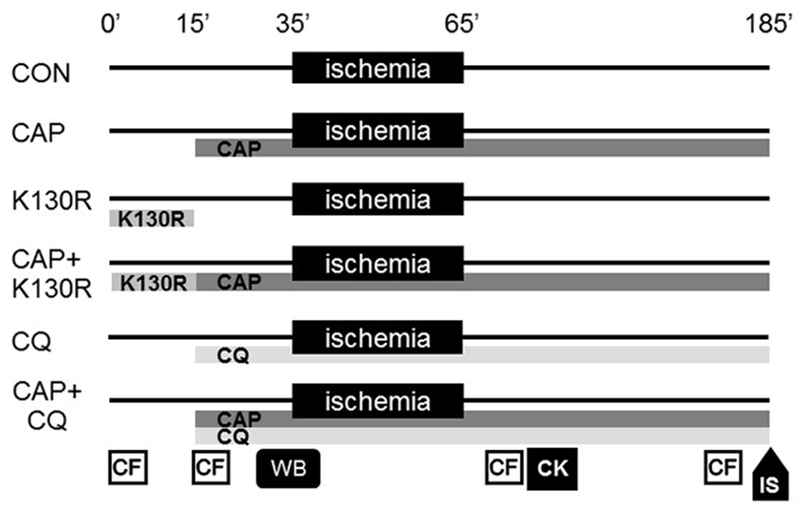
Experimental protocol. Chloramphenicol - CAP, Control - CON, TAT-HA-Atg5K130R - K130R, CK - Creatine kinase, CF – Coronary flow measurement, WB – Western blot, IS – Infarct size.
2.6. Measurement of infarct size, coronary flow and creatine kinase release
At the end of perfusion, hearts were sliced into 2mm-thick slices, and right ventricles were removed. Slices were immersed in 1% triphenyltetrazolium-chloride for 20 min, then in 4% formalin for 24 h and scanned. Weight of slices was measured. Necrotic area was evaluated by planimetry (ImageJ). Data is expressed as a percentage of left ventricular mass. Coronary flow rate was measured throughout the protocol by timed collection of coronary effluent. Creatine kinase (CK) release was measured in the coronary effluent collected from 10 min to 20 min after the onset of the reperfusion by a colorimetric assay (Stanbio™ CK Liqui-UV™ Test, Stanbio Laboratory, USA).
2.7. Western blotting
After 35 min aerobic perfusion, left atria or whole hearts were snap frozen. Samples were lysed in 500µL RIPA buffer with Tissuelyser LT (Quiagen, Germany) at 4°C. Lysates were centrifuged at 10,000×g at 4°C for 15 min. Supernatants were aliquoted and stored at −80°C. Protein concentration was assessed by BCA kit, then samples containing 40µg protein were prepared in Laemmli-buffer and electrophoresed on 4-12% polyacrylamide gels (Thermo Fisher Scientific, USA), then transferred to polyvinylidene difluoride membranes. Even loading was assessed with Ponceau staining. Membranes were blocked with 5% non-fat milk or 5% bovine serum albumin (BSA) in Tris-buffered saline with 0.05% Tween 20 (TBST) for 2 h. Antibodies against LC3 A/B, p-Akt, Akt, p-Erk1/2, Erk1/2 (Cell Signaling, USA) were diluted 1:1000 in 5% milk or 5% BSA in TBST and added to membranes and incubated for overnight at 4°C. After washing in TBST for 3×5 min, secondary antibody conjugated to horseradish peroxidase diluted 1:2000 in 5% milk or 5% BSA in TBST was added for 1 hour at room temperature. Signals were detected by enhanced chemiluminescence kit (Thermo Fisher Scientific, USA) and quantified with ImageQuant software (Bio-Rad, USA).
2.8. Statistical analysis
Differences between treatments were evaluated by One-way ANOVA using Fisher’s LSD as post-hoc test or non-parametric Kruskal-Wallis test (coronary flow data, 14-20 min timepoint) by Prism 5 (GraphPad, GraphPad Software, USA). Data are presented as means ± SEM.
3. Results
3.1. Pilot study: CAP induces autophagy in ex vivo perfused hearts but not in NRCMs and H9c2 cells
In a pilot study we aimed to compare the effect of CAP on autophagy in neonatal cardiomyocytes, in H9c2 cells and in ex vivo-treated hearts. Therefore, we used the most common autophagy marker LC3 to measure changes in autophagy. In isolated rat neonatal cardiomyocytes and in H9c2 cells there was no significant difference in LC3-II to I ratio between corresponding CON and CAP groups (Fig. 1A, 1B). Since CAP failed to induce autophagy in NRCMs and H9c2 cells, in our further experiments we used the isolated rat heart model. In the left atrium of hearts perfused with CAP, the ratio of LC3-II to LC3-I was significantly increased after 35 minutes compared to CON samples indicating that CAP induced cardiac autophagy (Fig. 1C). The discrepancy might be attributed to the difference in metabolic status of cultured cells and the ex vivo perfused hearts. Since insulin, fatty acids and other factors are present in cell culture medium due to the use of foetal calf serum, and since these factors are not included in the heart perfusion buffer, signaling pathways related to autophagy might be differentially modulated by CAP. Furthermore, the presence of cell types other than cardiomyocytes (endothelial cells, fibroblasts) in the heart might also contribute to the cardioprotective effects of CAP (see for a recent review: [20]).
3.2. Main study: TAT-HA-Atg5K130R blocks CAP-induced cardioprotection in isolated hearts
In order to assess whether the sequestration and/or degradation phases of autophagy are necessary for the cardioprotective effect of CAP, we used ex vivo Langendorff heart perfusion experiments. CAP treatment significantly reduced infarct size and CK release as compared to CON hearts (Fig. 3A, 3B). Pretreatment with TAT-HA-Atg5K130R abolished the infarct size limiting effect of CAP, while pretreatment with CQ did not interfere with CAP-induced cardioprotection.
Fig. 3.
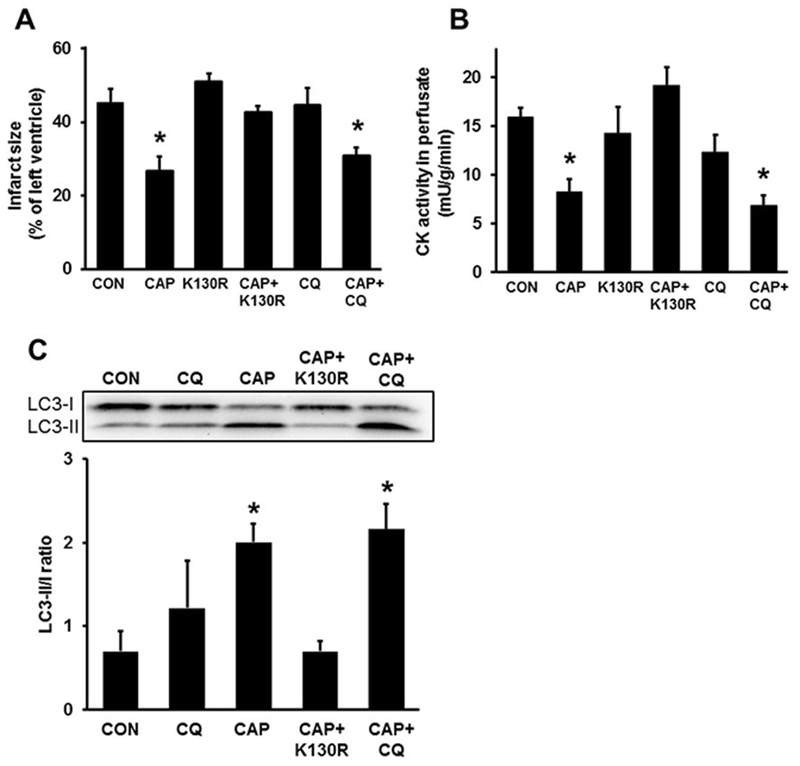
(A) Inhibition of autophagosome formation by TAT-HA-Atg5K130R abolishes CAP-induced cardioprotection (B) Inhibition of autophagosome formation (K130R) abolishes the CAP induced CK activity reduction in perfused rat hearts. n=3-8 (C) The effect of CAP on autophagy-related protein LC3 levels in isolated hearts n=3 *p <0.05 vs. CON Data are presented as means ± SEM. Chloramphenicol - CAP, Chloroquine – CQ, Control - CON, TAT-HA-Atg5K130R - K130R, Creatine kinase – CK
We also measured LC3 expression in isolated heart samples and the western blot results showed that CAP increased LC3-II/I ratio. Meanwhile, administration of TAT-HA-Atg5K130R but not CQ reduced the increase of LC3-II due to CAP administration (Fig. 3C). Since cardioprotection, as assessed by infarct size, and autophagosome formation, as assessed by LC3-II to LC3-I ratio, was lower in CAP+K130R group than in CAP group, these results indicate that CAP-induced cardioprotection requires the process of autophagosome formation but not autophagosomal clearance.
To characterize cardioprotective signaling mechanisms modulated by CAP, we assessed activation of proteins involved in the RISK/SAFE pathway. We found that the phosphorylation of Erk1/2 but not of Akt increased significantly due to CAP treatment (Fig. 4).
Fig. 4.
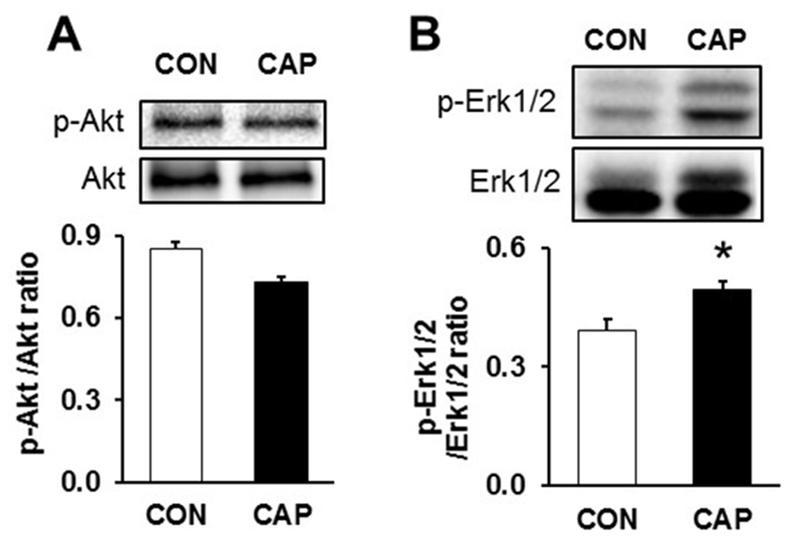
Assessment of cardioprotective pathways from ex vivo whole heart lysates. (A) Ratio of p-Akt to Akt signals (B) ratio of p-Erk1/2 to Erk1/2. n=5. Chloramphenicol - CAP, Control - CON
Coronary flow was significantly lower at the beginning of reperfusion (4-5 min) in CAP, CQ and CAP+CQ groups compared to CON group. Prior to ischemia (14-20 min) the coronary flow was lower in CQ and CAP+CQ. After the initiation of reperfusion (65-79 min) the coronary flow was decreased in K130R and CQ groups. At the end of the reperfusion time (169-185 min) the coronary flow was lower in CAP+CQ group compared to CON (Table 1).
Table 1 –
Measurement of coronary flow in ex vivo perfused hearts *p <0.05, vs. CON n=3-8 Data are presented as means ± SEM. Chloramphenicol - CAP, Chloroquine – CQ, Control - CON, TAT-HA-Atg5K130R - K130R
| Groups | Perfusion time (min) | |||
|---|---|---|---|---|
| 4-5 | 14-20 | 65-79 | 169-185 | |
| CON | 14±0.6 | 14±0.9 | 8.3±0.7 | 6.7±0.6 |
| CAP | 12±0.5* | 10±1.1 | 9.8±0.7 | 6.6±0.4 |
| K130R | 14.3±0.9 | 9.5±2.5 | 5.7±0.9* | 5.7±0.7 |
| CAP+K130R | 14.9±0.6 | 10.6±0.3 | 7.8±0.4 | 5.3±0.2 |
| CQ | 11±0.3* | 9.2±0.4* | 5±1.1* | 5.7±2.2 |
| CAP+CQ | 10.7±1.0* | 8.5±0.7* | 7.8±0.7 | 4±0.4* |
4. Discussion
Here we demonstrate that pretreatment with an inhibitor of autophagosome formation (TAT-HA-Atg5K130R functional null-mutant protein) abolished the infarct size limiting effect of CAP, while pretreatment with an inhibitor of autophagosome-lysosome fusion (CQ blocker of autophagosomal clearance) did not interfere with CAP-induced cardioprotection. Furthermore, CAP administration induced autophagy as assessed by an increased LC3-II/LC3-I ratio, which was inhibited by the administration of TAT-HA-Atg5K130R, but not of CQ. These results support that inhibition of autophagosome formation abolishes cardioprotection induced by CAP. Furthermore, this is the first demonstration that autophagosome formation but not autophagosomal clearance is required for cardioprotection afforded by the induction of autophagy (Fig. 5).
Fig. 5.
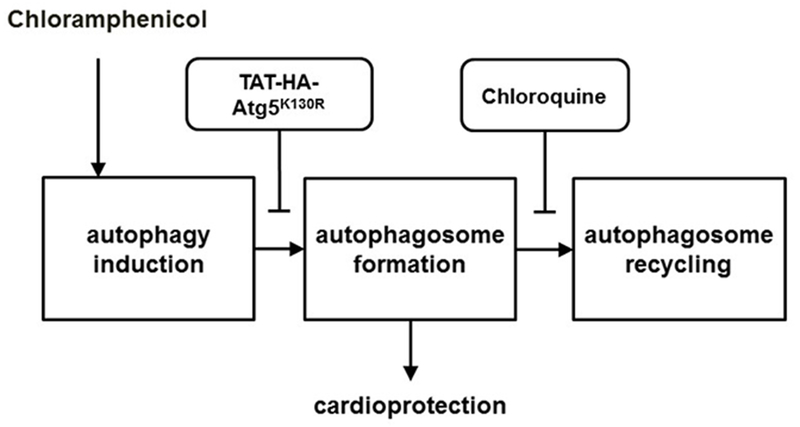
Chloramphenicol induced cardioprotection requires the process of autophagosome formation, but not autophagosome recycling.
In our present study, CAP significantly increased the LC3-II/I ratio in ex vivo hearts which indicates the induction of autophagy. Furthermore, CAP treatment reduced infarct size and creatine kinase activity in isolated rat hearts. These results are in agreement with those of Granville et al. [10] who showed that CAP reduced infarct size in an open chest rabbit model of regional ischemia. Moreover, similar results were obtained in a previous study with a more water soluble derivative of CAP, chloramphenicol succinate (CAPS), in a swine model of myocardial ischemia-reperfusion injury by Sala-Mercado et al. [12]. They found that CAPS, administered as a pre-treatment or before reperfusion, rendered the porcine heart resistant to ischemia-reperfusion injury, however, they did not investigate which stage of autophagy is necessary for cardioprotection. Interestingly, in our present ex vivo rat heart experiment, CAPS did not reduce infarct size (data not shown). Since CAPS is a prodrug activated by esterases in the blood [21] which are not present in isolated heart perfusion, our data suggests that in vivo metabolism of CAPS into CAP plays a significant role in its cardioprotective effect. In 2014 Shiomi et al. [22] demonstrated that in isolated guinea pig hearts sevoflurane reduced infarct size after 30 min, but not 45 min of cardiac ischemia. In this experiment, CAP pretreatment extended the time of ischemia after which sevoflurane preconditioning reduced infarct size. These results well demonstrate that CAP or its derivatives are able to induce or facilitate cardioprotection.
We further investigated whether CAP exerts cardioprotection via autophagy. Therefore, in the present study we determined whether autophagy is required for CAP-induced cardioprotection and which stage of autophagy is essential for the cardioprotective effect of pharmacologically induced autophagy. We used TAT-HA-Atg5K130R to inhibit autophagosome formation and CQ to investigate the role of lysosomal degradation. Atg5 is not known to participate in other pathways besides autophagy, and therefore TAT-HA-Atg5K130R is the most specific inhibitor available. In the present study we found that pretreatment with TAT-HA-Atg5K130R abolished the infarct size limiting effect of CAP. Similarly, Shiomi et al. found that a non-specific autophagy inhibitor, 3-methyladenine, abolished the CAP-induced restoration of sevoflurane preconditioning after prolonged ischemic insult [22]. These results demonstrate that the sequestration phase of autophagy is essential for CAP-induced cardioprotection against acute ischemia/reperfusion injury.
The role of autophagosomal clearance in cardioprotection has been investigated in several models. Guo et al. [23] showed that one hour after cardiac ischemia LC3-II/I ratio was significantly elevated, and that ischemic postconditioning limited the elevation in the LC3-II/I ratio evidencing that cardioprotective interventions might inhibit cardiac autophagy. However, the same study indicated that inhibition of autophagosomal clearance by CQ abolished cardioprotection by ischemic postconditioning, which suggests that inhibition of cardiac autophagy might be controversial in ischemic conditioning. Similarly, Wang et al. investigated the mechanisms underlying resveratrol-mediated cardioprotection, and found that chronic inhibition of autophagic clearance with bafilomycin A1 abrogated resveratrol-mediated cardiac protection in diabetic mice [24]. On the contrary, in our present study, acute treatment with CQ did not abolish CAP-induced cardioprotection in isolated healthy hearts. These findings suggest that although in vivo inhibition of autophagic clearance interferes with several types of cardioprotective stimuli, autophagic clearance is not likely to be necessary for cardioprotection triggered by pharmacologically induced autophagy in an ex vivo setting, the basis of which is unclear.
5. Conclusion
Here we have shown for the first time in the literature that the sequestration, but not the clearance phase of autophagy is necessary for the cardioprotective effect of CAP. Therefore, therapeutic tools developed on the basis of inducing autophagy sequestration might lead to novel therapeutic options against acute myocardial ischemia/reperfusion injury.
Acknowledgments
Funding
This work was supported by the European Foundation for the Study of Diabetes (EFSD 2013) and Hungarian Scientific Research Fund (OTKA K 109737 to Peter Ferdinandy), OTKA PD 109051 (Zoltan Giricz). Zoltan Giricz held a “János Bolyai Fellowship” from the Hungarian Academy of Sciences and Zoltan Varga was supported by the National Program of Excellence (TAMOP 4.2.4.A/1-11-1-2012-0001). Peter Ferdinandy was a Szentágothai Fellow of the National Program of Excellence (TAMOP 4.2.4.A/2-11-1-2012-0001). NIH P01-HL112730 supported Roberta A. Gottlieb (Principal Investigator) and Robert M. Mentzer, Jr. (Project Leader).
Footnotes
Declaration of Conflicting Interests
The authors declare that there is no conflict of interest.
6. References:
- 1.Gustafsson AB, Gottlieb RA: Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol 2008, 44:654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.He C, Klionsky DJ: Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009, 43:67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N: Autophagy: process and function. Genes & Development 2007, 21:2861–2873. [DOI] [PubMed] [Google Scholar]
- 4.Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM Jr., Gottlieb RA: Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res 2010, 3:365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giricz Z, Mentzer RM Jr., Gottlieb RA: Autophagy, myocardial protection, and the metabolic syndrome. J Cardiovasc Pharmacol 2012, 60:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andres AM, Hernandez G, Lee P, Huang C, Ratliff EP, Sin J, Thornton CA, Damasco MV, Gottlieb RA: Mitophagy is required for acute cardioprotection by simvastatin. Antioxid Redox Signal 2014, 21:1960–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shiomi M, Miyamae M, Takemura G, Kaneda K, Inamura Y, Onishi A, Koshinuma S, Momota Y, Minami T, Figueredo VM: Sevoflurane induces cardioprotection through reactive oxygen species-mediated upregulation of autophagy in isolated guinea pig hearts. J Anesth 2014, 28:593–600. [DOI] [PubMed] [Google Scholar]
- 8.Huang C, Liu W, Perry CN, Yitzhaki S, Lee Y, Yuan H, Tsukada YT, Hamacher-Brady A, Mentzer RM Jr., Gottlieb RA: Autophagy and protein kinase C are required for cardioprotection by sulfaphenazole. Am J Physiol Heart Circ Physiol 2010, 298:H570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prigione A, Cortopassi G: Mitochondrial DNA deletions and chloramphenicol treatment stimulate the autophagic transcript ATG12. Autophagy 2007, 3:377–380. [DOI] [PubMed] [Google Scholar]
- 10.Granville DJ, Tashakkor B, Takeuchi C, Gustafsson AB, Huang C, Sayen MR, Wentworth P Jr., Yeager M, Gottlieb RA: Reduction of ischemia and reperfusion-induced myocardial damage by cytochrome P450 inhibitors. Proc Natl Acad Sci U S A 2004, 101:1321–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He H, Chen M, Scheffler NK, Gibson BW, Spremulli LL, Gottlieb RA: Phosphorylation of mitochondrial elongation factor Tu in ischemic myocardium: basis for chloramphenicol-mediated cardioprotection. Circ Res 2001, 89:461–467. [DOI] [PubMed] [Google Scholar]
- 12.Sala-Mercado JA, Wider J, Undyala VV, Jahania S, Yoo W, Mentzer RM Jr., Gottlieb RA, Przyklenk K: Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia-reperfusion injury. Circulation 2010, 122:S179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li C, Liu Y, Liu H, Zhang W, Shen C, Cho K, Chen X, Peng F, Bi Y, Hou X, et al. : Impact of autophagy inhibition at different stages on cytotoxic effect of autophagy inducer in glioblastoma cells. Cell Physiol Biochem 2015, 35:1303–1316. [DOI] [PubMed] [Google Scholar]
- 14.Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, Cho DH, Choi B, Lee H, Kim JH, et al. : Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 2005, 280:20722–20729. [DOI] [PubMed] [Google Scholar]
- 15.Hamacher-Brady A, Brady NR, Gottlieb RA: Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem 2006, 281:29776–29787. [DOI] [PubMed] [Google Scholar]
- 16.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB: Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ 2007, 14:146–157. [DOI] [PubMed] [Google Scholar]
- 17.Kimura T, Takabatake Y, Takahashi A, Isaka Y: Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res 2013, 73:3–7. [DOI] [PubMed] [Google Scholar]
- 18.Gorbe A, Giricz Z, Szunyog A, Csont T, Burley DS, Baxter GF, Ferdinandy P: Role of cGMP-PKG signaling in the protection of neonatal rat cardiac myocytes subjected to simulated ischemia/reoxygenation. Basic Res Cardiol 2010, 105:643–650. [DOI] [PubMed] [Google Scholar]
- 19.Bell RM, Mocanu MM, Yellon DM: Retrograde heart perfusion: the Langendorff technique of isolated heart perfusion. J Mol Cell Cardiol 2011, 50:940–950. [DOI] [PubMed] [Google Scholar]
- 20.Hausenloy DJ, Garcia-Dorado D, Botker HE, Davidson SM, Downey J, Engel FB, Jennings R, Lecour S, Leor J, Madonna R, et al. : Novel targets and future strategies for acute cardioprotection: Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc Res 2017, 113:564–585. [DOI] [PubMed] [Google Scholar]
- 21.Ambrose PJ: Clinical pharmacokinetics of chloramphenicol and chloramphenicol succinate. Clin Pharmacokinet 1984, 9:222–238. [DOI] [PubMed] [Google Scholar]
- 22.Shiomi M, Miyamae M, Takemura G, Kaneda K, Inamura Y, Onishi A, Koshinuma S, Momota Y, Minami T, Figueredo VM: Induction of autophagy restores the loss of sevoflurane cardiac preconditioning seen with prolonged ischemic insult. Eur J Pharmacol 2014, 724:58–66. [DOI] [PubMed] [Google Scholar]
- 23.Guo L, Xu JM, Mo XY: Ischemic postconditioning regulates cardiomyocyte autophagic activity following ischemia/reperfusion injury. Mol Med Rep 2015, 12:1169–1176. [DOI] [PubMed] [Google Scholar]
- 24.Wang B, Yang Q, Sun YY, Xing YF, Wang YB, Lu XT, Bai WW, Liu XQ, Zhao YX: Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J Cell Mol Med 2014, 18:1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]