

Summary
The RING E3 ubiquitin ligase UHRF1 controls DNA methylation through its ability to target the maintenance DNA methyltransferase DNMT1 to newly replicated chromatin. DNMT1 recruitment relies on ubiquitylation of histone H3 by UHRF1; however, how UHRF1 deposits ubiquitin onto the histone is unknown. Here, we demonstrate that the ubiquitin-like domain (UBL) of UHRF1 is essential for RING-mediated H3 ubiquitylation. Using chemical crosslinking and mass spectrometry, biochemical assays, and recombinant chromatin substrates, we show that the UBL participates in structural rearrangements of UHRF1 upon binding to chromatin and the E2 ubiquitin conjugating enzyme UbcH5a/UBE2D1. Similar to ubiquitin, the UBL exerts its effects through a hydrophobic patch that contacts a regulatory surface on the “backside” of the E2 to stabilize the E2-E3-chromatin complex. Our analysis of the enzymatic mechanism of UHRF1 uncovers an unexpected function of the UBL domain and defines a new role for this domain in DNMT1-dependent inheritance of DNA methylation.
Keywords: UHRF1, chromatin, ubiquitylation, ubiquitin-like fold, DNA methylation, histone modification
Graphical Abstract
Highlights
-
•
The UBL domain of UHRF1 is required for its E3 ubiquitin ligase activity
-
•
A hydrophobic patch on the UBL is required to form a stable E2/E3/chromatin complex
-
•
The UHRF1 N terminus and UBL hydrophobic patch control targeted H3 ubiquitylation
-
•
DNMT1-mediated maintenance DNA methylation requires the UBL hydrophobic patch
Foster et al. identify a functional role for the ubiquitin-like domain of UHRF1 in its E3 ubiquitin ligase activity. Biochemical and cell-based assays reveal that a hydrophobic patch on the UBL domain controls targeted histone H3 ubiquitylation that is required for DNMT1 recruitment to newly replicated chromatin and subsequent maintenance of DNA methylation.
Introduction
DNA and histone modifications regulate chromatin function and mediate processes such as transcription, DNA repair, and DNA replication. Maintaining chromatin modifications after DNA replication is essential for chromatin homeostasis, especially for silenced regions of the genome, such as repetitive elements. The maintenance methyltransferase DNMT1 ensures that cytosine methylation at CpG dinucleotides is propagated to daughter cells. To achieve this, DNMT1 is recruited to newly replicated hemi-methylated DNA in order to copy CpG methylation marks to the newly synthesized DNA strand. The E3 ubiquitin ligase UHRF1 (ubiquitin-like containing PHD and RING finger domains protein 1) was found to play a critical role in this targeting process (Bostick et al., 2007, Sharif et al., 2007). UHRF1 is a multi-domain epigenetic regulator (Figure 1A) that reads histone H3 Lys-9 di- and tri-methylation (H3K9me2/3) marks in the context of an otherwise unmodified N terminus through its linked TTD-PHD (tandem-tudor domain and plant homeodomain) module (Arita et al., 2012, Rothbart et al., 2012) in addition to binding hemi-methylated CpGs via its SRA (SET and RING-associated) domain (Arita et al., 2008, Hashimoto et al., 2008). The recruitment of DNMT1 to newly replicated chromatin critically depends on the ubiquitylation of Lys-14, Lys-18, and/or Lys-23 in histone H3 that is catalyzed by the E3 ubiquitin ligase activity of UHRF1 harbored in its C-terminal RING domain (Nishiyama et al., 2013). The tandem mono-ubiquitin marks on histone H3 are recognized by DNMT1 via a ubiquitin interaction motif in its RFTS (replication foci targeting sequence) domain (Qin et al., 2015). In contrast to the targeting of DNMT1 to ubiquitylated histone H3, which has been elucidated in structural detail (Ishiyama et al., 2017), the enzymatic mechanism by which UHRF1 deposits ubiquitin onto the histone is not completely understood.
Figure 1.

The UBL Domain of UHRF1 Contacts Other Regions within the Protein
(A) Schematic of full-length mouse UHRF1 (Np95) indicating domain architecture and function. Red lines indicate known intra-molecular interactions.
(B) Crosslinking with mass spectrometry (XL-MS) profile of UHRF1. Crosslinks involving the UBL domain are shown in purple and other intra-protein crosslinks in red. Individual domains within UHRF1 are annotated.
(C) Proposed model of UHRF1 folding.
Recent studies indicated that SRA-mediated binding to hemi-methylated DNA can stimulate E3 activity in vitro, suggesting an allosteric regulation of UHRF1 by the chromatin substrate or by inter-domain contacts within the protein (Harrison et al., 2016, Vaughan et al., 2018). Such intra-molecular contacts between domains within UHRF1 have indeed been found to regulate its chromatin reader activity. Interactions of the polybasic region (PBR) in the linker between the SRA and the RING domain and of the linker between the TTD and the PHD domains with a groove in the TTD are thought to compete for binding to H3K9me2/3 marks (Gelato et al., 2014), and the PHD and SRA domains have also been shown to interact (Fang et al., 2016). These contacts are thought to be present in a “closed” auto-inhibited conformation of UHRF1, with binding of specific biological molecules, such as PI(5)P (Gelato et al., 2014), hemi-methylated DNA (Fang et al., 2016), LIG1 (Ferry et al., 2017), or USP7 (Zhang et al., 2015), shifting the conformation of UHRF1 to an “open” state competent to bind to its target modifications and to recruit DNMT1 to chromatin. Whether intra-molecular rearrangements are involved in the enzymatic mechanism of UHRF1, however, is not known. In addition, UHRF1 contains a conserved N-terminal ubiquitin-like domain (UBL) with so far elusive function.
Mechanistic insights into the biology of UHRF1 rely to a large extent on biochemical and structural studies using isolated domains or protein fragments in combination with modified histone peptides and oligonucleotides. These approaches, however, cannot recapitulate the numerous inter- and intra-molecular interactions and motions of the intact full-length protein when it engages with its chromatin substrate to exert its catalytic function. In order to probe global conformational changes and interactions of the full-length UHRF1 protein upon binding to different chromatin substrates, we applied an in vitro crosslinking and mass spectrometry (XL-MS) assay. Using this approach, we found that the UBL domain contacts other domains within UHRF1. Subsequent investigation of the function of the UBL domain revealed that it is not primarily involved in chromatin binding but in the RING-dependent E3 ubiquitin ligase activity of UHRF1 toward histone H3. We found that the UBL participates in distinct structural rearrangements within UHRF1 that are triggered by engagement of the protein with chromatin and the E2 ubiquitin conjugating enzyme UbcH5a. Removal of the whole domain or mutation of a single residue in a hydrophobic patch of the UBL domain interferes with the efficient recruitment of the E2 enzyme to chromatin. Similar to the regulatory interaction of ubiquitin with E2 enzymes of the UbcH5 family, we find that contacts between this hydrophobic patch and a regulatory “backside” surface on UbcH5a are necessary for efficient ubiquitin transfer from the E2 to histone H3. Mutation of the hydrophobic patch within UHRF1 in mouse embryonic stem cells results in reduced DNMT1 recruitment to newly replicated chromatin and loss of DNA methylation at repetitive elements. Our results demonstrate an essential function of the UBL domain for the UHRF1 E3 ligase activity in stabilizing the E2-E3-chromatin complex and explain its role in DNMT1-dependent inheritance of DNA methylation.
Results
The UBL Domain Makes Intra-molecular Contacts to Other Domains in UHRF1
Structures of the isolated domains of UHRF1, in complex with modified DNA oligonucleotide or peptide ligands, have been solved (Arita et al., 2008, Arita et al., 2012, Hashimoto et al., 2008, Rajakumara et al., 2011, Rothbart et al., 2012, Rothbart et al., 2013). However, due to its conformational flexibility and size, it is difficult to study the structure of the full-length UHRF1 protein by crystallography, electron microscopy (EM), or nuclear magnetic resonance approaches. To probe for intra-molecular interactions within UHRF1, we subjected recombinant mouse full-length UHRF1 (Np95) to a crosslinking and mass spectrometry approach (Figures 1B and S1A). Using a lysine-reactive crosslinker, we detected previously identified interactions within UHRF1, such as between the TTD and the PBR and between the PHD finger and the SRA domain. Other previously described contacts, such as between the TTD and TTD-PHD linker and between the PBR and the PHD-finger, were also detected, thus validating our approach. A surprising result from these XL-MS experiments was the presence of extensive crosslinks between the N-terminal UBL domain and multiple other regions within UHRF1. The crosslinks are likely to be intra-molecular, as UHRF1 forms a monomer in solution as demonstrated by size-exclusion chromatography and crosslinking at concentrations exceeding those used for the BS3 experiments (Figures S1B and S1C). These findings indicate that the UBL domain folds back onto the rest of the protein and is located in close proximity to the other domains of UHRF1 (Figure 1C).
The UBL Domain Is Required for the E3 Ubiquitin Ligase Activity of UHRF1
To investigate the contribution of the UBL domain to the function of UHRF1, we first performed nucleosome-binding experiments with full-length UHRF1 and truncated fragments (Figures S2A–S2C) to test an involvement in chromatin targeting. As previously reported, full-length recombinant UHRF1 had only little preference for mono-nucleosomes containing H3K9me3 marks and/or hemi-methylated CpGs in the linker DNA. In contrast, a fragment spanning the TTD, PHD, and SRA domains (amino acids [aas] 118–621) showed increased specificity for the H3K9me3/hemi-methylated DNA marks. Removal of the UBL domain, however, did not result in a reduction or altered specificity of nucleosome binding but rather to overall stronger interactions. The isolated UBL domain did not bind to nucleosomes, even at high molar ratios. It therefore seems that the UBL domain does not directly contribute to chromatin binding or to modulating the specificity of UHRF1 for modification marks.
UHRF1 is a RING-type E3 ubiquitin ligase. Having ruled out a function for the UBL domain in chromatin binding, we explored a possible role in the E3 ubiquitin ligase activity. We identified UbcH5a (UBE2D1) from a panel of E2 ubiquitin conjugating enzymes as the E2 showing the strongest stimulation of UHRF1 auto-ubiquitylation (Figure S3A) and used it to set up an in vitro ubiquitylation assay for UHRF1 (Figures S3B–S3E). Using this assay, we tested UHRF1 E3 ubiquitin ligase activity toward various unmodified nucleosomal substrates. UHRF1 displayed the highest E3 activity when 12 × 187 bp chromatin arrays (containing 12 nucleosomes regularly spaced by 601-nucleosome positioning sequences with 187 bp repeat length) were used as substrate, as evidenced by a faster rate of formation of di- and tri-ubiquitylated H3 species, whereas less E3 activity was detected with mono-nucleosomes or di- and tetra-nucleosomes (Figures 2A and S3F). Although mass spectrometric analysis of the modified histone substrate indicates specificity toward the N terminus of histone H3, with predominant ubiquitylation of Lys-18 and Lys-23 (Figure S3D; Data S1), as previously found, it is yet unclear how chromatin arrays enhance the activity.
Figure 2.

The UHRF1 E3 Ubiquitin Ligase Activity Is Stimulated by Longer Chromatin Arrays and Requires the UBL Domain
(A) Wild-type UHRF1 was used in E3 ubiquitin ligase assays in the presence of different chromatin substrates. An increase in the rate and amount of H3 ubiquitylation was observed in the presence of 12 × 187 bp chromatin arrays compared to mono-, di-, and tetra-nucleosomes (for quantification, see Figure S3F). Similarly, increased H3 ubiquitylation as well as poly-ubiquitin chain formation but minimal UHRF1 auto-ubiquitylation is detected in blots probed against the hemagglutinin (HA)-tagged ubiquitin.
(B) A RING finger point mutant (H730A) severely disrupts UHRF1-mediated H3 ubiquitylation and UHRF1 auto-ubiquitylation using 12 × 187 bp chromatin arrays as substrate. Removal of the UBL domain (ΔUBL) results in undetectable E3 activity toward histone H3 even over a long time course.
(C) E3 assays carried out with 0.4 μM UHRF1 (WT) and 1 μM UHRF1 ΔUBL and free UBL domain added in molar excess as indicated. Addition of increasing amounts of the free UBL domain to the ΔUBL fragment in trans did not result in stimulation of E3 activity.
(D) Equimolar mixtures of UHRF1 (wild-type, ΔUBL, or H730A) were tested in E3 assays to probe for inter-molecular complementation of E3 activity on chromatin arrays. Half-filled circles indicate the addition of 50% of the amount of UHRF1 compared to the standard assay conditions (filled circles).
Two recent studies found that UHRF1-mediated H3 ubiquitylation is stimulated in trans by short hemi-methylated DNA oligonucleotides (Harrison et al., 2016, Vaughan et al., 2018). We initially used unmodified nucleosome or chromatin substrates because interaction assays with mono-nucleosomes and full-length UHRF1 showed no substantial difference in binding between nucleosomes containing unmodified octamers, H3K9me3 octamers, or hemi- or fully-methylated CpG DNA (Figures S2A–S2C). In further tests with modified substrates (Figure S4A), the H3K9me3 modification had a stimulatory effect on the rate and extent of H3 ubiquitylation in the context of both mono-nucleosomes and chromatin arrays. Use of fully CpG methylated DNA led to an increase in UHRF1 auto-ubiquitylation rather than increased H3 ubiquitylation. The latter observation is in line with the reported inability of symmetrically CpG-methylated DNA oligonucleotides to stimulate the UHRF1 E3 ligase activity toward K9me2-modifed histone H3 N-terminal peptides (Harrison et al., 2016, Vaughan et al., 2018). Assays with mono-nucleosomes containing specifically positioned hemi-methylated CpGs in the linker DNA, which can be recognized by UHRF1 (Zhao et al., 2016), revealed intriguing ubiquitylation patterns (Figure S4B). Insertion of three hemi-methylated CpGs in the 3′ linker of the nucleosomal DNA resulted in a stark reduction in H3 ubiquitylation, and placing a single CpG in the 5′ linker resulted in H3 ubiquitylation equivalent to unmodified mono-nucleosomes. In both cases, UHRF1 auto-ubiquitylation was strongly stimulated compared to unmodified mono-nucleosomes. These results indicate that stimulation and targeting of the UHRF1 E3 activity within the chromatin context is a two-step process and that extent and position of pre-existing modifications on a nucleosome influence the enzymatic rate and the correct transfer of ubiquitin to histone H3. Without techniques to generate suitable long hemi-methylated chromatin substrates, these questions are currently difficult to address.
Using this in vitro assay and 12 × 187 bp chromatin arrays, we tested a mutant of UHRF1 lacking the UBL domain (ΔUBL). While a H730A point mutation that disrupts E3 activity by abolishing Zn2+ ion coordination in the RING finger drastically reduced H3 ubiquitylation, histone H3 ubiquitylation was almost absent in the ΔUBL mutant, with H3-Ub barely detectable even in long time courses (Figure 2B). To test whether addition of the UBL domain could rescue the UBL domain deletion in trans, we added a molar excess of purified UBL domain (Figure 2C) to wild-type and ΔUBL UHRF1 variants. No rescue of E3 activity of the ΔUBL mutant was observed even at high molar ratios nor was there enhanced activity for wild-type UHRF1. To test whether the UBL domain of a neighboring UHRF1 molecule, potentially positioned in close proximity within the same chromatin array, could substitute for the missing UBL domain in the ΔUBL mutant, we tested 1:1 mixtures of wild-type, ΔUBL, or H730A UHRF1 variants in E3 assays (Figure 2D). Crucially, no increase in activity was detected for a mixture of the ΔUBL and H730A mutants, indicating that a UBL domain from another UHRF1 molecule cannot rescue the E3 activity of the truncated ΔUBL mutant in trans. We conclude that the UBL domain is essential for the E3 ubiquitin ligase activity of UHRF1 and that it needs to be present within the context of the full-length protein to stimulate the E3 activity.
The N Terminus of UHRF1 Is Required for Targeted Ubiquitin Transfer from the E2 to the H3 Substrate
In initial experiments to test the functionality of UHRF1 mutants in mouse embryonic stem cells (mESCs), we found that N-terminally 3×FLAG-tagged UHRF1 was unable to rescue DNA methylation in Uhrf1−/− knockout cells, suggesting that the N terminus of UHRF1 might be functionally important. Data presented by DaRosa et al. (2018), in this issue of Molecular Cell, also indicated to us that mutation of Trp-2 to Ala resulted in diminished UHRF1-mediated histone H3 ubiquitylation. We initially used a version of UHRF1 lacking the first two amino acids (Met-Trp) in our in vitro experiments due to a BamHI restriction site at bp 5 in the UHRF1 cDNA that we used for cloning. Comparison of this mutant (UHRF1ΔMW) with wild-type UHRF1 (UHRF1WT) containing the native N terminus in E3 assays recapitulated this effect, with stark reduction in H3 ubiquitylation even over long time courses and drastically increased UHRF1 auto-ubiquitylation in the UHRF1ΔMW mutant (Figure 3A). Similar to UHRF1WT, E3 activity of UHRF1ΔMW was increased with 12 × 187 bp arrays compared to mono-, di-, or tetra-nucleosomes (Figure 3B). However, there was no appreciable difference in activity of UHRF1ΔMW on fully CpG-methylated chromatin arrays with or without H3K9me3 when compared to unmodified arrays (Figure S5A). This suggests that the very N-terminal residues in the UBL domain contribute to sensing the modifications and play a crucial role in directing the transfer of ubiquitin from E2∼Ub specifically to histone H3.
Figure 3.

The N Terminus of UHRF1 Is Required for Targeted Ubiquitin Transfer from the E2 to Histone H3
(A) E3 assays using 12 × 187 bp chromatin arrays as substrate. Deletion of Met-Trp at the very N terminus substantially decreases UHRF1-mediated H3 ubiquitylation while increasing UHRF1 auto-ubiquitylation as detected by higher molecular weight species in the anti-HA blot.
(B) UHRF1ΔMW auto-ubiquitylation and H3 ubiquitylation is stimulated in the presence of 12 × 187 bp arrays in E3 assays when compared against mono-, di-, and tetra-nucleosomes, where very little E3 activity is detected.
In the case of both UHRF1WT and UHRF1ΔMW, the use of a ubiquitin mutant in which all lysines are mutated to arginines (NoK) resulted in no detectable reduction in ubiquitylated histone H3 species (Figure S5B), indicating that H3 is modified by multiple mono-ubiquitins rather than poly-ubiquitin chains, confirming previous results and validating our assay.
The UBL Domain Is Involved in the Formation of the Enzyme/Substrate Complex
To further investigate the role of the UBL domain in the E3 activity of UHRF1, we probed the interaction between UHRF1 and the E2 enzyme UbcH5a/UBE2D1. In glutathione S-transferase (GST) pull-down experiments using GST-UbcH5a as bait, UHRF1 binding was barely detectable when added on its own. In the presence of 12 × 187 bp chromatin arrays, however, a complex between UbcH5a, UHRF1, and chromatin could form. In contrast, complex formation was reduced when adding mono-nucleosomes (Figure 4A). This is consistent with the increased UHRF1 E3 activity on chromatin arrays, but not with shorter chromatin templates. Interestingly, the 12 × 187 bp chromatin arrays could not bind to UbcH5a in the absence of UHRF1 (Figure 4B, lane 10), implying that UbcH5a preferentially recognizes chromatin-bound UHRF1 and only when all three components are present is a stable E2-E3-substrate complex formed, allowing ubiquitin transfer from E2∼Ub to the substrate. Removal of the UBL domain resulted in ∼50% reduction in complex formation compared to the UHRF1 protein (ΔMW variant), and the effect of removing the RING-linker region was even stronger (Figures 4B and 4C). Removal of both the UBL and the RING-linker domains had an additive effect with a fragment spanning aas 118–621 showing an almost complete loss of the ability to form the UbcH5a/UHRF1/chromatin complex (Figures 4B and 4C), demonstrating that the UBL and the RING-linker region cooperate in complex formation. Although the reduced binding of ΔUBL UHRF1 to UbcH5a in the presence of 12 × 187 bp arrays might explain a partially reduced E3 activity of UHRF1, it does not explain the almost complete loss of activity that we observe. Therefore, it is likely that the UBL domain aids in transferring ubiquitin from E2∼Ub to the histone H3 substrate in addition to its role in recruiting UbcH5a. The N terminus seems not to be involved in E2 binding, as both wild-type and the ΔMW mutant behave similarly in these assays (Figure 4A).
Figure 4.

The RING Finger and UBL Domain of UHRF1 Are Required for Stabilizing the E2/E3/Chromatin Complex
(A) GST-UbcH5a pull-down assays indicate that UHRF1 alone shows very little binding to UbcH5a. Binding of UHRF1 to GST-UbcH5a is stimulated in the presence of 12 × 187 bp chromatin arrays but less so by mono-nucleosomes. Similar results are seen when comparing UHRF1WT and UHRF1ΔMW.
(B) GST-pull-down assays using UHRF1 (ΔMW and the indicated mutants) in the absence or presence of 12 × 187 bp chromatin arrays. Binding was analyzed as in (A).
(C) The binding of UHRF1 variants in the presence of 12 × 187 bp arrays relative to GST-UbcH5a, shown in (B), was quantified using ImageJ in at least 3 independent experiments and normalized to the UHRF1ΔMW protein. The mean was plotted ± the SEM.
Changes in UHRF1 Conformation upon E2 and Substrate Binding
In order to probe potential conformational rearrangements within UHRF1 upon formation of the UbcH5a/UHRF1/chromatin complex, we set out to identify inter- and intra-molecular interactions using XL-MS experiments with UHRF1 and UbcH5a in the presence (Figure 5E) or absence (Figure 5D) of 12 × 187 bp chromatin arrays. As comparison, we tested UHRF1 interactions in the presence of the 12 × 187 bp arrays (Figure 5C) without UbcH5a and also UHRF1 with mono-nucleosomes (Figure 5B). These experiments indicate that UHRF1 binds to mono-nucleosomes in a state resembling the proposed closed conformation. Strikingly, there are far fewer crosslinks detected within UHRF1 when bound to mono-nucleosomes (Figure 5B) compared to free UHRF1 (Figure 5A). This could be due to “masking” of low abundant crosslinked UHRF1 peptides by the additional histone peptides in the XL-MS samples. Alternatively, this could indicate a change in flexibility of UHRF1. XL-MS captures different conformations over time and thus samples mixtures of multiple structural states, especially with flexible proteins, such as UHRF1. Fewer crosslinks could indicate that nucleosome binding stabilizes UHRF1 in the closed state, which reduces the conformational space available, limiting the crosslinks to a few available lysines stably positioned in close proximity. Although the overall conformation of UHRF1 when bound to 12 × 187 bp arrays appears similar to mono-nucleosomes, there are some changes in the crosslinks within UHRF1 and between UHRF1 and the histones. Most notably, the TTD-PBR interaction is no longer detected, indicating that UHRF1 engages differently with mono-nucleosomes and chromatin arrays. UbcH5a crosslinks to the SRA-PBR region of UHRF1 via its N and C termini (Figure 5D). In the UbcH5a/UHRF1/chromatin complex, UbcH5a also crosslinks to the UBL and TTD domains (Figure 5E). This implies a close proximity between the UBL, TTD, PHD, and SRA domains; the H3 tail; and UbcH5a and that E2 binding induces another conformation in UHRF1 distinct from the one bound to mono-nucleosomes or chromatin arrays alone. The low number of crosslinks within UHRF1 when complexed to UbcH5a and the 12 × 187 bp arrays also suggests that UHRF1 assumes a more stable conformation in the E2/E3/chromatin complex. As a comparison, we carried out XL-MS experiments with the UHRF1ΔMW mutant (Figures 5A–5E, right). Strikingly, binding of UHRF1ΔMW to the 12 × 187 bp arrays results in loss of “long-range” contacts between the UBL domain and the PHD, SRA, and PBR regions. This indicates that engaging with chromatin arrays forces an open conformation in UHRF1ΔMW. In addition, UHRF1ΔMW is generally slightly differently connected to the histones and to UbcH5a, possibly indicating a different overall positioning of nucleosomes, UHRF1ΔMW, and UbcH5a in the E2/E3/chromatin complex.
Figure 5.

XL-MS Experiments to Probe the Conformational Changes in UHRF1 upon Chromatin and E2 Binding
(A–E) XL-MS diagrams of UHRF1 in isolation (A), with mono-nucleosomes (B), 12 × 187 bp chromatin arrays (C), UbcH5a/UBE2D1 (D), and in the context of 12 × 187 bp arrays and UbcH5a (E). Intra-protein crosslinks are in red and inter-protein crosslinks in blue (histones) or green (UbcH5a). XL-MS experiments with UHRF1WT are on the left and UHRF1ΔMW on the right. Schematic representations of the proposed UHRF1 conformation in each experiment are shown with arrows indicating the predominant crosslinks from UHRF1 to histones and UbcH5a. UHRF1 domains are color coded as in Figure 1A, with histones in yellow and UbcH5a in green. Table S1 has the list of crosslinks identified in each experiment, and Data S1 has an example spectrum as output from StavRox.
Because XL-MS measurements are quite variable and not quantitative, we cannot draw firm conclusions as to definitive structural arrangements within the assemblies. Our XL-MS results rather give indications of proximities between protein regions and whether certain interactions are favored over others. In aggregate, we conclude that UHRF1 and the histones assume distinct structural conformations upon formation of the E2/E3/chromatin complex; that in this complex, the histone H3 N-terminal tail, the UBL, TTD, PHD, and SRA domains, and the E2 are all in close proximity; and that the very N terminus of UHRF1 influences these structural rearrangements. Interestingly, auto-ubiquitylation sites identified by mass spectrometry in UHRF1ΔMW (Figure S5C; Data S1) cluster in regions of UHRF1 close to E2 crosslinks (Figure 5E), consistent with these regions being next to the E2 active site and therefore prone to becoming ubiquitylated.
A Hydrophobic Patch on the UBL Domain Mediates the Stimulatory Effect on the UHRF1 E3 Activity
UbcH5 family E2 enzymes contain a regulatory surface positioned on the backside opposite the E2∼Ub thioester site (Brzovic et al., 2006) that can be bound by a second ubiquitin to stimulate transfer of the active site ubiquitin to the substrate (Buetow et al., 2015). This contact to the backside of UbcH5 family E2 enzymes was found to be mediated by a hydrophobic patch on ubiquitin centered on Ile-44 (Brzovic et al., 2006). Analysis of the sequence and of a PDB structure of the human UHRF1 UBL domain (PDB: 2FAZ) shows a ubiquitin fold and a similarly placed hydrophobic patch with a phenylalanine (Phe-46) occupying the position of Ile-44 (Figure 6A). To test whether this hydrophobic patch in the UBL domain plays a similar role in the E3 activity of UHRF1, we mutated Phe-46 to Ala (F46A). In E3 assays, the F46A UHRF1 mutant behaved like the ΔUBL UHRF1 mutant with strongly reduced H3 ubiquitylation (Figures 6B and S6A). Residual activity is detected with the F46A mutant, indicating that other residues within the UBL domain contribute to facilitating the ubiquitin transfer from the E2 to the substrate. Chromatin electrophoretic mobility shift assays (EMSAs) to ensure that the deletion of the UBL domain or the F46A mutation do not affect UHRF1 binding to the 12 × 187 bp arrays used in the E3 assays indicated no substantial difference in binding between the wild-type and the UBL domain mutants (Figures S6B and S6C). However, the formation of the complex between UHRF1, chromatin arrays, and GST-UbcH5a was reduced to similar levels observed for UHRF1 ΔUBL in the F46A mutant (Figures 4B, 4C, and 6C). The F46A mutant therefore behaves functionally equivalent to the deletion of the complete UBL domain, demonstrating that the hydrophobic patch of the UBL domain is indeed required to stimulate ubiquitin transfer from E2∼Ub to the histone H3 substrate. The regulatory surface on UbcH5a that binds to the hydrophobic patch of ubiquitin is concentrated around Ser-22 (Brzovic et al., 2006). Mutation of this Ser to Arg (S22R) in UbcH5a resulted in an almost complete loss of auto-ubiquitylation on UHRF1 (both wild-type [WT] and ΔMW) and loss of histone H3 ubiquitylation in our E3 assays (Figure 6B). In GST-UbcH5a pull-downs with chromatin arrays, the S22R mutant E2 reduced UHRF1 binding to similar levels as the UHRF1 F46A mutant (Figure 6D). Our results uncover that the UBL domain acts through the regulatory backside surface of UbcH5a via a hydrophobic patch analogous to the way ubiquitin regulates E2 enzymes of the UbcH5 family and that this interaction between the UBL and the E2 is essential for the E3 ligase activity of UHRF1 by stabilizing an active E2/E3/substrate complex (Figure 6E).
Figure 6.

The E3 Activity of UHRF1 Is Dependent on a Hydrophobic Patch on the UBL and a Regulatory Ubiquitin Binding Surface on the E2
(A) (Top panel) Atomic space-fill structures of ubiquitin (PDB: 1UBQ) and the UHRF1 UBL domain (PDB: 2FAZ) color coded by hydrophobicity. Orange-red represents more hydrophobic residues, whereas blue residues are more polar. (Bottom panel) Sequence alignment of human ubiquitin with the UBL domain of UHRF1 from various species and the UBL domain of human Parkin. F46 in the UHRF1 UBL and corresponding isoleucines in ubiquitin and Parkin are indicated in red.
(B) E3 ubiquitin ligase assays carried out with the indicated mutants over 1 hr at 25°C. H3 ubiquitylation is drastically reduced in the F46A mutant with some UHRF1 auto-ubiquitylation observed. The S22R mutation in UbcH5a abrogates ubiquitylation of the substrate (UHRF1 or histone H3) in the context of both UHRF1WT and UHRF1ΔMW.
(C) GST-UbcH5a pull-downs using UHRF1WT and the F46A mutant. Binding assays were carried out as described in Figure 4C. Quantification of three independent experiments indicates reduced binding of F46A UHRF1 to UbcH5a relative to the wild-type in the presence of 12 × 187 bp arrays. The mean was plotted ± the SEM.
(D) GST-UbcH5a pull-downs using UHRF1WT with wild-type UbcH5a and the S22R mutant in the presence of 12 × 187 bp arrays. Quantification of three independent experiments reveals reduced binding of UHRF1 to S22R UbcH5a compared with the wild-type. The mean was plotted ± the SEM.
(E) Proposed model for the contact between the hydrophobic patch on the UBL domain of UHRF1 (indicated in orange) and the regulatory region on UbcH5a centered on Ser-22.
The UBL Hydrophobic Patch Is Critical for the Maintenance Methylation Function of UHRF1
Lastly, we tested the contribution of the hydrophobic patch on the UBL domain to the maintenance methylation function of UHRF1 in a physiological setting. We introduced the F46A UBL mutation into mESCs via a CRISPR/Cas9-targeted knockin approach in addition to a H730A mutant to disrupt the RING-dependent E3 activity (Figure S7A). We first investigated whether the UBL domain mutation resulted in defective recruitment of the maintenance DNA methyl transferase DNMT1 to nuclear replication foci similar to ubiquitylation-deficient RING domain mutants of UHRF1 (Nishiyama et al., 2013). To this end, we labeled the Uhrf1F46A and Uhrf1H730A mutant mESCs with EdU to mark newly replicated DNA in cells undergoing S phase and then performed co-immunofluorescence microscopy by staining against DNMT1 and EdU (Figures 7A and S7B). Comparing the mutants to wild-type mESCs revealed that DNMT1 localization to EdU-labeled replication foci is drastically reduced in the F46A UBL mutant to almost the extent seen in the Uhrf1−/− knockout mESCs (Sharif et al., 2007), which show a delocalized nuclear DNMT1 signal. The H730A RING mutant shows a weaker effect with some detectable DNMT1 targeting to replication foci but increased diffuse nuclear DNMT1 staining compared to wild-type cells. We also assessed DNA CpG methylation levels at repetitive regions (LINE-1 elements) in four independent Uhrf1F46A and Uhrf1H730A mESC clones by bisulfite conversion of genomic DNA and subsequent deep sequencing and compared the degree of CpG methylation to Uhrf1WT and Uhrf1−/− mESCs (Figure 7B). Due to the defective DNMT1 recruitment, Uhrf1−/− mESCs have very low methylation levels (Sharif et al., 2007). As expected, DNA methylation is very low in the Uhrf1F46A mESCs similar to the Uhrf1−/− cells. In line with the DNMT1 localization experiments, the Uhrf1H730A mutant mESCs display a drastic reduction of DNA methylation, however, not as much as the Uhrf1F46A mESCs. The partial DNMT1 localization defect and slightly elevated CpG methylation levels of the Uhrf1H730A mutant compared to the Uhrf1F46A mutant or the knockout correspond to the residual ubiquitylation activity observed for this mutant in our in vitro E3 assays and suggest that some H3 ubiquitylation, and thereby DNA methylation, might still occur in the Uhrf1H730A mutant mESCs. Importantly, the F46A mutant still binds DNMT1 (Figure 7C). The observed effects of this point mutant in the UBL domain of UHRF1 on DNA methylation are therefore likely to be caused by defective histone H3 ubiquitylation and the resulting disruption of DNMT1 recruitment and not by defective direct recruitment of DNMT1 by UHRF1. Taken together, our results show that maintenance DNA methylation critically depends on histone H3 ubiquitylation by UHRF1 and that the hydrophobic patch on the UBL domain of UHRF1 is absolutely required for this activity in vivo in addition to the RING finger.
Figure 7.

A Hydrophobic Patch Mutant of the UBL Domain Does Not Support UHRF1-Dependent DNA Methylation in Mouse ESCs
(A) DNMT1 localization during DNA replication assessed by immunofluorescence in Uhrf1WT, Uhrf1−/−, and Uhrf1 mutant (Uhrf1H730A/clone 2C11 and Uhrf1F46A/clone 1D7) mESCs. Newly synthesized DNA in S phase cells was visualized with fluorescently labeled EdU. Scale bars for individual nuclei and pool of cells are 5 μm and 10 μm, respectively.
(B) Western blot analysis (top panel) of the expression of UHRF1 and DNMT1 in the mESCs used to measure the effect of UBL and RING domain point mutants on DNA CpG methylation (lower panel) and DNMT1 localization to nuclear replication foci shown in (A). DNA methylation levels at LINE-1 elements (%) as measured by targeted bisulfite sequencing (TaBA-seq) of n = 4 biological replicates of wild-type J1 (Uhrf1WT), Uhrf1−/−, Uhrf1F46A, and Uhrf1H730A mESCs are shown. Dashed red line indicates the median methylation level in WT J1 ESCs. Boxes indicate the 25th to the 75th percentile and whiskers represent 1.5-fold interquartile range (IQR). Outliers are indicated by dots above and below the whiskers. Welch two-sided t test: ∗∗∗p < 0.001.
(C) Co-immunoprecipitation experiments of C-terminally CFP-tagged UHRF1 and the indicated mutants with the mCherry-tagged RFTS domain of DNMT1. Constructs were co-transfected and immunoprecipitated from 293T cells.
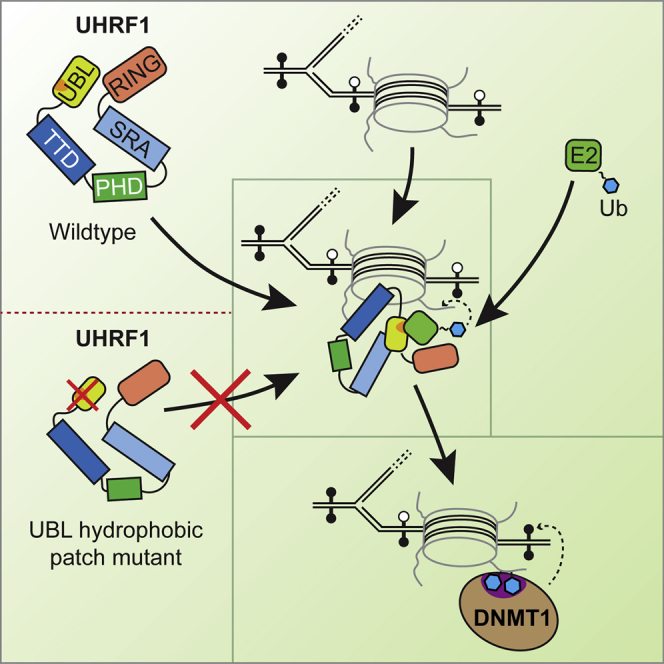
(D) Proposed model of the mechanism of UHRF1-mediated H3 ubiquitylation on chromatin. UHRF1 engages chromatin via an interaction of the TTD-PHD module with histone H3 methylated at Lys-9 (blue square) and SRA-mediated binding to a hemi-methylated CpG in the linker DNA (open circle). UbcH5a can bind to UHRF1 in this configuration with the UBL hydrophobic patch (orange) contacting the “backside” of UbcH5a. This complex is stabilized by neighboring nucleosomes. This positions the E2∼Ub for efficient transfer of ubiquitin to H3 (Lys-18 and Lys-23). DNMT1 binds the ubiquitylated H3 via ubiquitin-interacting motifs in its RFTS domain (purple) to maintain DNA CpG methylation.
Discussion
Owing to its essential function in DNA methylation, UHRF1 has received considerable attention over recent years, and the mechanisms by which UHRF1 recognizes chromatin modifications, such as H3K9me2/3 (Arita et al., 2012, Rothbart et al., 2012) and hemi-methylated CpG DNA (Arita et al., 2008), are well understood. In comparison, the enzymatic mechanism by which UHRF1 ubiquitylates histone H3, which is necessary for the recruitment of DNMT1 to replication forks, is largely uncharacterized. In this study, we identify a critical role for the N-terminal ubiquitin-like fold in the E3 ubiquitin ligase activity of UHRF1. Removal of the UBL domain or a single F46A point mutation abrogates histone H3 ubiquitylation on chromatin substrates in in vitro ubiquitylation assays, demonstrating that a hydrophobic patch on the UBL domain is involved in this process. Two recent studies found that UHRF1 mutants lacking the complete UBL domain cannot support maintenance methylation, but these mutants also displayed impaired DNMT1 binding (Li et al., 2018, Smets et al., 2017). Our results show that mutating the hydrophobic patch produces an equivalent effect in abrogating DNMT1 recruitment to nuclear replication foci, resulting in loss of DNA methylation, but without affecting the binding of UHRF1 to DNMT1 (Figures 7A–7C). We therefore propose that the UBL domain has an essential function in the recruitment of DNMT1 to newly replicated chromatin by controlling the targeted H3 ubiquitylation by the E2∼Ub enzyme (Figure 7D). The defects in DNMT1 recruitment and DNA methylation observed for UHRF1 mutants lacking the UBL domain are therefore probably a result of reduced H3 ubiquitylation.
Binding studies between the different UBL mutants and the E2-conjugating enzyme UbcH5a in the presence of chromatin suggest that the hydrophobic patch contributes to the formation of a stable E2-E3-substrate complex. However, the partial loss of binding observed in the F46A and ΔUBL mutants does not explain their almost complete loss of E3 activity. Therefore, it seems that the UBL domain has functions beyond simply recruiting the E2 enzyme. Binding of a second ubiquitin to the backside regulatory surface of UbcH5 family E2 enzymes via the hydrophobic patch (Brzovic et al., 2006) can stimulate transfer of ubiquitin from the E2∼Ub thioester site to the substrate (Buetow et al., 2015). Mutating the regulatory ubiquitin binding site of UbcH5a (S22R) dramatically reduced UHRF1 E3 activity, supporting the model that the UBL domain of UHRF1 acts like a “regulatory ubiquitin” in binding to the distal ubiquitin binding site of the E2 and facilitating ubiquitin transfer from the active site to the substrate. Whether the UBL domain allosterically activates the E2 similar to ubiquitin (Buetow et al., 2015) or whether it predominantly acts by positioning the E2∼Ub on the chromatin substrate for targeted ubiquitylation of the correct target lysines on histone H3 cannot be concluded from our results at present. What we can conclude, however, is that E2 recruitment, stimulation of the E3 activity of UHRF1, and correctly directing ubiquitylation toward histone H3 must be a multi-step process that differentially involves the hydrophobic patch, the UHRF1 N terminus, and the extent and position of DNA and histone modifications on the chromatin substrate. Data from DaRosa et al. (2018) argue against a function of the UBL domain in allosterically activating the E2 but rather support a role in recruiting the E2 and directing the transfer of ubiquitin from the E2 to histone H3.
Functions of UBL domains within other ubiquitylation systems have been described. Parkin, a Ring-between-Ring (RBR) E3 ligase, has an N-terminal UBL domain that is known to be auto-inhibitory (Trempe et al., 2013). Our analysis of the UHRF1 E3 mechanism reveals a stimulatory role of the UBL domain, demonstrating that this domain can have different activities depending on the context. Another more closely related mechanism is found in the PRC1 ubiquitylation module, where BMI1 (PCGF4), a protein required by Ring1a/1b and the E2 UbcH5c for H2AK119 ubiquitylation, has a predicted ubiquitin-like fold (Gray et al., 2016). It was reported that oligomerization of this UBL domain (alongside PHC2 as part of the PRC1 module) was required for ubiquitylation activity and possible targeting of the activity to neighboring nucleosomes (Isono et al., 2013). The UBL domain could play a similar role for UHRF1-mediated H3 ubiquitylation, given the enhanced activity on longer chromatin substrates. However, our results argue against any oligomerization of UHRF1 as it forms monomers in solution. Another interesting aspect of the PRC1 ubiquitylation module is its usage of UbcH5c, an E2 enzyme closely related to UbcH5a that we found to be optimal for UHRF1 in our experiments. Structural analyses of the PRC1 ubiquitylation module bound to a nucleosomal substrate have revealed how BMI1 and Ring1b precisely position the active site of the E2 on the nucleosome and thereby generate the specificity for H2AK119 ubiquitylation (McGinty et al., 2014). As discussed above, a similar mechanism might be operational between UHRF1 and UbcH5a to direct ubiquitylation to the histone H3 tail.
In the in vitro assays for our study, we mostly used a chromatin substrate made up of 12 repeats of the 601-nucleosome positioning sequence and unmodified octamers. The higher activity of UHRF1-mediated histone H3 ubiquitylation in the presence of these arrays, but not with shorter substrates, such as mono-, di-, or tetra-nucleosomes, suggests that chromatin structure might influence UHRF1 activity and that neighboring nucleosomes in particular positions within the array, which are not present in the shorter arrays, may stimulate activity. This is supported by our XL-MS experiments that show that UHRF1 engages differently with mono-nucleosomes and chromatin arrays and that binding of UHRF1 to the arrays shifts UHRF1 into distinct conformations, depending on the presence of the E2 enzyme and an intact UBL domain. Arrays might be better at inducing an active UHRF1 conformation or, alternatively, could position UHRF1, the E2, and histones in neighboring nucleosomes more favorably so that particular H3 N-terminal tails are available to serve as substrates for ubiquitin transfer from E2∼Ub. Another option is that this is a concentration effect. Data presented by Harrison et al. (2016) show that the interaction of UHRF1 with DNA and histone H3 N termini is regulated by positive mutual allostery leading to an “opening up” of the protein and that DNA and histone H3 tails can stimulate binding to their respective modified or unmodified counterparts, regardless of their methylation status, albeit with different Kd values. The very high local nucleosome concentration in the array, with concentrations of linker DNA and histone H3 N-terminal tails presumably far exceeding the Kd values that permit modification specificity, could “trap” UHRF1 and enable even unmodified DNA and histone H3 tails to bind and induce an open conformation competent for E2 recruitment, leading to ubiquitylation activity. Both mechanisms might be at play simultaneously.
In order to understand how UHRF1 and UbcH5a cooperate on chromatin to ubiquitylate histone H3, detailed structural analyses are required. How UHRF1 interacts with chromatin arrays compared to distinct mono-nucleosomes is currently unclear. Our XL-MS experiments serve as a useful first approximation for which regions of UHRF1 and UbcH5a interact in the context of a chromatin substrate (Figure 5E). Currently, there is detailed information for how the individual domains of UHRF1 bind to their respective ligands (Arita et al., 2008, Arita et al., 2012, Gao et al., 2018, Hashimoto et al., 2008, Hu et al., 2011, Nady et al., 2011, Rajakumara et al., 2011, Rothbart et al., 2012, Rothbart et al., 2013, Xie et al., 2012). However, how the full-length protein is configured on nucleosomes is as yet unclear. Our work clearly shows a crucial role for the N-terminal UBL domain in the E3 ligase activity of UHRF1 in addition to the C-terminal RING finger and demonstrates a requirement for the whole protein to achieve H3 ubiquitylation. It will be critical for future structural efforts to concentrate on full-length UHRF1, including the UBL and the RING finger, and the appropriate chromatin substrates to shed light on how the E2 enzyme is correctly positioned to achieve targeted ubiquitylation of histone H3 at precisely the lysine residues suitable for DNMT1 recruitment and subsequent DNA methylation.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-H3 | Abcam | Cat#Ab1791; RRID: AB_302613 |
| Anti-HA | SCBT | Cat#sc-7392; RRID: AB_627809 |
| Anti-ubiquitin | SCBT | Cat#sc-8017; RRID: AB_628423 |
| Anti-UHRF1 | SCBT | Cat#sc-373750; RRID: AB_10947236 |
| Anti-DNMT1 (WB) | NEB | Cat#5032S; RRID: AB_10548197 |
| Anti-DNMT1 (IF) | Abnova | Cat#PAB15590; RRID: AB_10678356 |
| Anti-mCherry | Smets et al., 2017 | N/A |
| Anti-GFP | Molecular Probes | Cat#A11122; RRID: AB_221569 |
| Anti-α-tubulin | Sigma | Cat#T-6199; RRID: AB_477583 |
| Anti-rabbit IgG (HRP) | Jackson ImmunoResearch | Cat#211-032-171; RRID: AB_2339149 |
| Anti-mouse IgG (HRP) | Dako | Cat#P0447; RRID: AB_2617137 |
| Anti-rat IgG (HRP) | Jackson ImmunoResearch | Cat#712-035-150; RRID: AB_2340638 |
| Anti-rabbit IgG (Alexa 488) | Jackson ImmunoResearch | Cat#711-547-003; RRID: AB_2340620 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| H3unmod (1-31, C-terminal thioester group) | Cambridge Peptide | N/A |
| H3K9me3 (1-31, C-terminal thioester group) | Cambridge Peptide | N/A |
| His6-UBE1 | Boston Biochem | Cat#E-304 |
| E2-conjugating enzymes | Boston Biochem | Cat#K-980B |
| Ubiquitin (WT) | Boston Biochem | Cat#U100-H |
| Ubiquitin (noK) | Boston Biochem | Cat#UB-NOK |
| HA-Ubiquitin | Boston Biochem | Cat#U-110 |
| GST-UbcH5a | Boston Biochem | Cat#E2-615 |
| Chicken albumin | Sigma | Cat#A7641-50mg |
| BS3 d0/d12 crosslinker | Creative Molecules | Cat#001SS |
| Trypsin | Promega | Cat#V5113 |
| Critical Commercial Assays | ||
| EZ DNA Methylation-Gold Kit | Zymo Research | Cat#D5005 |
| Quant-iT PicoGreen dsDNA Assay Kit | Thermo Fisher Scientific | Cat#P7589 |
| Deposited Data | ||
| Primary western blotting and immunofluorescence data deposited on Mendeley | This paper | https://doi.org/10.17632/vjnw35hm7d.1 |
| Mi-Seq data for LINE-1 CpG methylation | This paper | GEO: GSE119120 |
| Experimental Models: Cell Lines | ||
| HEK293T | Stefan Jentsch (Max Planck Institute of Biochemistry, Planegg-Martinsried) | N/A |
| J1 mouse ES cell line | Kerry Tucker (Ruprecht-Karls University, Heidelberg) | N/A |
| J1 UHRF1−/− | Mulholland et al., 2015 | N/A |
| J1 F46A UHRF1 | This paper | N/A |
| J1 H730A UHRF1 | This paper | N/A |
| Oligonucleotides | ||
| Primer: 3xhemi-meth DNA (3′) Forward: ATCGGAT CTTACATGCACAGG |
This paper | N/A |
| Primer: 1xhemi-meth DNA (5′) Forward: ATCGGATC TTACATGCACAGG |
This paper | N/A |
| Primer: 1xhemi-meth DNA (5′) Reverse: AATTCAGTACTAC GCGGCCGCCCTGG |
This paper | N/A |
| Primer: 3xhemi-meth DNA (3′) Reverse: AATTCAGTACTACG CGGCCGCCCTGG |
This paper | N/A |
| F46A gRNA forward: CACCGCTACAGAGACTCTTTTATAG | This paper | N/A |
| F46A gRNA reverse: aaacCTATAAAAGAGTCTCTGTAGC | This paper | N/A |
| H730A gRNA forward: CACCGACAGACGTTGTGCTGACACA | This paper | N/A |
| H730A gRNA reverse: aaacTGTGTCAGCACAACGTCTGTC | This paper | N/A |
| L1_biseq_for: ACACTCTTTCCCTACACGACGCTCTTCCG ATCTGTTGAGGTAGTATTTTTTGTGGGT |
This paper | N/A |
| L1_biseq_reverse: GTCTCGTGGGCTCGGAGATGTG TATAAGAGACAGTTCCAAAAACTATCAAATTCTCTAAC |
This paper | N/A |
| Recombinant DNA | ||
| Human H3.1Δ1-31 T32C, cloned into pET28a | Bartke et al., 2010 | pTB070 |
| Human H2A, cloned into pET21 | Bartke et al., 2010 | pTB037 |
| Human H2B, cloned into pET21 | Bartke et al., 2010 | pTB008 |
| Human H3, cloned into pET21 | Bartke et al., 2010 | pTB013 |
| Human H4, cloned into pET21 | Bartke et al., 2010 | pTB009 |
| Mouse UHRF1ΔMW (3-782), cloned into modified pET24 with N-terminal His6-SUMO tag | This paper | pTB781 |
| Mouse UHRF1ΔMW (3-782) F46A point mutant, in modified pET24 with N-terminal His6-SUMO tag | This paper | pTB780 |
| Mouse UHRF1ΔMW (3-782) H730A point mutant, in modified pET24 with N-terminal His6-SUMO tag | This paper | pTB783 |
| Mouse UHRF1 (1-782), cloned into modified pET24 with N-terminal His6-SUMO tag | This paper | pTB875 |
| Mouse UHRF1 (1-782) F46A point mutant, in modified pET24 with N-terminal His6-SUMO tag | This paper | pTB881 |
| Mouse UHRF1 (1-782) H730A point mutant, in modified pET24 with N-terminal His6-SUMO tag | This paper | pTB880 |
| Mouse UHRF1 short fragment (118-621), into modified pET24 with N-terminal His6-SUMO tag | This paper | pTB799 |
| Mouse UHRF1 ΔUBL (118-782), into modified pET24 with N-terminal His6-SUMO tag | This paper | pTB778 |
| Mouse UHRF1 ΔRING/linker (3-621), into modified pET24 with N-terminal His6-SUMO tag | This paper | pTB777 |
| Mouse UHRF1 His6-tagged UBL-domain (1-117), cloned into pET28 | This paper | pTB820 |
| Mouse UHRF1 UBL-domain (1-78) cloned into pET24 | This paper | pTB877 |
| Human UBE2D1/UbcH5a, cloned into pRSFDuet1 | This paper | pTB852 |
| Human UBE2D1/UbcH5a S22R, in pRSFDuet1 | This paper | pTB853 |
| Human UBE2D1/UbcH5a, cloned into pGEX-6P1 | This paper | pTB886 |
| Human UBE2D1/UbcH5a S22R, in pGEX-6P1 | This paper | pTB887 |
| Human HP1α cloned into pGEX-2T | Bartke et al., 2010 | pTB163 |
| Mouse UHRF1 C-terminal CFP-tag (1-782), cloned into pECFP-N1 | This paper | pTB816 |
| Mouse UHRF1 C-terminal CFP-tag ΔUBL (118-782), in pECFP-N1 | This paper | pTB814 |
| Mouse UHRF1 C-terminal CFP-tag F46A (1-782), in pECFP-N1 | This paper | pTB815 |
| Mouse UHRF1 C-terminal CFP-tag H730A (1-782), in pECFP-N1 | This paper | pTB817 |
| mCherry-RFTS | Smets et al., 2017 | pc1375 |
| pUC19-16x601 | Bartke et al., 2010 | pTB050 |
| pUC19-8xdi601 | This paper | pTB518 |
| pUC19-4xtetra601 | This paper | pTB685 |
| pUC19-MMTVA | Thomas Schalch | pTB027 |
| pWM530-12x187bp | Guohong Li | pTB703 |
| M. SssI DNA CpG methyltransferase cloned into pBAD24 | Bartke et al., 2010 | pTB120 |
| SpCas9-T2A-Puromycin/gRNA vector (px459) | Addgene | 62988 |
| Software and Algorithms | ||
| StavRox | Götze et al., 2012; https://www.stavrox.com/ | Version 3.6.6 |
| xVis | Grimm et al., 2015; http://xvis.genzentrum.lmu.delogin.php | Web Interface |
| GraphPad Prism | https://www.graphpad.com/scientific-software/prism/ | Version 7.0c (Mac) |
| ImageJ | https://imagej.nih.gov/ij/ | Version 1.51S |
| UCSF Chimera | https://www.cgl.ucsf.edu/chimera/ | Version 1.12 |
| Trim Galore | https://github.com/FelixKrueger/TrimGalore | v.0.3.1 |
| bsmap | https://github.com/zyndagj/BSMAPz; Xi and Li, 2009 | v.2.90 |
| methylKit R package | https://github.com/al2na/methylKit; Akalin et al., 2012 | v.0.9.5 |
| MaxQuant | http://www.coxdocs.org/doku.php?id=maxquant:start | v.1.5.8.3 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Till Bartke (till.bartke@helmholtz-muenchen.de).
Experimental Model and Subject Details
We used the following cell lines:
-
•
HEK293T. Human cell line, female. Grown in DMEM/10% FBS. Not authenticated for this work.
-
•
J1. Mouse ES cell line, male, derived from mouse strain 129S4/SvJae. Grown in DMEM/16%FBS/0.1 mM β-mercaptoethanol/2 mM L-glutamine/1 × MEM Non-essential amino acids/100 U/mL penicillin/100 μg/mL streptomycin/homemade recombinant LIF/2i (1 μM PD032591 and 3 μM CHIR99021). Not authenticated for this work.
Method Details
Purification of Recombinant Proteins
The cDNA encoding amino acids 1-782 for mouse UHRF1 (Np95) was cloned into a modified pET24 vector (pCA528) in-frame and immediately proceeding an N-terminal His6-SUMO tag that can be cleaved off with His6-Ulp1 SUMO-specific protease. Transformed E. coli BL21(DE3)/RIL were grown to O.D. 0.6 and induced with 0.1 mM IPTG overnight at 20°C. Cells were harvested by centrifugation and re-suspended in lysis buffer (20 mM HEPES-KOH pH 7.5, 300 mM NaCl, 10% glycerol, 20 mM imidazole, 1 mM PMSF, 1x EDTA-free protease inhibitors (Roche)). Cells were lysed with lysozyme and sonication on ice and clarified by centrifugation at 20,000 g for 45 min at 4°C. Cell lysate was syringe-filtered with 0.45 μm filters and was incubated with Ni-Sepharose beads (GE Healthcare). Beads were washed (20 mM HEPES-KOH pH 7.5, 300 mM NaCl, 10% glycerol, 20 mM imidazole) and eluted (20 mM HEPES-KOH pH 7.5, 300 mM NaCl, 10% glycerol, 300 mM imidazole) before immediate removal of the imidazole by dialysis. Eluted protein was incubated with Ulp1 SUMO-specific protease (made in-house) before concentrating the protein to ∼2 mL using 10 kDa MWCO spin concentrators (Vivaspin, Sartorius) and further purified by size-exclusion chromatography (SEC) using a Superdex 200 16/60 column (GE Healthcare) in 20 mM HEPES-KOH pH 7.5, 300 mM NaCl, 10% glycerol, 1 mM DTT. Pure fractions (> 90% by Coomassie Brilliant Blue-stained 10% SDS-PAGE) were pooled and concentrated to ∼1.5 mg/ml before snap freezing aliquots in liquid nitrogen and stored at −80°C. Other UHRF1 fragments and mutants were purified in an equivalent manner. For the UHRF1ΔMW variants, UHRF1 (3-782) was cloned into the pCA528 vector in-frame with the N-terminal His6-SUMO tag using a BamHI site at bp 5 of the murine UHRF1 cDNA resulting in a RPDP linker between the SUMO-cleavage site and the UHRF1 open reading frame. Point mutants were obtained by site-directed mutagenesis (Quikchange mutagenesis, Agilent) and behaved similar to the wild-type protein. UHRF1 protein was purified from bacteria >4 times and all protein preparations were functional and behaved similarly. His6-UBL was expressed from a pET28a(+) vector and purified in a similar manner using Ni-Sepharose beads and SEC but the N-terminal tag was not cleaved off. The untagged UHRF1 UBL-domain (1-78) was cloned into a pET24b(+) vector backbone. Cells were lysed as above and cleared lysate was injected onto a 5 mL HiTrap SP HP column using a gradient from 100 mM to 1000 mM NaCl in 20 mM Tris pH 7.0 and 1 mM DTT. Fractions containing the UBL-domain were pooled, concentrated, and injected onto a Superdex 75 16/60 column (GE Healthcare) and eluted by isocratic elution in 20 mM Tris pH 7.5, 100 mM NaCl, 10% glycerol (v/v) and 1 mM DTT. Purified fractions were pooled and aliquots were flash frozen in liquid nitrogen and stored at −80°C.
Wild-type human UbcH5a (UBE2D1) was synthesized by Genscript and cloned into a pRSFDuet1 vector with a Prescission protease cleavable N-terminal His6-tag. The S22R mutant was made by Quikchange site-directed mutagenesis (Quikchange mutagenesis, Agilent). Transformed E. coli BL21(DE3)/RIL cells were grown to an OD600 of 0.6 and protein expression was induced with IPTG at a final concentration of 0.5 mM for 3 hr at 37°C. Cells were harvested by centrifugation and re-suspended in lysis buffer (20 mM Tris-HCl pH 8.0, 300 mM NaCl, 20 mM imidazole, 1x complete EDTA-free protease inhibitors and 0.5 mM PMSF). Cells were lysed with lysozyme and sonication on ice and clarified by centrifugation at 20,000 g for 45 min at 4°C. Cell lysate was syringe-filtered with 0.45 μm filters and was incubated with Ni-Sepharose beads (GE Healthcare). Beads were washed with wash buffer (20 mM Tris-HCl pH 8.0, 300 mM NaCl, 20 mM imidazole) and protein was eluted with wash buffer containing 300 mM imidazole. The imidazole was removed by dialysis into 20 mM Tris-HCl pH 8.0, 100 mM NaCl and the N-terminal tag removed by incubation with Prescission protease (GE Healthcare) for 16 hr at 4°C. The protein was finally purified by size-exclusion chromatography using a Superdex 75 16/60 column (GE Healthcare) in 20 mM Tris-HCl pH 8.0, 100 mM NaCl, 10% glycerol (v/v) and 1 mM DTT. Pure fractions were pooled and concentrated to ∼200 μM or 25 μM working aliquots before snap freezing aliquots in liquid nitrogen and storage at −80°C. For the GST-tagged UbcH5a, human UbcH5a (wild-type and S22R mutant) was sub-cloned into the pGEX-6P1 vector in-frame with an N-terminal GST-tag and Prescission protease site. Transformed E. coli BL21(DE3)/RIL cells were grown, induced, and lysed as above but clarified lysate was incubated with Glutathione Sepharose 4B beads for 2 hr at 4°C. Beads were washed with wash buffer as above and protein eluted with wash buffer supplemented with 10 mM reduced glutathione. Reduced glutathione was removed by dialysis and the protein was further purified by size-exclusion chromatography as above for the untagged UbcH5a before storage in 25 μM working aliquots.
Preparation of Modified Recombinant Nucleosomes
Recombinant histone proteins were expressed from pET21b(+) (Novagen) vectors and purified by denaturing gel filtration and ion exchange chromatography as described (Bartke et al., 2010). The purification for hH4 was altered. Recombinant hH4 was purified in 1 M NaCl for the size-exclusion chromatography (SEC) step and utilized a linear gradient from SAU-200 (20 mM NaAcetate pH 5.2, 200 mM NaCl, 7 M urea, 1 mM EDTA, 5 mM 2-mercaptoethanol) to TU-1000 (20 mM Tris HCl pH 7.5, 1 M NaCl, 7 M urea, 1 mM EDTA, 5 mM 2-mercaptoethanol) for the denaturing ion-exchange chromatography (IEX) step after diluting the NaCl of the SEC fractions to 200 mM. Truncated hH3.1 Δ1-31 T32C protein was generated in vivo by expressing a hH3.1 Δ1-31 T32C precursor in the presence of TEV-protease. For this, E. coli cells harboring the pET28a(+)-AraC-pBAD-His6TEV/pro-hH3.1 Δ1-31 T32C plasmid were grown in LB medium supplemented with 0.25% L-arabinose to keep TEV-protease induced. At OD600 of 0.6 the expression of pro-hH3.1 Δ1-31 T32C was induced for 3 hr at 37°C with 50 μM IPTG. TEV-protease processes the precursor hH3.1 into tail-less hH3.1 Δ1-31 T32C. The insoluble protein was extracted from inclusion bodies with solubilisation buffer (20 mM Tris HCl pH 7.5, 7 M guanidine HCl and 100 mM DTT) for 1 hr at RT and passed over a Sephacryl S-200 gel filtration column (GE Healthcare) in SAU-200 (20 mM NaAcetate pH 5.2, 7 M urea, 200 mM NaCl and 1 mM EDTA) without any reducing agents. Positive fractions were directly loaded onto a reversed-phase Resource RPC column (GE Healthcare) and eluted with a gradient of 0%–65% B (A: 0.1% TFA in water; B: 90% acetonitrile, 0.1% TFA) over 20 column volumes. Fractions containing pure hH3.1 Δ1-31 T32C were pooled and lyophilised. All histone proteins were stored lyophilised at −80°C.
Native chemical ligations were carried out in 500 μL of degassed NCL buffer (200 mM KPO4 pH 7.9, 2 mM EDTA, 6 M Guanidine HCl) containing 1 mg of modified H3.1 amino acids 1-31 thioester peptide (Cambridge Peptides), 4 mg of truncate H3.1 Δ1-31 T32C, 12.5 mg 4-Mercaptophenylacetic acid (MPAA, Sigma 653152-1G) and 10 mg TCEP (Sigma, C4706-2G) as a reducing agent at pH 7.5. The reactions were incubated overnight at 40°C and quenched by the addition of 60 μL 1 M DTT and 700 μL 0.5% acetic acid. After a centrifugation step to remove any precipitates the ligation reactions were directly loaded and purified on a reversed-phase chromatography column (Perkin Elmer Aquapore RP-300 C8 250x4.6 mm i.d.) using a gradient of 45%–55% B (Buffer A: 0.1% TFA in water; B: 90% acetonitrile, 0.1% TFA) over 10 column volumes. Positive fractions containing ligated full-length histone H3.1 were then combined and lyophilised. Histone octamers were refolded from purified histone proteins. Individual histones were re-suspended in unfolding buffer (7 M GuHCl, 20 mM Tris-HCl pH 7.5, 10 mM DTT) for 1 hr at RT before mixing in approximately stoichiometric amounts (for hH3.1 and hH4 but a slight excess of hH2A and hH2B) and dialysis against refolding buffer (2 M NaCl, 10 mM Tris-HCl pH 7.5, 1 mM EDTA, 5 mM 2-mercaptoethanol). The first dialysis was carried out for >6 hr with two further changes into fresh refolding buffer. Histone octamers were purified away from (H3-H4)2 tetramers and H2A-H2B dimers by gel filtration over a Superdex 200 column (GE Healthcare). Nucleosomal DNAs containing either one (mono-nucleosomes), two (di-nucleosomes) or four (tetra-nucleosomes) 601 nucleosome positioning sequences separated by a 50 bp linker (for di- and tetra-nucleosomes) were prepared as described (Bartke et al., 2010). In short, nucleosomal 601 DNA was excised from the purified plasmid DNAs (Plasmid Giga kit, QIAGEN) by digestion with EcoRV and separated from the vector by PEG precipitation. Di- (2x601) and tetra- (4x601) nucleosomal DNAs were prepared by a similar method with optimization of the PEG-amount to best separate the nucleosomal DNA from the plasmid backbone. For the larger array DNA (12x187bp, the pWM530-12x187 was a kind gift from Guohong Li), purified plasmid DNAs prepared as above were digested with EcoRV with the plasmid backbone containing multiple EcoRV sites, enabling purification of the larger array DNA by PEG precipitation as above. Methylated nucleosomal DNA was made by incubation with M. SssI CpG methylatransferase (made in-house) at 37°C for 5 hr and then purified by phenol/chloroform extraction and ethanol precipitation. Complete methylation was verified by BstUI digestion (NEB). Hemi-methylated mono-nucleosome DNA used in nucleosome EMSAs and E3 assays (3x hemi-meth DNA 3′ and 1x hemi-meth DNA 5′) was prepared by PCR using oligonucleotide primers (Forward primer: ATCGGATCTTACATGCACAGG, Reverse primer: AATTCAGTACTACGCGGCCGCCCTGG, purchased from Integrated DNA Technologies, the methylated cytosines are in bold and underlined with the 3′ hemi-meth having hemi-methylated CpGs in the reverse primer and 5′ hemi-meth DNA having a hemi-methylated CpG in the forward primer) and purified by ion exchange chromatography (HiTrap Q, GE Healthcare). Nucleosomes were assembled by mixing refolded octamers with nucleosomal DNAs into high salt buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 2 M NaCl, 1 mM DTT) and salt deposition dialysis from 2 M NaCl to 100 mM NaCl. The molar ratio of octamer:DNA used was checked (by 5% native PAGE in 0.2x TBE for mono-/di-nucleosomes and 0.7% agarose in 0.2x TB for tetra-nucleosomes and 12x187bp arrays) empirically for each batch of purified octamer and each batch of nucleosomal DNA. Di- and tetra-nucleosomes and 12x187bp chromatin arrays were assembled in the presence of MMTV A competitor DNA and a slight excess of octamers for longer chromatin arrays to ensure saturation of the 601 repeats (Dorigo et al., 2003). 12x187bp chromatin arrays were purified away from free DNA and competitor MMTVA DNA/nucleosomes by incubation with MgCl2 (5 mM final concentration) on ice for 30 min and centrifugation at 16,100 g for 30 min at 4°C. Chromatin arrays were re-suspended in MgCl2-free buffer. All reconstituted nucleosomes and chromatin were prepared fresh and stored at 4°C for 1-2 weeks.
In Vitro Ubiquitylation Assays
Ubiquitylation assays were typically performed in 50 μL reactions containing 200 nM His6-UBE1 (Boston Biochem, E-304), 1 μM E2 UBE2D1 (UbcH5a, Boston Biochem or purified in-house), 5 mM MgCl2, 5 mM ATP, 20 mM Tris-HCl pH 7.5, 100 mM NaCl, 10 μM ubiquitin (Boston Biochem), 0.4 μM UHRF1WT (1 μM UHRF1ΔMW) and 2.5 μg octamer-containing nucleosome or chromatin. Assays were performed at 25°C and quenched after 30 min (unless stated otherwise in the figure legends) with SDS-PAGE loading buffer (2% SDS, 10% glycerol, 5% 2-mercaptoethanol added fresh, 62.5 mM Tris-HCl pH 6.8, 0.01% bromophenol-blue). For E3 assay time courses, samples were taken at the indicated time points and quenched with SDS-PAGE loading buffer. Reactions were run on 8% (for UHRF1 auto-poly-ubiquitylation) or 15% SDS-PAGE (for histone H3 and H3-Ub), transferred to PVDF membranes and visualized by western blot probed with anti-H3 and anti-ubiquitin or anti-HA antibodies.
In Vitro Ubiquitylation Assays LC-MS/MS
For identification of ubiquitylated substrates three different experiments were carried out. In the first experiment (b015p026), two E3 assays were set up using wild-type using UHRF1ΔMW, unmodified chromatin substrate (12x187bp) and ∼1/5 of the quenched reaction was run on a 15% SDS-PAGE gel. Coomassie-stained bands at 25 kDa and ∼35 kDa were cut out, destained, and treated with two consecutive rounds of propionylation with propionic anhydride (Sigma, 240311) followed by in-gel trypsin digestion overnight at 37°C. Peptides were extracted with acid and analyzed by LC/MS-MS. Experiment number 3 (b015p056) was carried out in a similar manner except UHRF1WT was used these assays and bands were cut out at 25 kDa, ∼35 kDa and ∼43 kDa. The samples were processed as above. Experiment number 2 (b015p047) cut out bands at ∼100 kDa and 25 kDa but did not protect lysine residues with propionic anhydride although cysteine residues were reduced with DTT and capped with iodoacetamide prior to in-gel trypsin digestion as described. The experimental setup is briefly visualized in Data S1.
LC-MS/MS Experiment 1 (b015p026) and 3 (b015p056)
Dried digested peptides were solubilised in 15 μL 0.1% trifluoroacetic acid (TFA) and clarified solution was transferred to auto sampler vials for LC-MS analysis. Peptides were separated using an Ultimate 3000 RSLC nano liquid chromatography system (Thermo Scientific) coupled to a LTQ Orbitrap Velos mass spectrometer (Thermo Scientific) via an EASY-Spray source. 5 μL sample in technical duplicate was loaded onto a trap column (Acclaim PepMap 100 C18, 100 μm x 2 cm) at 8 μl/min in 2% acetonitrile, 0.1% TFA, 5% DMSO. Peptides were eluted on-line to an analytical column (EASY-Spray PepMap C18, 75 μm x 50 cm). Peptides were separated using a stepped 120 min gradient, 4%–25% buffer B for 45 min, 25%–45% buffer B for 30 min (buffer B is 80% acetonitrile, 0.1% formic acid, 5% DMSO). Eluted peptides were analyzed by the Velos operating in positive polarity using a data-dependent acquisition mode. Ions for fragmentation were determined from an initial MS1 survey scan at 70,000 resolution (at m/z 200) followed by HCD (Higher-energy collisional dissociation) of the top 12 most abundant ions at a resolution of 17,500. MS1 and MS2 scan AGC targets set to 1e6 and 1e5 for a maximum injection time of 50 ms and 110 ms, respectively. A survey scan m/z range of 350 – 1800 m/z was used, with a normalized collision energy set to 27%, underfill ratio – 1%, charge state exclusion enabled for unassigned, +1, +8 and > +8 ions.
Data were processed using MaxQuant software platform (v1.5.8.3) with database searches carried out by the in-built Andromeda search engine against the Swissprot Mus musculus database. A reverse decoy database was used at a 1% false-discovery rate (FDR) for peptide spectrum matches and protein identification. Search parameters included: two missed cleavages, a fixed modification of cysteine carbamidomethylation and variable modification of methionine oxidation, GlyGly, LRGG and propionylation of lysine. Label-free quantification was enabled with an LFQ minimum ratio count of 2. ‘Match between runs’ function was used with match and alignment time limits of 1 and 20 min, respectively. Protein and peptide identification and relative quantification outputs from MaxQuant were further processed in Microsoft Excel, with hits to the ‘reverse database’, ‘potential contaminants’ (peptide list only) and ‘Only identified by site’ fields removed.
LC-MS/MS Experiment 2 (b015p047)
Dried digested peptides were solubilised in 15 μL 0.1% trifluoroacetic acid (TFA) and clarified solution was transferred to auto sampler vials for LC-MS analysis. Peptides were separated using an Ultimate 3000 RSLC nano liquid chromatography system (Thermo Scientific) coupled to a LTQ Orbitrap XL mass spectrometer (Thermo Scientific) via a Proxeon nano-spray source. 5 μL sample in technical duplicate was loaded onto a trap column (Acclaim PepMap 100 C18, 100 μm x 2 cm) at 8 μl/min in 2% acetonitrile, 0.1% TFA. Peptides were eluted on-line to an analytical column (EASY-Spray PepMap C18, 75 μm x 50 cm). Peptides were separated using a stepped 60 min gradient, 4%–25% buffer B for 45 min, 25%–45% buffer B for 15 min (buffer B is 80% acetonitrile, 0.1% formic acid). Eluted peptides were analyzed by the LTQ Orbitrap XL operating in positive polarity using a data-dependent acquisition mode. Ions for fragmentation were determined from an initial MS1 survery scan at 30,000 resolution (at m/z 200), followed by ion Trap CID (collisional induced dissociation) of the top 6 most abundant ions. MS1 and MS2 scan AGC targets set to 1e6 and 1e4 for a maximum injection time of 500 ms and 100 ms, respectively. A survey scan m/z range 350 – 1800 was used, with a normalized collision energy set to 35%, charge state rejection enabled for +1 ions and a minimum threshold for triggering fragmentation of 500 counts.
Data were processed using MaxQuant software platform (v1.5.8.3) with database searches carried out by the in-built Andromeda search engine against the Swissprot Mus musculus database. A reverse decoy database was used at a 1% false-discovery rate (FDR) for peptide spectrum matches and protein identification. Search parameters included: two missed cleavages, a fixed modification of cysteine carbamidomethylation and variable modification of methionine oxidation, protein N-terminal acetylation, GlyGly and LRGG. Label-free quantification was enabled with an LFQ minimum ratio count of 2. ‘Match between runs’ function was used with match and alignment time limits of 1 and 20 min, respectively. Protein and peptide identification and relative quantification outputs from MaxQuant were further processed in Microsoft Excel, with hits to the ‘reverse database’, ‘potential contaminants’ (peptide list only) and ‘Only identified by site’ fields removed.
GST Pull-Downs
For GST-UbcH5a pull-downs, 10 μM ubiquitin, 1 μM UHRF1, 2.5 μg octamer-containing nucleosomes or chromatin and 1 μM GST-UBE2D1 (Boston Biochem or made in-house) or GST (made in-house) were incubated for 1 hr at 25°C in 20 mM Tris-HCl pH 7.5, 100 mM NaCl, 5 mM MgCl2, 5 mM ATP. 5 μL equilibrated glutathione Sepharose 4B beads (GE Healthcare, 17075601) were added and rotated for 2 hr at RT in 20 mM Tris-HCl pH 7.5, 10 mM NaCl, 0.1% NP40, 1 mM DTT. Beads were washed 3 times and bound proteins were eluted in SDS-PAGE loading buffer, boiled at 95°C for 5 min and analyzed by 15% SDS-PAGE followed by silver staining. 20% of the input and 50% of the pull-down was loaded. Bands corresponding to UHRF1 were quantified using ImageJ and normalized to the band intensity of GST-UBE2D1 in each assay. The binding of each UHRF1 fragment was quantified in this way in >3 independent experiments and normalized to the binding of wild-type UHRF1. Bar charts were made using GraphPad Prism with the mean plotted and the error bars represent the standard error of the mean (SEM).
EMSA
0.1 μM octamer-containing mono-nucleosomes (kept constant) were incubated with increasing concentrations of UHRF1 in 10 μL 20 mM Tris-HCl pH 7.5, 1 mM EDTA, 100 mM NaCl, 1 mM DTT for 5 min at 4°C. Samples were analyzed by 5% native PAGE in 0.2x TBE at 4°C and nucleosome bands were visualized with ethidium bromide or SYBR safe (Thermo Scientific, S33102). The apparent binding of UHRF1 to mono-nucleosomes was determined by measurement of the disappearance of the band corresponding to the unbound nucleosome by integration of the band intensity of this band using ImageJ (http://imagej.nih.gov/ij/). The ratio of the intensity of free nucleosome in each lane over the intensity of the nucleosome-only samples was calculated and used for the normalization of the percentage of unbound mono-nucleosome in each lane. Binding analysis was repeated in triplicate and mean measurements were plotted using GraphPad Prism. Binding at 50% substrate saturation gives a level of quantification with respect to nucleosome binding (Table S2).
For the chromatin EMSA, an equivalent method was used with 0.1 μM octamer-containing 12x187bp chromatin arrays. Binding was analyzed using 0.7% agarose in 0.2x TB but buffer conditions and analysis was similar to that for mono-nucleosome EMSAs described above.
Crosslinking with Mass Spectrometry
Samples were incubated for 15 min at RT in 10 mM HEPES-KOH pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM DTT. A 1 mg aliquot of isotopically-coded BS3 d0/d12 crosslinker (Creative Molecules, 001SS) was reconstituted to 25 mM in water and immediately added to the mixture in equimolar amounts to the number of moles of lysine residues present in the sample. The crosslinking reaction was incubated for 30 min at 25°C with mild agitation and the reaction was quenched with 50 mM NH4HCO3 for 20 min at 25°C. The samples were reduced in volume in a speedvac and re-suspended in 50 μL 7.2 M urea, 100 mM NH4HCO3 and incubated for 15 min at 25°C. Cysteines were reduced with 5 mM DTT for 1 hr at 51°C and protected with 15 mM iodoacetamide (Sigma, I1149-25G) for 45 min in the dark at 25°C. The reaction was quenched with DTT and urea was diluted to < 1 M with 50 mM NH4HCO3 before adding MS-grade trypsin (Promega, V5113) in a ratio of 1:25 (trypsin:protein) and incubated for 16 hr at 37°C. The reaction was stopped by the addition of acetic acid (2.5% final concentration, v/v) and peptides were purified using C18 stage tips (Glygen). Peptides were eluted in 60% acetonitrile, 0.1% formic acid and dried and stored at −20°C.
Crosslinking with Mass Spectrometry LC-MS/MS
Sample preparation, peptide loading and chromatography were performed as stated above (LC-MS/MS for Experiment 1 and 3) and analyzed using an Orbitrap Velos mass spectrometer with the following scan settings. Eluted peptides were analyzed by the LTQ Orbitrap Velos operating in positive polarity using a data-dependent acquisition mode. Ions for fragmentation were determined from an initial MS1 survey scan at 30,000 resolution (at m/z 200), followed by ion trap CID (collisional induced dissociation) of the top 10 most abundant ions. MS1 and MS2 scan AGC targets set to 1e6 and 1e4 for a maximum injection time of 500 ms and 100 ms, respectively. A survey scan m/z range 350 – 1800 was used, with a normalized collision energy set to 35%, charge state rejection enabled for +1 ions and a minimum threshold for triggering fragmentation of 500 counts.
Raw files from experiments on the Orbitrap Velos instrument were converted to mgf files using MSConvert on proteowizard and crosslinked peptides were identified using Stavrox (Götze et al., 2012). Light/heavy-labeled BS3 was used as a cross-linker. A FDR of < 5% was used to filter out low-confidence crosslinks and subsequent higher-confidence crosslinks were visualized using xVis (Grimm et al., 2015). For some experiments (UHRF1ΔMW only, UHRF1 ΔMW + mono-nucleosome and UHRF1 ΔMW + UbcH5a + 12x187bp array), replicates of identified crosslinked peptides were pooled and visualized. Excel tables of the crosslinked sites identified can be found in Table S1. An example spectrum of the StavRox data analysis output can be seen at the end of Data S1.
Size-Exclusion Chromatography
Size-exclusion chromatography (SEC) experiments were carried out on an AKTA FPLC systems (GE Healthcare) using a Superdex 200 16/60 column (GE Heathcare) that was calibrated using molecular weight markers (GE Healthcare). The protein was clarified by centrifugation at maximum speed in a bench top centrifuge at 4°C, before loading onto the column. Protein and protein complexes were eluted using isocratic elution in 20 mM Tris-HCl pH 7.5, 300 mM NaCl, 10% glycerol (v/v), 1 mM DTT at 0.5 ml/min.
In-Gel Crosslinking
20 μM of protein (UHRF1WT, UHRF1ΔMW, HP1α and chicken egg-white albumin) was treated with a dilution series of glutaraldehyde (0%, 0.005%, 0.01%, 0.02% and 0.05% final concentration (v/v), Sigma 10575873) incubated for 5 min at RT in 10 μL volume. 5x SDS-PAGE loading buffer was added, samples were boiled at 95°C for 5 min and loaded onto a 10% SDS-PAGE gel in Tris-Glycine buffer and stained with Coomassie Brilliant Blue.
SDS-PAGE and Western Blot Analysis
SDS-PAGE was used to separate proteins according to their molecular weight. Samples were boiled and denatured for 5-10 min at 95°C in sample buffer (62.5 mM Tris-HCl pH 6.8, 10% glycerol, 2% SDS, 0.01% bromophenol blue and 5% 2-mercaptoethanol) before loading onto an appropriate percentage polyacrylamide gel (8% up to 17.5%, homemade) and running at 200 V at RT in Tris-Glycine buffer (3% (w/v) Tris, 15% glycine, 1% SDS). For protein immunodetection, samples were first separated by SDS-PAGE and transferred from the gel to pre-activated (in 100% methanol) PVDF membrane (0.45 μm; Millipore, IPVH00010) using a wet transfer blotting system (Bio-rad) at a constant current (350 mA) for 75 mins at 4°C in transfer buffer (10X in 2 L: 151.5 g Tris, 721 g glycine and 50 g SDS supplemented with 20% methanol or ethanol). PVDF membrane was blocked in 5% milk in TBS-T (150 mM Tris-HCl pH 7.5, 50 mM NaCl supplemented with 0.1% or 0.5% Tween-20) for 1 hr at RT. The membrane was probed with primary antibody diluted in 5% milk in TBS-T and incubated for 2 hr at RT or overnight at 4°C on a rotating wheel. The membrane was subsequently washed three times for 5 min each at RT in TBS-T followed by incubation with HRP-conjugated secondary antibody diluted in 5% milk in TBS-T for 1 hr at RT. The membrane was washed three times in TBS-T ad incubated for 5 min with ECL Western Blotting Substrate (Thermo Scientific, 34080) and exposed and developed on X-ray films (Fujifilm Super RX, 4741019236) or imaged using a ChemiDoc system (Bio-rad).
Protein Staining of Polyacrylamide Gels
Protein samples were separated by SDS-PAGE and proteins were visualized by Coomassie Brilliant Blue or silver staining depending on the amount of protein loaded. Gels were immersed in Coomassie Brilliant Blue solution (Expedeon, ISB1L) for 1 hr at RT and de-stained with at least 3 changes of water. For gels with lower amounts of protein loaded, silver staining was carried out. The gel was fixed by incubating in fixing solution (30% ethanol, 15% acetic acid) for 1 hr at RT before immersing in incubation solution (0.5 M sodium acetate, 25% ethanol, 0.1% glutaraldehyde, 0.2% sodium thiosulphate) for 1 hr at RT. The gel was washed three times with water before staining in 0.1% silver nitrate. Protein bands are revealed with a solution containing 2% sodium carbonate, 0.05% formaldehyde and the reaction stopped with EDTA when required.
Cloning and Plasmid DNA Purification
Newly recombined DNA constructs were generated by either directly sub-cloning a target vector into a donor vector by restriction enzyme digests (NEB or Roche restriction enzymes), by PCR amplification strategy or by Gibson Assembly (NEB, as manufacturer’s instructions). Digestion was carried out at 37°C for 2 hr and the vector was dephosphorylated with 5 U of Calf intestine phosphatase (CIP) at 37°C for 1 hr to prevent re-ligation. DNA fragments were run on a 1% agarose gel and gel-purified using a kit (QIAGEN) as the manufacturer’s instructions and ligated with NEB Quick Ligation Kit. Ligation reactions were transformed directly into XL-10 Gold cells. Plasmid DNA was extracted and purified from 5 or 250 mL bacterial culture using Miniprep or Maxiprep kits (QIAGEN), following the manufacturer’s instructions. The DNA concentration was measured using the Nanodrop ND-1000 spectrophotometer and checked by restriction digest and sequencing as performed by the genomics core facility on site at the MRC London Institute of Medical Sciences or GATC Biotech.
Cell Culture
For CRISPR-assisted cell line generation, immunostaining and TaBa sequencing, mouse ESCs were maintained on 0.2% gelatin-coated dishes in Dulbecco’s modified Eagle’s medium (Sigma) supplemented with 16% fetal bovine serum (FBS, Sigma), 0.1 mM β-mercaptoethanol (Invitrogen), 2 mM L-glutamine (Sigma), 1 × MEM Non-essential amino acids (Sigma), 100 U/mL penicillin, 100 μg/mL streptomycin (Sigma), homemade recombinant LIF tested for efficient self-renewal maintenance, and 2i (1 μM PD032591 and 3 μM CHIR99021 (Axon Medchem, Netherlands)). All cell lines were regularly tested for Mycoplasma contamination by PCR.
HEK293T cells for the UHRF1-CFP and mCherry-RFTS co-transfection were maintained in Dublbecco’s modified Eagle’s medium (Sigma) supplemented with 10% fetal bovine serum (FBS, Sigma), 100 U/ml penicillin and 100 μg/ml streptomycin.
Co-immunoprecipitation
Cells were seeded at 1x107 cells per T75 flask the day before transfection, which was carried out using Lipofectamine 2000 (Thermo-Fisher, as the manufactrurer’s protocol) with the plasmid amounts adapted to have mCherry-RFTS in at least 3-molar excess over the UHRF1-CFP. Cells were harvested after 48 hr, washed twice in ice-cold PBS and re-suspended in 200 μL lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.1 mM MgCl2, 0.1% NP40, 1x EDTA-free complete protease inhibitors (Roche), 2 mM PMSF and 1 mg/ml DNaseI) with incubation on ice for 30 min and extensive pipetting every 10 min. Cell lysate was spun at 17,000 g for 10 min at 4°C and the supernatant was transferred to fresh tubes. 300 μL of wash/dilution buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA) was added before carrying out the co-immunoprecipitation. 20 μL was saved as an input for western blot analysis. 25 μL of GFP-Trap_A beads (Chromotek) per IP were washed three times in wash/dilution buffer and added to the lysate with incubation for 2 hr at 4°C with rotation end over end. Beads were spun at 2500 g for 2 min at 4°C and the supernatant discarded before washing the beads three times in wash/dilution buffer. The beads were re-suspended in 100 μL 2x SDS-sample buffer, boiled for 10 min at 95°C and spun at 2500 g for 2 min before loading onto a 10% SDS-PAGE gel for subsequent western blot analysis.
CRISPR/Cas9 Gene Editing
For generation of the Uhrf1 mutants, Uhrf1-specific gRNAs were cloned into a modified version of the SpCas9-T2A-Puromycin/gRNA vector (px459 - Addgene plasmid #62988; Ran et al., 2013) where we fused truncated human Geminin (hGem) to SpCas9 for increasing homology-directed repair efficiency (Gutschner et al., 2016). A 200 bp ssDNA oligonucleotide harboring the H730A and F46A mutations and ∼100 bp of homology to the genomic locus was synthesized (IDT, Coralville, IA, USA). For targeting in wild-type J1 ESCs, cells were transfected with a 4:1 ratio of donor oligo and Cas9/gRNA construct. After 2 days transfection and subjected to a transient puromycin selection (1 μg/mL) for 48 h. Colonies were allowed to grow for 6 days, at which point they were picked into 96-well plates and screened using restriction-fragment length polymorphism (RFLP) analysis. Cell lysis in 96-well plates, PCR on lysates, and restriction digest were performed as previously described (Mulholland et al., 2015). For all cell lines, Uhrf1 mutation was confirmed by Sanger sequencing. Knockout of Uhrf1 in mESCs was performed as described recently (Mulholland et al., 2015).
EdU-Labeling Combined with Immunostaining
For immunostaining, ESCs were grown on coverslips coated with Geltrex (Life Technologies) diluted 1:100 in DMEM/F12 (Life Technologies). The Cells were incubated in 10 μM EdU (5-Ethynyl-2′-Deoxyuridin) in growth medium for 10 min at 37°C 5% CO2, rinsed for four times with PBS (pH 7.4; 140 mM NaCl, 2.7 mM KCl, 6.5 mM Na2HPO4, 1.5 mM KH2PO4) prewarmed to 37°C. All following steps during immunostaining were performed at room temperature. The cells fixed for 10 min with 2% paraformaldehyde (pH 7.0; prepared from paraformaldehyde powder (Merck) by heating in PBS up to 60°C; store at −20°C), washed three times for 10 min with PBS-T (PBS, 0.01% Tween20), permeabilized for 5 min in PBS supplemented with 0.5% Triton X-100, and washed two times for 10 min with PBS. Cells were then incubated in blocking solution (PBS-T, 4% BSA) for 1 h, afterward incubated for 30 min in the EdU reaction mix (0.1 M Tris-HCl pH 7, 4 mM CuSO4, 50 mM Na-ascorbate and 20 μM Alexa 647 azide dye) and rinsed four times with PBS-T. Primary and secondary antibodies were diluted in blocking solution. Coverslips were incubated with primary and secondary antibody solutions in dark humid chambers for 1 h and washed three times for 10 min with PBS-T after primary and secondary antibodies. For DNA counterstaining, coverslips were incubated 10 min in PBS-T containing a final concentration of 2 μg/mL DAPI (Sigma-Aldrich) and washed three times for 10 min with PBS-T. Coverslips were mounted in antifade medium (Vectashield, Vector Laboratories) and sealed with colorless nail polish.
Targeted Bisulfite Amplicon (TaBA) Sequencing
Genomic DNA was isolated from 2x106 cells using the PureLink Genomic DNA Mini Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The EZ DNA Methylation-Gold Kit (Zymo Research) was used for bisulfite conversion according to the manufacturer’s instructions with 500 ng of genomic DNA used as input and the modification that bisulfite converted DNA was eluted in 2 × 20 μL Elution Buffer. The sequences of the LINE1 (an abundant transposon class within the mouse genome) specific primers were appended with Illumina TruSeq and Nextera compatible overhangs. The amplification of bisulfite converted DNA was performed in 25 μL PCR reaction volumes containing 0.4 μM each of forward and reverse primers, 2 mM Betaine (Sigma-Aldrich, B0300-1VL), 10 mM Tetramethylammonium chloride solution (Sigma-Aldrich T3411-500ML), 1x MyTaq Reaction Buffer, 0.5 units of MyTaq HS (Bioline, BIO-21112), and 1 μL of the eluted bisulfite converted DNA (∼12.5 ng). The following cycling parameters were used: 5 min for 95°C for initial denaturation and activation of the polymerase, 40 cycles (95°C for 20 s, 58°C for 30 s, 72°C for 25 s) and a final elongation at 72°C for 3 min. Agarose gel electrophoresis was used to determine the quality and yield of the PCR. For purifying amplicon DNA, PCR reactions were incubated with 1.8x volume of CleanPCR beads (CleanNA, CPCR-0005) for 10 min. Beads were immobilized on a DynaMag-96 Side Magnet (Thermo Fisher, 12331D) for 5 min, the supernatant was removed, and the beads washed 2x with 150 μL 70% ethanol. After air drying the beads for 5 min, DNA was eluted in 15 μL of 10 mM Tris-HCl pH 8.0. Amplicon DNA concentration was determined using the Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher, P7589) and then diluted to 0.7 ng/μL. Thereafter, indexing PCRs were performed in 25 μL PCR reaction volumes containing 0.08 μM (1 μL of a 2 μM stock) each of i5 and i7 Indexing Primers, 1x MyTaq Reaction Buffer, 0.5 units of MyTaq HS (Bioline, BIO-21112), and 1 μL of the purified PCR product (diluted to 0.5 ng/μl) from the previous step. The following cycling parameters were used for the indexing PCR: 5 min for 95°C for initial denaturation and activation of the polymerase, 15 cycles (95°C for 10 s, 55°C for 30 s, 72°C for 40 s) and a final elongation at 72°C for 5 min. Agarose gel electrophoresis was used to determine the quality and yield of the PCR. An aliquot from each indexing reaction (5 μL of each reaction) was then pooled and purified with CleanPCR magnetic beads as described above and eluted in 1 μL x Number of pooled reactions. Concentration of the final library was determined using PicoGreen and the quality and size distribution of the library was assessed with a Bioanalyzer. Dual indexed TaBA-seq libraries were sequenced on an Illumina MiSeq in 2x300 bp output mode.
TaBA-Seq Alignment and Analysis
Raw, paired-end TaBA-seq reads were first quality filtered and the adaptor sequences removed using Trim Galore (v.0.3.1) with the ‘–paired’ parameter and adaptor sequences to be trimmed manually defined as “-a CTGTCTCTTATA -a2 AGATCGGAAGAGC.” Alignments were carried out to the mouse genome (mm10) using bsmap (v.2.90) using the following parameters ‘-v 5 -r 2 -I 8 -p 10’. CpG-methylation calls were extracted from the mapping output using the methratio.py function in bsmap (Xi and Li, 2009). Quantification of methylation levels was performed using the R package, methylKit (Akalin et al., 2012). Analysis was restricted to CpGs with a minimal coverage >20 and no more than the 99.9th percentile of coverage. Only CpGs meeting these coverage criteria and detected in all replicates were used for further analysis R.
Quantification and Statistical Analysis
The details of quantification and statistical analysis methods used can be found with each figure legend or in the method details above.
Data and Software Availability
The primary western blotting and imaging data for this paper is deposited on Mendeley data available at https://doi.org/10.17632/vjnw35hm7d.1. The accession number for the Mi-Seq data for the LINE-1 CpG methylation bisulfate sequencing reported in this paper is GEO: GSE119120.
Acknowledgments
We thank members of the Bartke group for input and discussions. We also thank Guohong Li, Ernest Laue, Aaron Alt, and Timothy Richmond for providing plasmids; Carol Wooding for technical support; and Agnieszka Gambus, Sonja Lorenz, Robert Schneider, Heinrich Leonhardt, and Brenda Schulman for valuable advice and support. We are grateful to Joseph Harrison, Paul DaRosa, Brian Kuhlman, and Rachel Klevit for sharing unpublished results. Funding for B.M.F. and T.B. was provided from the Medical Research Council (grant no. MC_UP_1102/2) and the European Research Council (ERC grant no. 309952). Funding for P.S., C.B.M., and S.B. was provided from the Deutsche Forschungsgemeinschaft (DFG grants SFB1064/A22). P.S. is a fellow of the Integrated Research Training Group (IRTG-1064).
Author Contributions
B.M.F. conceived the study, performed experiments, analyzed data, and wrote the manuscript; P.S. and C.B.M. performed experiments, analyzed data, and contributed to writing the manuscript; A.M. and H.K. performed mass spectrometry experiments and analyses; S.B. supervised experiments, analyzed data, and contributed to writing the manuscript; and T.B. supervised the project, analyzed data, and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: November 1, 2018
Footnotes
Supplemental Information includes seven figures, two tables, and one data file and can be found with this article online at https://doi.org/10.1016/j.molcel.2018.09.028.
Supplemental Information
Excel tables listing crosslinked residues identified for each set of substrates. The proteins crosslinked, the residue number for each and the StavRox score and FDR are shown.
Tables of the apparent Kd values (+/− SEM) for the mono-nucleosome and chromatin EMSAs are shown.
The experimental setup for the identification of ubiquitylated species (e.g., gel bands used) and a table of identified species from each experiment. Example spectra from each experiment with the modified peptide and identified b- and y-ions. An example spectral output from the StavRox data analysis software for the XL-MS data. A crosslinked peptide between UHRF1 and UBE2D1 is shown.
References
- Akalin A., Kormaksson M., Li S., Garrett-Bakelman F.E., Figueroa M.E., Melnick A., Mason C.E. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita K., Ariyoshi M., Tochio H., Nakamura Y., Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818–821. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- Arita K., Isogai S., Oda T., Unoki M., Sugita K., Sekiyama N., Kuwata K., Hamamoto R., Tochio H., Sato M. Recognition of modification status on a histone H3 tail by linked histone reader modules of the epigenetic regulator UHRF1. Proc. Natl. Acad. Sci. USA. 2012;109:12950–12955. doi: 10.1073/pnas.1203701109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartke T., Vermeulen M., Xhemalce B., Robson S.C., Mann M., Kouzarides T. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell. 2010;143:470–484. doi: 10.1016/j.cell.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick M., Kim J.K., Estève P.O., Clark A., Pradhan S., Jacobsen S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–1764. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- Brzovic P.S., Lissounov A., Christensen D.E., Hoyt D.W., Klevit R.E. A UbcH5/ubiquitin noncovalent complex is required for processive BRCA1-directed ubiquitination. Mol. Cell. 2006;21:873–880. doi: 10.1016/j.molcel.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Buetow L., Gabrielsen M., Anthony N.G., Dou H., Patel A., Aitkenhead H., Sibbet G.J., Smith B.O., Huang D.T. Activation of a primed RING E3-E2-ubiquitin complex by non-covalent ubiquitin. Mol. Cell. 2015;58:297–310. doi: 10.1016/j.molcel.2015.02.017. [DOI] [PubMed] [Google Scholar]
- DaRosa P.A., Harrison J.S., Zelter A., Davis T.N., Brzovic P., Kuhlman B., Klevit R.E. A bifunctional role for the UHRF1 UBL domain for controlling hemi-methylated DNA-dependent histone ubiquitylation. Mol. Cell. 2018;72:753–765. doi: 10.1016/j.molcel.2018.09.029. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorigo B., Schalch T., Bystricky K., Richmond T.J. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J. Mol. Biol. 2003;327:85–96. doi: 10.1016/s0022-2836(03)00025-1. [DOI] [PubMed] [Google Scholar]
- Fang J., Cheng J., Wang J., Zhang Q., Liu M., Gong R., Wang P., Zhang X., Feng Y., Lan W. Hemi-methylated DNA opens a closed conformation of UHRF1 to facilitate its histone recognition. Nat. Commun. 2016;7:11197. doi: 10.1038/ncomms11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferry L., Fournier A., Tsusaka T., Adelmant G., Shimazu T., Matano S., Kirsh O., Amouroux R., Dohmae N., Suzuki T. Methylation of DNA ligase 1 by G9a/GLP recruits UHRF1 to replicating DNA and regulates DNA methylation. Mol. Cell. 2017;67:550–565.e5. doi: 10.1016/j.molcel.2017.07.012. [DOI] [PubMed] [Google Scholar]
- Gao L., Tan X.-F., Zhang S., Wu T., Zhang Z.-M., Ai H.-W., Song J. An intramolecular interaction of UHRF1 reveals dual control for its histone association. Structure. 2018;26:304–311.e3. doi: 10.1016/j.str.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelato K.A., Tauber M., Ong M.S., Winter S., Hiragami-Hamada K., Sindlinger J., Lemak A., Bultsma Y., Houliston S., Schwarzer D. Accessibility of different histone H3-binding domains of UHRF1 is allosterically regulated by phosphatidylinositol 5-phosphate. Mol. Cell. 2014;54:905–919. doi: 10.1016/j.molcel.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Götze M., Pettelkau J., Schaks S., Bosse K., Ihling C.H., Krauth F., Fritzsche R., Kühn U., Sinz A. StavroX--a software for analyzing crosslinked products in protein interaction studies. J. Am. Soc. Mass Spectrom. 2012;23:76–87. doi: 10.1007/s13361-011-0261-2. [DOI] [PubMed] [Google Scholar]
- Gray F., Cho H.J., Shukla S., He S., Harris A., Boytsov B., Jaremko Ł., Jaremko M., Demeler B., Lawlor E.R. BMI1 regulates PRC1 architecture and activity through homo- and hetero-oligomerization. Nat. Commun. 2016;7:13343. doi: 10.1038/ncomms13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm M., Zimniak T., Kahraman A., Herzog F. xVis: a web server for the schematic visualization and interpretation of crosslink-derived spatial restraints. Nucleic Acids Res. 2015;43(W1):W362–W369. doi: 10.1093/nar/gkv463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutschner T., Haemmerle M., Genovese G., Draetta G.F., Chin L. Post-translational regulation of Cas9 during G1 enhances homology-directed repair. Cell Rep. 2016;14:1555–1566. doi: 10.1016/j.celrep.2016.01.019. [DOI] [PubMed] [Google Scholar]
- Harrison J.S., Cornett E.M., Goldfarb D., DaRosa P.A., Li Z.M., Yan F., Dickson B.M., Guo A.H., Cantu D.V., Kaustov L. Hemi-methylated DNA regulates DNA methylation inheritance through allosteric activation of H3 ubiquitylation by UHRF1. eLife. 2016;5:1400–1406. doi: 10.7554/eLife.17101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H., Horton J.R., Zhang X., Bostick M., Jacobsen S.E., Cheng X. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008;455:826–829. doi: 10.1038/nature07280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L., Li Z., Wang P., Lin Y., Xu Y. Crystal structure of PHD domain of UHRF1 and insights into recognition of unmodified histone H3 arginine residue 2. Cell Res. 2011;21:1374–1378. doi: 10.1038/cr.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama S., Nishiyama A., Saeki Y., Moritsugu K., Morimoto D., Yamaguchi L., Arai N., Matsumura R., Kawakami T., Mishima Y. Structure of the Dnmt1 reader module complexed with a unique two-mono-ubiquitin mark on histone H3 reveals the basis for DNA methylation maintenance. Mol. Cell. 2017;68:350–360.e7. doi: 10.1016/j.molcel.2017.09.037. [DOI] [PubMed] [Google Scholar]
- Isono K., Endo T.A., Ku M., Yamada D., Suzuki R., Sharif J., Ishikura T., Toyoda T., Bernstein B.E., Koseki H. SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev. Cell. 2013;26:565–577. doi: 10.1016/j.devcel.2013.08.016. [DOI] [PubMed] [Google Scholar]
- Li T., Wang L., Du Y., Xie S., Yang X., Lian F., Zhou Z., Qian C. Structural and mechanistic insights into UHRF1-mediated DNMT1 activation in the maintenance DNA methylation. Nucleic Acids Res. 2018;46:3218–3231. doi: 10.1093/nar/gky104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty R.K., Henrici R.C., Tan S. Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature. 2014;514:591–596. doi: 10.1038/nature13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland C.B., Smets M., Schmidtmann E., Leidescher S., Markaki Y., Hofweber M., Qin W., Manzo M., Kremmer E., Thanisch K. A modular open platform for systematic functional studies under physiological conditions. Nucleic Acids Res. 2015;43:e112. doi: 10.1093/nar/gkv550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nady N., Lemak A., Walker J.R., Avvakumov G.V., Kareta M.S., Achour M., Xue S., Duan S., Allali-Hassani A., Zuo X. Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J. Biol. Chem. 2011;286:24300–24311. doi: 10.1074/jbc.M111.234104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A., Yamaguchi L., Sharif J., Johmura Y., Kawamura T., Nakanishi K., Shimamura S., Arita K., Kodama T., Ishikawa F. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature. 2013;502:249–253. doi: 10.1038/nature12488. [DOI] [PubMed] [Google Scholar]
- Qin W., Wolf P., Liu N., Link S., Smets M., La Mastra F., Forné I., Pichler G., Hörl D., Fellinger K. DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res. 2015;25:911–929. doi: 10.1038/cr.2015.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakumara E., Wang Z., Ma H., Hu L., Chen H., Lin Y., Guo R., Wu F., Li H., Lan F. PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol. Cell. 2011;43:275–284. doi: 10.1016/j.molcel.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart S.B., Krajewski K., Nady N., Tempel W., Xue S., Badeaux A.I., Barsyte-Lovejoy D., Martinez J.Y., Bedford M.T., Fuchs S.M. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol. 2012;19:1155–1160. doi: 10.1038/nsmb.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart S.B., Dickson B.M., Ong M.S., Krajewski K., Houliston S., Kireev D.B., Arrowsmith C.H., Strahl B.D. Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev. 2013;27:1288–1298. doi: 10.1101/gad.220467.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif J., Muto M., Takebayashi S., Suetake I., Iwamatsu A., Endo T.A., Shinga J., Mizutani-Koseki Y., Toyoda T., Okamura K. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–912. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- Smets M., Link S., Wolf P., Schneider K., Solis V., Ryan J., Meilinger D., Qin W., Leonhardt H. DNMT1 mutations found in HSANIE patients affect interaction with UHRF1 and neuronal differentiation. Hum. Mol. Genet. 2017;26:1522–1534. doi: 10.1093/hmg/ddx057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempe J.-F., Sauvé V., Grenier K., Seirafi M., Tang M.Y., Ménade M., Al-Abdul-Wahid S., Krett J., Wong K., Kozlov G. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science. 2013;340:1451–1455. doi: 10.1126/science.1237908. [DOI] [PubMed] [Google Scholar]
- Vaughan R.M., Dickson B.M., Cornett E.M., Harrison J.S., Kuhlman B., Rothbart S.B. Comparative biochemical analysis of UHRF proteins reveals molecular mechanisms that uncouple UHRF2 from DNA methylation maintenance. Nucleic Acids Res. 2018;46:4405–4416. doi: 10.1093/nar/gky151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y., Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009;10:232. doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S., Jakoncic J., Qian C. UHRF1 double tudor domain and the adjacent PHD finger act together to recognize K9me3-containing histone H3 tail. J. Mol. Biol. 2012;415:318–328. doi: 10.1016/j.jmb.2011.11.012. [DOI] [PubMed] [Google Scholar]
- Zhang Z.-M., Rothbart S.B., Allison D.F., Cai Q., Harrison J.S., Li L., Wang Y., Strahl B.D., Wang G.G., Song J. An allosteric interaction links USP7 to deubiquitination and chromatin targeting of UHRF1. Cell Rep. 2015;12:1400–1406. doi: 10.1016/j.celrep.2015.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q., Zhang J., Chen R., Wang L., Li B., Cheng H., Duan X., Zhu H., Wei W., Li J. Dissecting the precise role of H3K9 methylation in crosstalk with DNA maintenance methylation in mammals. Nat. Commun. 2016;7:12464. doi: 10.1038/ncomms12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Excel tables listing crosslinked residues identified for each set of substrates. The proteins crosslinked, the residue number for each and the StavRox score and FDR are shown.
Tables of the apparent Kd values (+/− SEM) for the mono-nucleosome and chromatin EMSAs are shown.
The experimental setup for the identification of ubiquitylated species (e.g., gel bands used) and a table of identified species from each experiment. Example spectra from each experiment with the modified peptide and identified b- and y-ions. An example spectral output from the StavRox data analysis software for the XL-MS data. A crosslinked peptide between UHRF1 and UBE2D1 is shown.