

Abstract
Background and Objectives
The helicase-primase inhibitor amenamevir (ASP2151) is a novel therapeutic agent which has been approved for the treatment of herpes zoster. The present study examined the pharmacokinetic profile of amenamevir in rodents and compared it with data from the literature of past and current established therapies (acyclovir and valaciclovir) to provide additional data to facilitate drug discovery and proper drug use.
Methods
In situ absorption, blood and plasma radioactivity concentrations, tissue distribution, and excretion were determined using liquid scintillation counting. Plasma amenamevir concentrations were measured using a validated chromatographic method. Chemical structures of in vivo metabolites were investigated using liquid chromatography-mass spectrometry and nuclear magnetic resonance spectroscopy.
Results
Amenamevir, after single intravenous administration to mice, had an elimination half-life of 2 h. Bioavailability was 40% after single oral administration. In situ absorption data indicated that amenamevir is mainly absorbed in the small intestine. The main component in mouse plasma was amenamevir, accounting for 87.9% of amenamevir-derived components. Our results suggest that the main elimination pathway in mice is oxidative metabolism at a methyl group and a 1,2,3-trisubstituted benzene ring followed by biliary and fecal excretion. Following oral administration of 14C-amenamevir to mice, 100.63% of the dose (10.06% in urine and 90.46% in feces) was excreted by 96 h post-dose.
Conclusions
The underlying mechanism of the improved pharmacokinetic profile of amenamevir was linked to an improved absorption ratio (not hepatic availability) compared to acyclovir, and qualitative differences in elimination (slow metabolism of amenamevir vs rapid urinary excretion of acyclovir/valaciclovir).
Electronic supplementary material
The online version of this article (10.1007/s13318-018-0481-y) contains supplementary material, which is available to authorized users.
Key Points
| Amenamevir showed bioavailability of 40% and slow elimination by oxidative metabolism in rodents | |
| The improved pharmacokinetic profile of amenamevir in comparison with those of past and current established agents was ascribed to its good absorption and slow elimination |
Introduction
Knowledge of the differences between an established drug and a new drug candidate (or newly approved drug) is important for facilitating drug discovery and proper drug use. In the past, the established drug for herpes simplex infection was acyclovir, an acyclic nucleoside analog that targets viral deoxyribonucleic acid polymerase. Issues associated with acyclovir include (1) its low bioavailability (15–20%), which limits efficacy even at high doses [1, 2], and (2) its short elimination half-life (t1/2: 2.5–3.3 h), which requires frequent dosing (3–5 times daily) [2]. A valine ester of acyclovir, valaciclovir, which has a bioavailability of 54% in humans [3], replaced acyclovir, and has been an established drug for herpes simplex infection for decades. However, the issue of a short elimination half-life remained. The t1/2 of valaciclovir is similarly as short as 2.5 h [3], thereby requiring frequent doses for the treatment of herpes zoster, initial episode of genital herpes, and recurrent episodes of genital herpes [4].
The helicase-primase is a promising target for new herpes simplex infection drugs [5]. Amenamevir (ASP2151; Fig. 1) inhibits helicase-primase activity at 0.1 μmol/L (48.3 ng/mL) or greater and herpes simplex virus type-1 replication with a 50% effective concentration of 0.036 μmol/L (17.4 ng/mL) in vitro [6]. In a mouse model of herpes simplex virus type-1 infection, the 50% effective oral doses of amenamevir and valaciclovir were 1.9 mg/kg and 27 mg/kg twice daily, respectively [6]. The pharmacokinetic-pharmacodynamic relationships of amenamevir in mice, guinea pigs, and humans have been studied extensively [7–9]. According to the Clinical Trials registry, the clinical development of amenamevir has always been associated with a once daily dosing regimen, and its efficacy in phase III study was satisfactory [10]. Given these promising nonclinical and clinical study results, amenamevir has been approved for the treatment of herpes zoster in Japan. In addition, we have not examined a prodrug of amenamevir because its good pharmacokinetic profile eliminates the need for more frequent dosing and prodrug synthesis. Indeed, the t1/2 of amenamevir in humans is approximately 8 h [11], and pharmacokinetic-pharmacodynamic analysis revealed that a once daily administration of 200 mg to humans was enough to maintain efficacious plasma concentration (200 ng/mL) for a necessarily long duration (15–21 h) [8]. These characteristics (no prodrug, infrequent administration, and good pharmacokinetic profile) make amenamevir highly favorable over acyclovir. To our knowledge, however, the underlying mechanism of this improved pharmacokinetic profile has not been investigated.
Fig. 1.

Chemical structure of 14C-amenamevir. *Position of 14C label
Here, we used unlabeled and 14C-labeled amenamevir (14C-amenamevir) to determine the (1) absorption profile (bioavailability, absorption sites, and absorption ratio), (2) distribution to target organs and other tissues, and (3) main elimination pathways of this drug in rodents. Rodents were selected because they are routinely used in these types of studies. In addition, bioavailability and elimination pathways of acyclovir are similar between rodents and humans [1, 2, 12], whereas dogs show much higher bioavailability [13] and monkeys show distinct and extensive metabolism [14]. Further, at the time that these studies were conducted, there was no evidence of any significant species differences in absorption and metabolism between rodents and humans, suggesting that comparison of the pharmacokinetics of acyclovir and amenamevir in rodents would be translatable to humans.
Materials and methods
Figure 2 shows an overview of the experiments.
Fig. 2.

Overview of experiments in the present study
Chemicals
14C-amenamevir (Fig. 1, specific activity 2.23 MBq/mg and radiochemical purity > 99%) was synthesized at EaglePicher Pharmaceutical Services (Lenexa, KS, USA). Unlabeled amenamevir, its solid dispersion (amenamevir:hydroxypropylmethylcellulose 2910 [TC-5 E] = 1:3), internal standard (AS1709867-00, D6-amenamevir), and metabolites R1 and R5 were supplied by Astellas Pharma (Tokyo, Japan). All organic solvents for high-performance liquid chromatography (HPLC) were of HPLC grade. Carbon dioxide absorbent (Carbo-Sorb E) and scintillation cocktails (Hionic-Fluor, PermaFluor E+, and Pico-Fluor 40) were purchased from PerkinElmer (Waltham, MA, USA). All other reagents were of analytical grade.
Animals
Male CD1 mice and F344 rats were purchased from Charles River Laboratories Japan (Kanagawa, Japan). In an in situ absorption study, pharmacokinetic studies, tissue distribution study, and excretion studies in mice, animal numbers were selected to mitigate the risk of drawing wrong conclusions due to inter-individual differences and random errors. In a metabolite identification study in rats, the number of animals was established to obtain the necessary amounts of metabolites. At the initiation of dosing, animals were aged 8–9 weeks and weighed 28–36 g for mice and 164–195 g for rats. Animals were given food and water ad libitum except when fasted overnight for at least 12 h before and 4–6 h after administration, unless otherwise stated. The animal rooms were maintained at a temperature of 20–26 °C, with a relative humidity of 40%–70%. Animals were acclimated for about 1 week prior to drug administration.
Dose Preparation
For all in vivo and in situ experiments using 14C-amenamevir, the compound was dissolved in N,N-dimethylformamide and diluted tenfold with 20% (w/v) (2-hydroxypropyl)-β-cyclodextrin solution to prepare a formulation of 14C-amenamevir at 0.3 mg/0.669 MBq/mL (final concentration) for oral doses and an in situ experiment, and at 0.6 mg/1.338 MBq/mL (final concentration) for intravenous doses.
For the pharmacokinetic study using unlabeled amenamevir, the compound was dissolved in N,N-dimethylformamide and diluted tenfold with 20% (w/v) (2-hydroxypropyl)-β-cyclodextrin solution to prepare formulations of amenamevir at 0.1, 0.3, and 1.0 mg/mL (final concentration) for 1, 3, and 10 mg/kg oral doses, and at 0.6 mg/mL (final concentration) for a 3 mg/kg intravenous dose.
The metabolite identification study in rats using unlabeled amenamevir required a higher dose to obtain biological matrices with higher metabolite concentrations, and therefore adequate analytical data for structure estimation. Amenamevir solid dispersion was suspended in dosing vehicle (5% [w/v] TC-5 E in Japanese Pharmacopeia second fluid) using a pestle and agate mortar to prepare a formulation of amenamevir at 20 mg/mL.
In Situ absorption study in mice
Bile ducts of 15 fasted mice were ligated under ether anesthesia to prevent bile from flowing into the intestine. Gastric loops, proximal-small intestinal loops, middle-small intestinal loops, distal-small intestinal loops, and large intestinal loops were prepared in the digestive tract (n = 3 per loop). For the gastric loop, ligation was made at the cardia and the pylorus. For the proximal-small intestinal loop, the first ligation was made at the pylorus. The second ligation was then done to form a 4-cm loop. A 4-cm middle-small intestinal loop was made by placing first and second ligation 4-cm apart. For the distal-small intestinal loop, the first ligation was made at the ileocecal junction. The second ligation was then done to prepare a 4-cm loop. For the large intestinal loop, the first ligation was done at the ileocecal junction. The second ligation was then done at the caudal region of the large intestine in order to make the loop as long as possible. For all loops, additional ligation was placed outside of the loop to ensure tight ligation.
After injection of 14C-amenamevir (0.03 mg/0.1 mL/loop) into each ligated loop, the gastrointestinal tract was returned to the abdominal cavity in the normal position, and the surrounding muscle and skin were sutured. Anesthesia was then discontinued. At 1 h post-injection, the mice were again anesthetized and the ligated sites were removed and weighed, before being homogenized in distilled water using a homogenizer to prepare 10% (w/v) homogenate. The total radioactivity of the homogenates was measured by the combustion method.
Pharmacokinetic Study Using Unlabeled Amenamevir in Mice
After single intravenous administration of amenamevir at 3 mg/kg or single oral administration of amenamevir at 1, 3, and 10 mg/kg to fasted mice, blood samples were collected at 0.1, 0.25, 0.5, 1, 2, 3, 4, 6, and 8 h post-dose (n = 3). No food was given after drug administration. Blood samples were collected using syringes containing heparin sodium from the inferior vena cava of three animals per time point under ether anesthesia, after which animals were sacrificed by exsanguination. Blood samples were centrifuged to obtain plasma and stored frozen in a freezer. Plasma amenamevir concentration was measured using a previously established method [15] with slight modification (i.e. while the previously established method used filtration in removal of insoluble endogenous substances, the present study used centrifugation [21,800×g, 4 °C, 10 min]). The bioanalytical method is summarized as follows. Internal standard, D6-labeled amenamevir, was weighed, dissolved, and diluted to prepare internal standard working solution (final solvent composition: acetonitrile/water [20:80, v/v], final D6-labeled amenamevir concentration: 250 ng/mL). The internal standard working solution (0.05 mL) and acetonitrile/water (20:80, v/v) (0.05 mL) were added to 0.1 mL of each animal plasma sample. After centrifugation to remove insoluble endogenous substances, the supernatants (0.05 mL) were injected onto a column-switching liquid chromatography-tandem mass spectrometry (LC–MS/MS) system, consisting of Waters Alliance 2695 model (Milford, MA, USA), Waters 515 HPLC pump, Shiseido 3011 six-port valve (Tokyo, Japan), and Thermo Finnigan TSQ triple quadrupole mass spectrometer with performance kit (San Jose, CA, USA). The lower limit of quantitation (LLOQ) was 5 ng/mL.
Pharmacokinetic Study Using 14C-Amenamevir in Mice
After single intravenous or oral administration of 14C-amenamevir at 3 mg/kg to mice, blood samples were collected at 0.1, 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h post-dose (n = 3). Blood samples were collected using syringes containing heparin sodium from the inferior vena cava of three animals per time point under ether anesthesia without cannulation or fluid replacement, after which animals were sacrificed by exsanguination. Blood samples (typically 0.8 mL per animal per time point) were cooled on ice and then divided into three aliquots. One aliquot was sampled with a hematocrit capillary tube and then used to determine the hematocrit value. The second aliquot was used for analysis of total radioactivity. The remaining aliquot was centrifuged to obtain plasma. Total radioactivity in plasma and blood was measured by the combustion method. The distribution ratio into blood cells was calculated from the hematocrit value and the concentration of radioactivity in the blood and plasma.
Tissue Radioactivity Concentration in Mice
After single oral administration of 14C-amenamevir at a dose of 3 mg/kg to mice, 3 animals per time point were sacrificed by exsanguination under ether anesthesia at 0.5, 1, 4, and 24 h post-dose. Blood and plasma samples were collected using the same procedure as described in Sect. 2.6. Tissues (brain, spinal cord, hypophysis, eyes, Harderian glands, submaxillary glands, thyroid glands, thymus, heart, lungs, liver, spleen, pancreas, adrenals, kidneys, epididymides, testes, skeletal muscle, skin, fat, femur, urinary bladder, stomach, small intestine, and large intestine) were isolated and weighed. Total radioactivity in biological samples was measured by the combustion method.
In Vivo Metabolite Identification in Rats
Mice were considered too small to be able to isolate adequate amounts of drug metabolites, even at high doses. Rats were more appropriate, due to their larger size. Rat metabolites were therefore isolated and identified in the first step. In the next step (see Sect. 2.9), mouse metabolites were identified using the identified rat metabolites.
Matrix
A relatively high dose (200 mg/kg) was designated to allow the isolation of adequate amounts of drug metabolites. After single oral administration of unlabeled amenamevir at a dose of 200 mg/kg to intact rats, blood samples were collected via the inferior vena cava of three animals per time point under ether anesthesia without fluid replacement at 1, 2, and 4 h post-dose, after which animals were sacrificed by exsanguination. The blood samples (typically 7 mL per animal per time point) were centrifuged (1500×g, 15 min, 4 °C) to obtain plasma. Urine samples were collected 0–6 h and 6–24 h after single oral administration of unlabeled amenamevir at a dose of 200 mg/kg to 19 intact rats. Animals were housed individually in urine collection cages and sacrificed after last collection (24 h after dosing). Bile samples were collected 0–6 h and 6–24 h after single oral administration of unlabeled amenamevir at a dose of 200 mg/kg to 10 bile duct-cannulated rats. Bile duct cannulation was carried out under ether anesthesia. Animals were sacrificed after last sample collection (24 h after dosing). These matrices were pooled per matrix per time point before further processing.
Isolation
A preliminary experiment indicated that bile samples contained amenamevir and its metabolites R1–R10, while plasma and urine samples contained amenamevir and R5. Bile was therefore selected as the matrix from which to isolate amenamevir metabolites.
R1 to R9 were isolated from the pooled bile sample collected 0–6 h post-dose as follows. An aliquot (40 mL) of the pooled bile sample was mixed with 4 mL of 1 mol/L hydrochloride solution and 400 mL of water and centrifuged at 1500×g for 5 min. The supernatant was processed by solid phase extraction (SPE) using an Oasis HLB 35 cc 6 g LP (Waters) and separated into two fractions: an acetonitrile-eluted fraction (R1 to R8-containing fraction) and an ethyl acetate-eluted fraction (R9-containing fraction).
The acetonitrile-eluted fraction was concentrated under reduced pressure, and then lyophilized to a dry powder. After reconstitution in 0.05% formic acid/methanol (95:5, v/v) and filtration using a syringe filter, the sample was injected onto a preparative HPLC system (SCL-10Avp system controller, LC-6AD pump, and SPD-10Avp UV detector; Shimadzu, Kyoto, Japan) coupled with a DC-1200 fraction collector (Eyela; Tokyo, Japan). Chromatography was carried out on an Ultra Pack ODS-S-50C (Yamazen; Osaka, Japan) with a solvent gradient of methanol, tetrahydrofuran, and 0.05% formic acid aqueous solution with a flow rate of 5.0 mL/min. The eluate was collected at 1-min intervals. A group of fractions containing R1 to R3 and another containing R4 to R8 were separately pooled for further processing.
The pooled fraction containing R1, R2, and R3 was concentrated under reduced pressure, and then lyophilized to a dry powder. After reconstitution in water/acetonitrile (50:50, v/v), the sample was injected onto a preparative HPLC system under HPLC condition 1 (Table 1). The R1-, R2-, and R3-containing fractions were separately concentrated under reduced pressure. Each remaining aqueous solution was loaded onto an Oasis HLB 6 cc 0.2 g. After washing with water, the sample was eluted with acetonitrile, and the eluate was evaporated to dryness under reduced pressure. The isolated R1 (2.70 mg), R2 (1.20 mg), and R3 (0.43 mg) were analyzed to estimate their chemical structures.
Table 1.
Preparative HPLC conditions used to isolate rat metabolites
| Condition ID | Metabolite | Column | Flow rate (mL/min) | Mobile phase component |
|---|---|---|---|---|
| 1 | R1–R3 | C18 columna | 4.70 | FA, MeOH, THF, H2O |
| 2 | R4–R8 | C30 columnb | 4.70 | AcONH4, MeOH, H2O |
| 3 | R4–R8 | C30 columnb | 2.36 | ACN, H2O |
| 4 | R9 | C30 columnb | 2.36 | FA, ACN, H2O |
| 5 | R9 | C30 columnb | 2.36 | FA, ACN, H2O |
| 6 | R10 | C30 columnb | 2.00 | FA, ACN, H2O |
| 7 | R10 | C18 columna | 4.70 | AcONH4, ACN, H2O |
Column temperature: 50 °C, Detection: UV275 nm, HPLC system: SCL-10A system controller, LC-10AD pump, CTO-10A column oven, SIL-10A autosampler, and SPD-10AV UV detector (Shimadzu) coupled with a SF-2120 fraction collector (Advantec Toyo)
ACN acetonitrile, AcONH4 ammonium acetate, FA formic acid, MeOH methanol, THF tetrahydrofuran
aInertsil ODS-3 Prep Guard Cartridge (7.6 mm I.D. × 30 mm, 5 μm) and Inertsil ODS-3 (10 mm I.D. × 250 mm, 5 μm) (GL Sciences, Tokyo, Japan)
bDevelosil C30-UG-5 Guard Column (8 mm I.D. × 10 mm, 5 μm) and Develosil C30-UG-5 (10 mm I.D. × 250 mm, 5 μm) (Nomura Chemical)
The pooled fraction containing R4 to R8 was concentrated under reduced pressure and lyophilized to a dry powder. After reconstitution in water/acetonitrile (50:50, v/v), the sample was injected onto a preparative HPLC system under HPLC condition 2 (Table 1). Further separation was conducted under HPLC condition 3 (Table 1). The R4-, R5-, R6-, R7-, and R8-containing fractions were concentrated under reduced pressure, and the remaining aqueous solutions were lyophilized. The isolated R4 (0.92 mg), R5 (2.97 mg), R6 (0.38 mg), R7 (0.31 mg), and R8 (0.50 mg) were analyzed to estimate their chemical structures.
The ethyl acetate-eluted fraction (R9-containing fraction) was evaporated to dryness under reduced pressure, dissolved in water/methanol (5:24, v/v), and purified under HPLC condition 4 (Table 1). The fraction was further purified under HPLC condition 5 (Table 1). The R9-containing fractions were concentrated under reduced pressure, and the remaining aqueous solutions were lyophilized. The isolated R9 (0.29 mg) was analyzed to estimate its chemical structure.
R10 was isolated from the pooled bile sample collected 6–24 h post-dose as follows. An aliquot (60 mL) of the pooled bile sample was mixed with 6 mL of 1 mol/L hydrochloride solution and 600 mL of 10 mmol/L ammonium acetate and centrifuged at 1500×g for 5 min. The supernatant was loaded onto an Oasis HLB 35 cc 6 g LP and washed with water. Fractions were collected by stepwise elution with water/methanol = 7/3, 6/4, 5/5, 4/6, 3/7, 2/8, 1/9, and methanol. The fraction eluted using water/methanol = 3/7 contained R10, and was concentrated under reduced pressure and lyophilized to a dry powder. After reconstitution in 1.5 mL of methanol and 1.0 mL of water, the sample was injected onto a preparative HPLC system under HPLC condition 6 (Table 1). Further separation was conducted under HPLC condition 7 (Table 1). The R10-containing fractions were concentrated under reduced pressure, and the remaining aqueous solutions were lyophilized. The isolated R10 (0.4 mg) was analyzed to estimate its chemical structure.
Identification
The isolated metabolites were analyzed using a nuclear magnetic resonance (NMR) spectrometer Varian INOVA600 (Palo Alto, CA, USA) to obtain 1H-NMR spectra. When further NMR analysis was needed to estimate a chemical structure, Heteronuclear Multiple-Bond Coherence (HMBC), Heteronuclear Single Quantum Coherence (HSQC), Heteronuclear Single Quantum Coherence ADiabatic (HSQCAD), Nuclear Overhauser and Exchange SpectroscopY (NOESY), TOtally Correlated SpectroscopY (TOCSY), and decoupling experiments were conducted. In addition to NMR analysis, liquid chromatography-mass spectrometry (LC/MS) analysis was conducted using a Shiseido NANOSPACE SI-2 system coupled to a Thermo Fisher Scientific TSQ Quantum Ultra AM (Waltham, MA, USA) with an electrospray ionization probe. To compare mass spectra and retention times (RT) between isolated metabolites and authentic standards using LC/MS, chromatography was performed on a Develosil C30-UG-5 (4.6 mm I.D × 250 mm, 5 μm; Nomura Chemical, Seto, Japan) at 50 °C with a mixture of 0.05% formic acid aqueous solution and acetonitrile at a flow rate of 0.5 mL/min. The mass spectrometer was operated in the positive and negative ion mode with capillary temperature of 350 °C, sheath gas of nitrogen at 80 units, and auxiliary gas of nitrogen at 20 units.
In Vivo Metabolite Identification in Mice
Mouse plasma samples were obtained from the above pharmacokinetic study using 14C-amenamevir. Mouse bile and urine samples were obtained from an excretion study using 14C-amenamevir, which are mentioned below. Mouse metabolites were identified by comparing RT of radioactive peaks in mouse samples with those of amenamevir and R1 to R10. To identify an unknown peak of RT 43 min in mouse bile, the HPLC fractions were collected, pooled, and dried under reduced pressure. The residue was dissolved in 1.5 mL of 0.2 mol/L phosphate buffer (pH 6.8). To obtain an aglycone of the unknown peak, an aliquot (0.4 mL) of the sample was mixed with 50 μL of 0.2 mol/L phosphate buffer (pH 6.8) and 50 μL of 10 mg/mL β-glucuronidase solution (from Escherichia coli; purchased from Sigma-Aldrich, St. Louis, MO, USA), and incubated at 37 °C for 2 h. The reaction was stopped by adding 200 μL of acetonitrile, then the mixture was mixed well and centrifuged. The supernatant was filtered and injected onto an HPLC system (SCL-10Avp system controller, LC-10ADvp pump, CTO-10Avp column oven, and SIL-10ADvp autosampler [Shimadzu] coupled with a SF-2120 fraction collector [Advantec Toyo; Tokyo, Japan]). Chromatography was performed on a Develosil C30-UG-5 (4.6 mm I.D × 250 mm, 5 μm) at 50 °C with a mixture of 0.05% formic acid aqueous solution and acetonitrile at a flow rate of 0.5 mL/min. The eluent was collected at 1-min intervals and the total radioactivity was measured by direct method. The aglycone was identified by comparing the RT of the aglycone to those of R1–R10.
In Vivo Metabolic Profiling in Mice
Mouse plasma samples were obtained from an above pharmacokinetic study using 14C-amenamevir. The collection times were 0.1, 0.25, 0.5, 1, 2, 3, 4, 6, and 8 h post-dose. Aliquots (0.1 mL each) of the samples were pooled per time point, deproteinized with an equal volume of acetonitrile, and centrifuged. The supernatant was analyzed using the same HPLC system and condition as those used in vivo metabolite identification in mice. RT and peak areas of each radioactive component were used to identify the component and determine the concentration ratio, respectively.
Mouse bile and urine samples were obtained from an excretion study using 14C-amenamevir, as mentioned below. The collection times were 0–6, 6–24, 24–48, 48–72, and 72–96 h post-dose for urine, and 0–6, 6–24, and 24–48 h post-dose for bile. After pooling the individual samples per time point in the ratios of their excretion volumes, the samples were directly injected onto the HPLC system under the same separation and detection condition as the plasma samples.
Excretion in Mice
Following single oral administration of 14C-amenamevir at 3 mg/kg, urine and feces were collected from four intact mice, and bile and urine were collected from four bile duct-cannulated mice with ligation at the gallbladder. The ligation and bile duct cannulation were carried out under sodium pentobarbital anesthesia. Samples were collected in containers on dry ice. From the intact mice, urine samples were collected at 0–6, 6–24, 24–48, 48–72, and 72–96 h post-dose and fecal samples at 0–24, 24–48, 48–72, and 72–96 h post-dose. From the bile duct-cannulated mice, excreta samples were collected at 0–6, 6–24, and 24–48 h post-dose. At the end of sample collection from intact mice, cages were rinsed with a small volume of distilled water and acetonitrile, and the rinse solutions were collected as cage wash. Total radioactivity in urine samples, bile samples, and cage wash was measured by the direct measurement method, whereas that of feces samples was measured by the combustion method.
Enterohepatic Circulation in Mice
Four bile duct-cannulated mice with ligation at the gallbladder received a bolus intra-duodenal injection of a 0.1-mL aliquot of pooled bile sample collected for 0–24 h in the same manner as in the biliary excretion experiment above. Samples were collected and analyzed in the same manner as in the biliary excretion experiment.
Radioactivity Measurement
The liquid scintillation counters (LSC) Tri-carb 2100TR (PerkinElmer; formerly Packard Instrument), Tri-carb 2700TR (PerkinElmer), and LS 6500 (Beckman, Brea, California, USA) were used for radioanalysis.
Gastrointestinal loop homogenates, blood, plasma, tissues, and feces were analyzed by the combustion method. A sample was dried in a drying oven and combusted in a sample oxidizer. The resulting 14CO2 was trapped using a PermaFluor E+ scintillation cocktail and Carbo-Sorb E carbon dioxide absorbent, and analyzed by LSC.
Urine, bile, and cage wash were analyzed by the direct measurement method. A sample was mixed with the Hionic-Fluor scintillation cocktail and analyzed by LSC.
HPLC fractions were analyzed by the direct measurement method. A sample was mixed with the Pico-Fluor 40 scintillation cocktail and analyzed by LSC.
Data Analysis
Concentration of total radioactivity was expressed as ng eq./mL or ng eq./g based on LSC counts and the specific activity of the dose formulation. Pharmacokinetic parameters (maximum concentration [Cmax], time to maximum concentration [Tmax], area under the concentration versus time curve from time zero to infinity [AUCinf], elimination half-life [t1/2], total body clearance [CLtotal], and volume of distribution at steady state [Vdss]) were calculated using a non-compartment model in WinNonlin version 4.1 (Pharsight, Mountain View, CA, USA).
Absorption ratios (Fa) were calculated using the following equation [16, 17]:
Absolute bioavailability (F) were calculated using the following equation [18]:
Distribution of radioactivity into blood cells was calculated using the following equation [19–21]:
where
Cp: plasma radioactivity concentration (ng eq./mL)
Cb: blood radioactivity concentration (ng eq./mL)
Ht: hematocrit value (%)
Plasma concentrations and the abundance of amenamevir and its metabolites in metabolite fingerprinting were calculated using the following equations:
Urinary and biliary excretion and abundance of amenamevir and its metabolites in metabolite fingerprinting were calculated using the following equations:
Renal clearance (CLr) in mice was calculated using the following equation:
Allometric scaling of clearances (CL) was conducted using the following equation. Body weights of mice and humans were 20 g and 70 kg.
Hepatic availability (Fh) in humans was calculated using the following equation. Blood/plasma ratio (Rb) was assumed to be 1. Hepatic blood flow (Qh) in humans was 1450 mL/min [22].
Oral clearance (CLoral) in humans was calculated using the following equation:
Results
Pharmacokinetics in Mice
The concentration–time curves of amenamevir and total radioactivity are shown in Figs. 3 and 4, respectively. Pharmacokinetic parameters after oral administration are summarized in Table 2.
Fig. 3.

Plasma concentrations of unchanged drug after single intravenous (iv) and oral (po) administration of unlabeled amenamevir to mice. Doses are 3 mg/kg for intravenous administration and 1, 3, and 10 mg/kg for oral administration. Data represent mean + SD of three animals
Fig. 4.

Blood and plasma concentrations of radioactivity after single intravenous (iv) and oral (po) administration of 14C-amenamevir to mice at 3 mg/kg. Data represent mean + SD of three animals
Table 2.
Pharmacokinetic parameters after single oral or intravenous administration of unlabeled amenamevir or 14C-amenamevir to mice
| Dose (mg/kg) and dosing route | Matrix | Analyte |
C
max
(ng/mL) |
T
max
(h) |
CLtotal (L/h/kg) |
Vdss (L/kg) |
AUCinf (μg × h/mL) |
F or Fa (%) |
|---|---|---|---|---|---|---|---|---|
| 1a, po | Plasma | Amenamevir | 72 | 1.0 | N.A. | N.A. | 0.35 | 40.5e |
| 3a, po | Plasma | Amenamevir | 265 | 1.0 | N.A. | N.A. | 1.05 | 40.5e |
| 10a, po | Plasma | Amenamevir | 893 | 1.0 | N.A. | N.A. | 3.97 | 46.1e |
| 3a, iv | Plasma | Amenamevir | N.A. | N.A. | 1.16 | 3.14 | 2.59 | N.A. |
| 3b, po | Plasma | Radioactivity | 429c | 1.0 | N.A. | N.A. | 1.82d | 40.1f |
| 3b, iv | Plasma | Radioactivity | N.A. | N.A. | N.A. | N.A. | 4.55d | N.A. |
| 3b, po | Blood | Radioactivity | 363c | 1.0 | N.A. | N.A. | 1.61d | 38.8f |
| 3b, iv | Blood | Radioactivity | N.A. | N.A. | N.A. | N.A. | 4.14d | N.A. |
Each parameter was calculated from the mean concentration–time profile of three animals
N.A. Not applicable, iv intravenous, po oral, Cmax maximum concentration, Tmax time to reach Cmax, CLTotal apparent toal clearance, Vdss volume of distribution at steady state, AUCinf area under the concentration-time curve from time 0 to infinity, F absoute bioavailability, Fa absorption ratio
aUnlabeled amenamevir was administered, b 14C-amenamevir was administered, cng eq./mL, dμg eq. × h/mL, eF, fFa
After single intravenous administration of unlabeled amenamevir to mice at 3 mg/kg, plasma concentrations of unchanged drug decreased with a t1/2 of 2.25 h. The CLtotal of 1.16 L/h/kg suggested relatively low clearance, and the Vdss of 3.14 L/kg suggested extensive tissue distribution of amenamevir in mice. After single oral administration of unlabeled amenamevir to mice at 1, 3, and 10 mg/kg, a Tmax of 1 h was observed, indicating rapid absorption. Plasma concentrations of unchanged drug increased almost dose-proportionally and the F was about 40%, regardless of dose.
Following single intravenous administration of 3 mg/kg of 14C-amenamevir to mice, the radioactivity concentrations in blood and plasma decreased with t1/2 of 6.7 and 3.2 h, respectively. AUCinf values in blood and plasma were 4137 and 4548 ng eq. × h/mL, respectively. Following single oral administration of 3 mg/kg of 14C-amenamevir to mice, blood and plasma radioactivity levels peaked at 1 h post-dose. Fa values in the blood and plasma were 38.8% and 40.1%, respectively.
The hematocrit values were 40.3%–49.5%. The distribution of radioactivity into blood cells was 31.5%–48.8% between 0.25 and 8 h after both oral and intravenous administration.
In Situ Absorption in Mice
After 14C-amenamevir was injected into the proximal-, middle- and distal-small intestinal loops of mice, the radioactivity absorption ratios were 32.05 ± 6.38%, 21.29 ± 5.12% and 16.48 ± 10.59%, respectively (mean ± standard deviation [SD], n = 3). The absorption ratios at the gastric and large intestinal loops were 4.36 ± 2.57% and − 0.17 ± 3.00%, respectively. These data suggest that amenamevir is absorbed in the small intestine.
Tissue Distribution of Radioactivity in Mice
Following single oral administration of 3 mg/kg of 14C-amenamevir to mice, radioactivity was highest in most tissues at 1 h post-dose (Table 3), suggesting relatively rapid distribution to tissues. The concentration of radioactivity in tissues at 1 h after dosing was highest in the liver, followed by small intestine, Harderian glands, kidneys, and large intestine. Low radioactivity levels were found in the femur, testes, spinal cord, and brain. While concentrations in the brain (18.0–32.6 ng eq./g) were lower than that in plasma, they were comparable to the EC50 value (17.4 ng/mL) for herpes simplex virus type-1 replication. Radioactivity distribution in the skin, where amenamevir is expected to exhibit efficacy, was moderate. Radioactivity disappeared from most tissues as the blood radioactivity concentration decreased. Tissue to plasma concentration ratios increased after the first sampling time point in the testes (0.1, 0.2, and 0.5 at 0.5, 1, and 4 h after dosing, respectively) and large intestine (1.4, 2.9, and 18.4, respectively) while the tissue to plasma concentration ratios decreased in the stomach (16, 2.5, and 5.0, respectively; Online Source 1). In the other tissues, tissue to plasma concentration ratios showed smaller changes. Note that tissue to plasma concentration ratios at 24 h after dosing are not available as plasma radioactivity concentrations were below the limit of quantification. At 24 h after dosing, radioactivity concentrations in all tissues were less than 2% of their respective maximum levels.
Table 3.
Tissue concentrations of radioactivity after single oral administration of 14C-amenamevir to mice at 3 mg/kg
| Tissue | Mean concentration (ng eq./g or mL) | |||
|---|---|---|---|---|
| 0.5 h | 1 h | 4 h | 24 h | |
| Blood | 305 | 365 | 165 | 7.08 |
| Plasma | 349 | 422 | 186 | BLQ |
| Brain | 25.9 | 32.6 | 18.0 | BLQ |
| Spinal cord | 33.3 | 41.6 | 20.0 | BLQ |
| Hypophysis | 865 | 921 | 590 | BLQ |
| Eyes | 153 | 202 | 103 | BLQ |
| Harderian glands | 1890 | 2330 | 1010 | 13.4 |
| Submaxillary glands | 793 | 969 | 387 | BLQ |
| Thyroid glands | 959 | 896 | 372 | BLQ |
| Thymus | 387 | 526 | 233 | BLQ |
| Heart | 824 | 987 | 419 | 3.53 |
| Lungs | 644 | 681 | 333 | 2.04 |
| Liver | 8850 | 8450 | 3980 | 21.2 |
| Spleen | 527 | 587 | 275 | BLQ |
| Pancreas | 1130 | 1230 | 570 | BLQ |
| Adrenals | 750 | 1020 | 333 | BLQ |
| Kidneys | 1880 | 2080 | 988 | 4.23 |
| Epididymides | 316 | 402 | 283 | BLQ |
| Testes | 32.1 | 75.0 | 95.6 | BLQ |
| Skeletal muscle | 388 | 450 | 210 | BLQ |
| Skin | 210 | 251 | 140 | BLQ |
| Fat | 334 | 417 | 180 | BLQ |
| Femur | 63.8 | 117 | 44.8 | BLQ |
| Urinary bladder | 319 | 426 | 288 | BLQ |
| Stomach | 5480 | 1020 | 827 | 5.35 |
| Small intestine | 5010 | 6370 | 1670 | BLQ |
| Large intestine | 500 | 1240 | 3060 | BLQ |
Each value represents the mean of three animals
BLQ below the limit of quantitation
In Vivo Metabolite Identification in Rats and Mice
Following single oral administration of 200 mg/kg of unlabeled amenamevir to rats, plasma, urine, and bile were collected and analyzed. As all metabolites found in plasma and urine were also detected in bile, bile samples were used to isolate rat metabolites. Ten metabolites were successfully isolated, and were named R1–R10. Their chemical structures were estimated using LC/MS and NMR spectra (Fig. 5). The details of the chemical structure estimation are provided in Online Source 2. After authentic standards of R1 and R5 became available, their RT and mass spectra were compared with those of isolated R1 and R5. As the data were matched very well, the chemical structures of R1 and R5 could be identified.
Fig. 5.
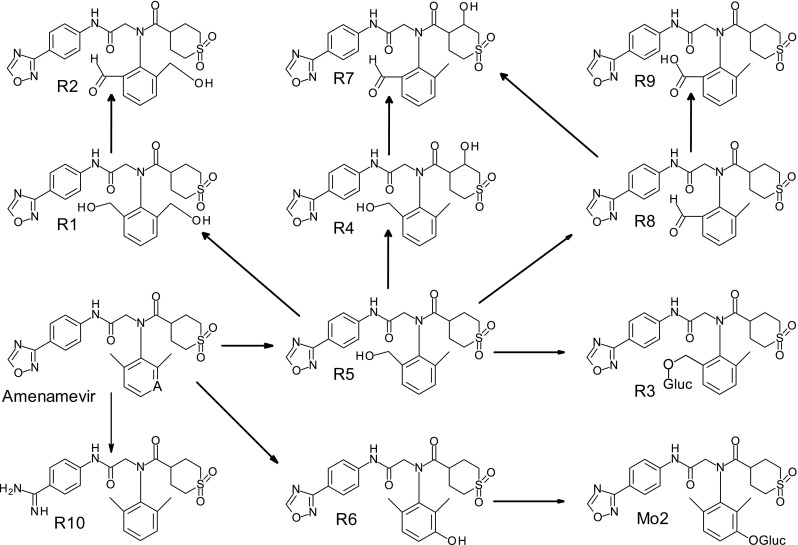
Postulated metabolic pathways of amenamevir in mice and rats. Gluc glucuronic acid moiety
Leftover samples in the mouse pharmacokinetic and excretion studies were analyzed to examine metabolites that were not present in rats. Comparison of radioactive peak RT of mouse samples with those of amenamevir and rat metabolites (R1 to R10) revealed three unknown metabolites (RT: 6, 9, and 43 min). The unknown peak of RT 6 min was observed in mouse plasma and bile, while the unknown peak of RT 9 min was in mouse urine. As these two metabolites were not observed in exploratory human metabolite fingerprinting (manuscript in preparation) and we lacked a sufficient sample amount to perform chemical structure analysis, we did not identify their chemical structures. The unknown peak of RT 43 min was abundant in mouse urine and bile, and was named Mo2. Mo2 was incubated with β-glucuronidase and injected onto an HPLC system. A radioactive peak was observed at the same RT as R6, suggesting that Mo2 might be a glucuronide of R6.
In Vivo Metabolic Profiles in Mice
Metabolites in mouse plasma, urine, and bile were semi-quantitatively profiled (Table 4). The main radioactive peak in mouse plasma was unchanged amenamevir, accounting for 87.9% (as AUC0−8h) of amenamevir-derived components in plasma. The most abundant components in mouse urine were amenamevir, R10, and Mo2, which cumulatively accounted for 50.0, 21.2, and 12.5%, respectively, of amenamevir-derived components in urine. In mouse bile, Mo2, R5, and R3 cumulatively comprised 30.6, 19.3, and 17.8%, respectively, of amenamevir-derived components. Rat metabolite R2, R4, R7, and R8 were not detected in mouse plasma, urine, or bile. Based on these results, the metabolic pathways of amenamevir in rats and mice were postulated (Fig. 5).
Table 4.
Metabolite profiles in mouse plasma, urine, and bile
| Metabolite ID or retention time (min) | Plasma (0–8 h) | Urine (0–96 h) | Bile (0–48 h) | |||
|---|---|---|---|---|---|---|
| AUC0−8h (ng eq. × h/mL) | % in plasmaa | % of dosea | % in urinea | % of dosea | % in bilea | |
| 6 min | 136 | 8.6 | N.D. | N.D. | 2.38 | 8.5 |
| 9 min | N.D. | N.D. | 0.79 | 8.3 | 0.04 | 0.1 |
| R10 | 1 | 0.1 | 2.01 | 21.2 | 0.66 | 2.3 |
| R1 | N.D. | N.D. | N.D. | N.D. | 1.66 | 5.9 |
| Mo2 | N.D. | N.D. | 1.19 | 12.5 | 8.60 | 30.6 |
| R3 | N.D. | N.D. | 0.75 | 7.9 | 5.01 | 17.8 |
| R5 | 54 | 3.4 | N.D. | N.D. | 5.42 | 19.3 |
| R6 | N.D. | N.D. | N.D. | N.D. | 1.56 | 5.6 |
| R9 | N.D. | N.D. | N.D. | N.D. | 0.05 | 0.2 |
| Amenamevir | 1390 | 87.9 | 4.75 | 50.0 | 2.71 | 9.6 |
N.D. Not detected, AUC0–8 area under the concentration-time curve from 0 to 8 h
aSee Sect. 2.14
Excretion in Mice
The cumulative urinary, biliary, and fecal excretion after a single oral dose of 14C-amenamevir (3 mg/kg) to mice is summarized in Table 5. Within 96 h after dosing, 100.63% of the dose (10.06% in urine and 90.46% in feces) was recovered, suggesting that all radioactivity was excreted from the body and that fecal excretion was predominant. CLr was calculated to be 0.136 L/h/kg. Within 48 h after dosing, bile duct-cannulated mice excreted 11.79% and 32.04% of the dose into urine and bile, respectively. This suggests that 14C-amenamevir is well absorbed in mice and that fecal excretion via bile is important for the excretion of 14C-amenamevir-derived radioactivity.
Table 5.
Excretion of radioactivity after single oral administration of 14C-amenamevir to mice at 3 mg/kg
| Treatment | Time (h) | Excretion of radioactivity (% of dose) | |||
|---|---|---|---|---|---|
| Urine | Bile | Feces | Total | ||
| Bile duct- cannulated | 0–6 | 5.43 ± 2.50a | 21.14 ± 10.19 | NS | 25.21 ± 13.40 |
| 0–24 | 10.32 ± 2.28 | 31.50 ± 10.48 | NS | 41.82 ± 12.73 | |
| 0–48 | 11.79 ± 1.43 | 32.04 ± 9.99 | NS | 43.83 ± 11.13 | |
| Intact | 0–6 | 5.31 ± 4.08 | NS | NS | NC |
| 0–24 | 8.87 ± 5.34 | NS | 88.26 ± 5.65 | 97.12 ± 4.40 | |
| 0–48 | 9.64 ± 5.38 | NS | 90.07 ± 5.50 | 99.72 ± 3.74 | |
| 0–72 | 9.94 ± 5.48 | NS | 90.38 ± 5.34 | 100.32 ± 3.40 | |
| 0–96 | 10.06 ± 5.47 | NS | 90.46 ± 5.32 | 100.63 ± 3.17b | |
Each value represents the mean ± SD of four animals
NC not calculated, NS not sampled, SD standard deviation
aMean ± SD of three animals as one animal did not void urine
bIncludes radioactivity in cage wash solvent. (0.11 ± 0.12%)
To assess enterohepatic recirculation, bile was collected from mice that had been dosed orally with 14C-amenamevir and injected directly into the duodenum of bile duct-cannulated (recipient) mice. Within 48 h after dosing, the recovery of radioactivity in bile and urine was 17.87% and 4.22%, respectively, indicating the presence of enterohepatic circulation.
Allometric Scaling
CLtotal and CLr in humans were predicted to be 10.6 L/h and 1.24 L/h. With the assumption that CLh = CLtotal − CLr, human CLh and Fh were calculated to be 9.32 L/h and 0.893, respectively. With the assumption that Fa is the same between mice and humans, CLoral in humans was calculated to be 29.5 L/h.
Previously, our research group reported that CLoral and CLr in humans were 22.37 L/h and 2.22 L/h, respectively, after repeated oral administration of amenamevir to humans at a dose of 300 mg/person for a week [11]. Allometric scaling accurately predicted the clearances in humans. CLr accounted for a small portion of clearance in both mice and humans.
Discussion
In the present study, the absorption, tissue distribution, and main elimination pathways of amenamevir were investigated in rodents. Oral administration of unlabeled amenamevir to mice resulted in a moderate F of approximately 40%. Plasma concentrations of unchanged drug increased almost dose-proportionally in mice at oral doses of 1, 3, and 10 mg/kg, which are close to the 50% effective oral dose of amenamevir in mice (i.e. 1.9 mg/kg twice daily [6]), suggesting the F might be constant around the therapeutic dose range. The underlying mechanism of this moderate F could be explained by using the data obtained in this study as follows. F is in proportion to Fa × Fh. Fa was 40.1% based on radioactivity AUC in plasma (Table 2), which was supported by the total radioactivity recovery of 43.8% in our biliary and urinary excretion experiment. As CLtotal of amenamevir was 1.16 L/h/kg, which is much lower than the hepatic blood flow in mice (5.88 L/h/kg [23]), this suggests that the Fh value must be high. Therefore, the observed F of amenamevir (approximately 40%) is due to a moderate Fa (approximately 40%) and high Fh value. We expect that this moderate Fa in fasted mice can be improved by optimization of formulation for two reasons. First, a clinical study showed slight (14%) alternation in AUC after formulation change [24]. Second, an in vitro membrane permeability test revealed high membrane permeability of amenamevir (data on file), suggesting that an improvement in solubility in the gastrointestinal tract might lead to an improvement in Fa.
The F of acyclovir in rats and humans are 19% [1] and 15–20% [2], respectively. The main elimination pathway of acyclovir in these species is urinary excretion as unchanged drug [2, 12], suggesting a high Fh value. Therefore, the low F of acyclovir may be associated with a low Fa and high Fh value. Valaciclovir was developed to improve the low F of acyclovir [1]. The F of valaciclovir in rats and humans are 63% [1] and 54% [3] as acyclovir, respectively. The main elimination pathway of valaciclovir in these species is hydrolysis to acyclovir, followed by urinary excretion [3, 25]. Further metabolism of acyclovir after oral administration of valaciclovir is very minor [3, 25], suggesting a high Fh value. Therefore, the moderate F of valaciclovir may be associated with a moderate Fa and high Fh value. Comparison of amenamevir with acyclovir and valaciclovir indicates that the Fa value is the differentiating factor for the F of these drugs in rodents and humans.
The in situ absorption experiment demonstrated that amenamevir is more efficiently absorbed from the small intestine than the stomach and large intestine in mice. To our knowledge, the absorption site of acyclovir has not been reported. Similar to amenamevir, valaciclovir also showed more efficient absorption in the small intestine than the colon in mice [26].
Drug-derived radioactivity was rapidly distributed in organs and tissues after oral administration of 14C-amenamevir (Table 3). The high distribution of radioactivity into the liver and kidneys favors biliary and renal excretion, which are excretion pathways for 14C-amenamevir-derived components. The tissue-to-plasma concentration ratios of 14C-amenamevir-derived radioactivity in the skin and brain were similar to those for acyclovir [12] and valaciclovir [25].
Concentrations of drug-derived radioactivity in most tissues decreased at a similar rate to the blood concentration (Table 3). There was no evidence of prolonged retention of radioactivity in any tissue. This complete elimination of radioactivity from tissues is in line with findings from our excretion study, where almost 100% recovery of the administered dose was observed by 96 h post-dose (Table 5).
Metabolic pathways of amenamevir were postulated based on the findings in the metabolite identification study. Of the metabolites shown in Fig. 5, R1, R3, R5, R6, R9, R10, and Mo2 were detected in mice. Most were oxidative metabolites, suggesting that oxidation plays an important role in the metabolism of amenamevir. Glucuronidation appears to play a role in subsequent metabolism of hydroxyl metabolites (glucuronidation of R5 and R6 produced R3 and Mo2, respectively).
Direct oxidation of amenamevir produced R5 (hydroxylation of a methyl group), R6 (hydroxylation of a 1,2,3-trisubstituted benzene ring), and R10 (breakdown of an oxadiazole ring). R5 and its subsequent metabolites (R1, R3, and R9) comprised 0.75% and 12.14% of the dose in urinary and biliary excretion, respectively. R6 and its subsequent metabolite Mo2 accounted for 1.19% (urine) and 10.16% (bile) of the dose. R10 accounted for 2.01% (urine) and 0.66% (bile) of the dose. Therefore, production of R5 and R6 is more important than that of R10 in the overall metabolism of amenamevir.
Amenamevir accounted for 87.9% of radioactivity in plasma (Table 4), suggesting that a large proportion of the pharmacologically active component is delivered to tissues and organs to exert its pharmacological actions.
After oral administration of 14C-amenamevir to intact or bile duct-cannulated mice, fecal or biliary excretion was greater than urinary excretion (Table 5), suggesting that amenamevir-derived radioactivity is excreted into bile followed by feces. Taking the main metabolic pathway and metabolite disposition into consideration, the predominant elimination pathway of amenamevir after oral absorption in mice is as follows: amenamevir is oxidatively metabolized to R5 or R6. Then R5, R6, and their subsequent metabolites are excreted into bile followed by feces.
Metabolites in rodents were similar to those in humans (manuscript in preparation). In addition, metabolism is the main elimination pathway in both rodents and humans (manuscript in preparation). Allometric scaling results also revealed the similarity in pharmacokinetics between rodents and humans. While elimination by metabolism is often associated with plasma concentration changes in patients with impaired hepatic function, such changes were not observed with amenamevir [27]. Metabolism-mediated drug–drug interaction of amenamevir was reported by our group [28] and is described in the package insert in Japan [29] to facilitate proper use.
Unlike the elimination profile of amenamevir, the predominant elimination pathway of acyclovir is renal excretion as unchanged drug, while that of valaciclovir is metabolism to acyclovir followed by renal excretion. It is important to point out that the very different chemical structures of amenamevir and acyclovir/valaciclovir lead to, not only differences in elimination pathways, but also elimination rates. The t1/2 of amenamevir in plasma was 2 h in rodents whereas that of acyclovir is 1 h [12]. In humans, the t1/2 of amenamevir is 8 h [11] whereas that of acyclovir is approximately 2.5–3.3 h [2]. This explains why a once daily dose was enough for amenamevir to exhibit excellent efficacy in humans [10], whereas 2–5 doses per day are necessary for acyclovir and valaciclovir [2, 4].
Conclusions
The oral bioavailability of amenamevir in mice was 40%, which may be attributed to a moderate absorption ratio and high hepatic availability. Amenamevir was primarily absorbed from the small intestine and 14C-amenamevir-derived component(s) were distributed to target organs (skin and brain). In the main elimination pathways in mice, amenamevir is oxidatively metabolized to R5 by hydroxylation of a methyl group or R6 by hydroxylation of a 1,2,3-trisubstituted benzene ring. R5, R6, and their subsequent metabolites are then excreted into bile followed by feces.
The mechanism underlying the improved pharmacokinetic profile of amenamevir (i.e. no need for a prodrug to enhance bioavailability or frequent administration due to a longer elimination half-life) is associated with an improved absorption ratio (not hepatic availability) compared to acyclovir, and qualitative differences in elimination (slow metabolism of amenamevir vs rapid urinary excretion of acyclovir/valaciclovir).
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank K. Kato and K. Suzumura of Astellas for their helpful discussions and T. Aoki, T. Yokoi, and M. Funatsu of Astellas and T. Shibahara of Nemoto Science for their scientific and technical support. We gratefully acknowledge K. Tabata of Astellas for his supervision.
Funding
This research was funded by Astellas.
Conflict of Interest
Y. Ohtsu and Y. Susaki are employees of Astellas. K. Noguchi was an employee of Astellas during the planning, experiment, and data interpretation of this research. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in, or financial conflict with, the subject matter or materials discussed in the manuscript other than those disclosed.
Ethical Approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. All procedures performed in studies involving animals were approved by animal care and use committees where the studies were conducted. This article does not contain any studies with human participants performed by any of the authors.
References
- 1.Beauchamp LM, Orr GF, de Miranda P, Bumette T, Krenitsky TA. Amino acid ester prodrugs of acyclovir. Antivir Chem Chemother. 1992;3(3):157–164. doi: 10.1177/095632029200300305. [DOI] [Google Scholar]
- 2.GlaxoSmithKline. Product monograph for acyclovir oral suspension (zovirax). 2016. https://ca.gsk.com/media/592844/zovirax.pdf. Accessed 17 Apr 2018.
- 3.Soul-Lawton J, Seaber E, On N, Wootton R, Rolan P, Posner J. Absolute bioavailability and metabolic disposition of valaciclovir, the L-valyl ester of acyclovir, following oral administration to humans. Antimicrob Agents Chemother. 1995;39(12):2759–2764. doi: 10.1128/AAC.39.12.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.GlaxoSmithKline. Product monograph for valacyclovir caplets (valtrex). 2015. https://ca.gsk.com/media/593038/valtrex.pdf. Accessed 17 Apr 2018.
- 5.James S, Larson K, Acosta E, Prichard M. Helicase-primase as a target of new therapies for herpes simplex virus infections. Clin Pharmacol Ther. 2015;97(1):66–78. doi: 10.1002/cpt.3. [DOI] [PubMed] [Google Scholar]
- 6.Chono K, Katsumata K, Kontani T, Kobayashi M, Sudo K, Yokota T, et al. ASP2151, a novel helicase-primase inhibitor, possesses antiviral activity against varicella-zoster virus and herpes simplex virus types 1 and 2. J Antimicrob Chemother. 2010;65(8):1733–1741. doi: 10.1093/jac/dkq198. [DOI] [PubMed] [Google Scholar]
- 7.Katsumata K, Chono K, Kato K, Ohtsu Y, Takakura S, Kontani T, et al. Pharmacokinetics and pharmacodynamics of ASP2151, a helicase-primase inhibitor, in a murine model of herpes simplex virus infection. Antimicrob Agents Chemother. 2013;57(3):1339–1346. doi: 10.1128/AAC.01803-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takada A, Katashima M, Kaibara A, Sawamoto T, Zhang W, Keirns J. Statistical analysis of amenamevir (ASP2151) between pharmacokinetics and clinical efficacies with non-linear effect model for the treatment of genital herpes. Clin Pharmacol Drug Dev. 2014;3(5):365–370. doi: 10.1002/cpdd.108. [DOI] [PubMed] [Google Scholar]
- 9.Takada A, Katashima M, Kaibara A, Chono K, Katsumata K, Sawamoto T, et al. Integrative pharmacokinetic-pharmacodynamic modeling and simulation of amenamevir (ASP2151) for treatment of recurrent genital herpes. Drug Metab Pharmacokinet. 2016;31(4):323–332. doi: 10.1016/j.dmpk.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Kawashima M, Nemoto O, Honda M, Watanabe D, Nakayama J, Imafuku S, et al. Amenamevir, a novel helicase-primase inhibitor, for treatment of herpes zoster: a randomized, double-blind, valaciclovir-controlled phase 3 study. J Dermatol. 2017;44(11):1219–1227. doi: 10.1111/1346-8138.13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohtsu Y, van Trigt R, Takama K, Groenendaal D, Takada A, Nakamura T, et al. Quantification of ASP2151 in human plasma and urine: a pitfall associated with supersaturation of analyte in urine. Chromatographia. 2017;80(2):217–227. doi: 10.1007/s10337-016-3236-4. [DOI] [Google Scholar]
- 12.de Miranda P, Krasny HC, Page DA, Elion GB. The disposition of acyclovir in different species. J Pharmacol Exp Ther. 1981;219(2):309–315. [PubMed] [Google Scholar]
- 13.Krasny HC, de Miranda P, Blum MR, Elion GB. Pharmacokinetics and bioavailability of acyclovir in the dog. J Pharmacol Exp Ther. 1981;216(2):281–288. [PubMed] [Google Scholar]
- 14.Good SS, de Miranda P. Metabolic disposition of acyclovir in the guinea pig, rabbit, and monkey. Am J Med. 1982;73(1A):91–95. doi: 10.1016/0002-9343(82)90071-7. [DOI] [PubMed] [Google Scholar]
- 15.Ohtsu Y, Otsuka S, Nakamura T, Noguchi K. Regulated bioanalysis of conformers - A case study with ASP2151 in dog plasma and urine. J Chromatogr B. 2015;997:56–63. doi: 10.1016/j.jchromb.2015.05.028. [DOI] [PubMed] [Google Scholar]
- 16.Krone V. Absorption: in vivo tests (radiolabeled) In: Vogel HG, Maas J, Hock FJ, Mayer D, editors. Drug discovery and evaluation: safety and pharmacokinetic assays. Berlin: Springer; 2013. pp. 799–834. [Google Scholar]
- 17.FDA. Draft redbook II toxicological principles for the safety assessment of direct food additives and color additives used in food, chapter V.B. metabolism and pharmacokinetic studies. 1993. https://www.fda.gov/Food/GuidanceRegulation/GuidanceDocumentsRegulatoryInformation/IngredientsAdditivesGRASPackaging/ucm078717.htm. Accessed 17 Apr 2018.
- 18.Schoenwald RD. Basic principles. In: Schoenwald RD, editor. Pharmacokinetics in drug discovery and development. London: CRC Press; 2002. pp. 3–30. [Google Scholar]
- 19.Ling J, Johnson KA, Miao Z, Rakhit A, Pantze MP, Hamilton M, et al. Metabolism and excretion of erlotinib, a small molecule inhibitor of epidermal growth factor receptor tyrosine kinase, in healthy male volunteers. Drug Metab Dispos. 2006;34(3):420–426. doi: 10.1124/dmd.105.007765. [DOI] [PubMed] [Google Scholar]
- 20.Abe Y, Ota E, Endo T, Murakami M, Kobayashi M. Absorption, disposition, metabolism, and excretion of ritobegron (KUC-7483), a novel selective beta3-adrenoceptor agonist, in rats. Pharmazie. 2014;69(12):881–888. [PubMed] [Google Scholar]
- 21.Takeuchi K, Shibata M, Kamiyama E, Umehara K. Expression levels of multidrug resistance-associated protein 4 (MRP4) in human leukemia and lymphoma cell lines, and the inhibitory effects of the MRP-specific inhibitor MK-571 on methotrexate distribution in rats. Exp Ther Med. 2012;4(3):524–532. doi: 10.3892/etm.2012.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10(7):1093–1095. doi: 10.1023/A:1018943613122. [DOI] [PubMed] [Google Scholar]
- 23.Sarin SK, Sabba C, Groszmann RJ. Splanchnic and systemic hemodynamics in mice using a radioactive microsphere technique. Am J Physiol. 1990;258(3):G365–G369. doi: 10.1152/ajpgi.1990.258.3.G365. [DOI] [PubMed] [Google Scholar]
- 24.Kusawake T, Keirns JJ, Kowalski D, den Adel M, Groenendaal-van de Meent D, Takada A, et al. Pharmacokinetics and safety of amenamevir in healthy subjects: analysis of four randomized phase 1 studies. Adv Ther. 2017;34(12):2625–2637. doi: 10.1007/s12325-017-0642-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burnette TC, de Miranda P. Metabolic disposition of the acyclovir prodrug valaciclovir in the rat. Drug Metab Dispos. 1994;22(1):60–64. [PubMed] [Google Scholar]
- 26.Yang B, Smith DE. Significance of peptide transporter 1 in the intestinal permeability of valacyclovir in wild-type and PepT1 knockout mice. Drug Metab Dispos. 2013;41(3):608–614. doi: 10.1124/dmd.112.049239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kusawake T, Kowalski D, Takada A, Kato K, Katashima M, Keirns JJ, et al. The influence of hepatic and renal impairment on the pharmacokinetics of a treatment for herpes zoster, amenamevir (ASP2151): phase 1, open-label, single-dose, parallel-group studies. Adv Ther. 2017;34(12):2612–2624. doi: 10.1007/s12325-017-0643-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kusawake T, den Adel M, Groenendaal-van de Meent D, Garcia-Hernandez A, Takada A, Kato K, et al. Pharmacokinetic evaluation of the interactions of amenamevir (ASP2151) with ketoconazole, rifampicin, midazolam, and warfarin in healthy adults. Adv Ther. 2017;34(11):2466–2480. doi: 10.1007/s12325-017-0634-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maruho. Amenamevir package insert (Japanese). 2017. http://www.info.pmda.go.jp/downfiles/ph/PDF/730155_6250046F1028_1_02.pdf. Accessed 17 Apr 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.