
Figure 9. Representative geometries for 1,2,3,4-tetraphenylbenzene (top row) and the bridgehead problem case of Figure 2e (bottom row).
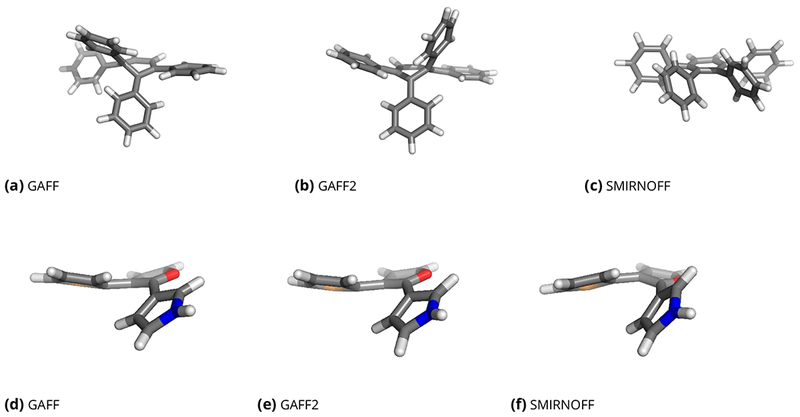
Here, we show representative geometries from short gas-phase simulations with each force field for each molecule. Depending on what force field is applied, the typical geometry varies considerably. In the case of 1,2,3,4-tetraphenylbenzene, GAFF and GAFF2 (a-b) lead to incorrect buckling of the central aromatic ring, whereas the prototype force field introduced here, SMIRNOFF99Frosst, keeps the aromatic ring remains planar, as expected (c). For the molecule in the bottom row, GAFF and GAFF2 (d, e) make the bond connecting the five-membered aromatic rings be non-rotatable, resulting in a slight buckling of the aromatic rings. In SMIRNOFF99Frosst (f), the connecting bonds are rotatable so no buckling occurs.