

Invasive group A streptococcus (GAS) infections include necrotizing soft tissue infections (NSTI) and streptococcal toxic shock syndrome (STSS). We have previously shown that host HLA class II allelic variations determine the risk for necrotizing fasciitis (NF), a dominant subgroup of NSTI, and STSS by modulating responses to GAS superantigens (SAgs).
KEYWORDS: FoxP3/GARP/LAP, group A streptococcus, HLA-II, T regulatory cells
ABSTRACT
Invasive group A streptococcus (GAS) infections include necrotizing soft tissue infections (NSTI) and streptococcal toxic shock syndrome (STSS). We have previously shown that host HLA class II allelic variations determine the risk for necrotizing fasciitis (NF), a dominant subgroup of NSTI, and STSS by modulating responses to GAS superantigens (SAgs). SAgs are pivotal mediators of uncontrolled T-cell activation, triggering a proinflammatory cytokine storm in the host. FoxP3-expressing CD4+ CD25+ T regulatory cells (Tregs) comprise phenotypically and functionally heterogeneous subsets with a profound ability to suppress inflammatory responses. Specifically, activated Tregs, which express glycoprotein A repetitions predominant (GARP) and display latent transforming growth factor β1 (TGF-β1) complexes (latency-associated peptide [LAP]), exhibit strong immunosuppressive functions. The significance of Tregs that may participate in suppressing inflammatory responses during NSTI is unknown. Here, we phenotypically characterized FoxP3/GARP/LAP-expressing Tregs in GAS-infected or SAg (SmeZ)-stimulated splenocytes from transgenic (tg) mice expressing human HLA-II DRB1*15 (DR15 allele associated with nonsevere NF/STSS-protective responses) or DRB1*0402/DQB1*0302 (DR4/DQ8 alleles associated with neutral risk for combined NF/STSS). We demonstrated both in vivo and in vitro that the neutral-risk allele upregulates expression of CD4+ CD25+ activated effector T cells, with a significantly lower frequency of Foxp3+/GARP+ LAP− but higher frequency of Foxp3− LAP+ Tregs than seen with the protective allele. Additional in vitro studies revealed that the presentation of SmeZ by the neutral-risk allele significantly increases proliferation and expression of effector cytokines gamma interferon (IFN-γ) and interleukin-2 (IL-2) and upregulates CD4+ CD25+ T cell receptors (TCRs) carrying specific Vβ 11 chain (TCRVβ11+) T cells and Th1 transcription factor Tbx21 mRNA levels. Our data suggest that neutral-risk alleles may drive Th1 differentiation while attenuating the induction of Tregs associated with suppressive function.
INTRODUCTION
Streptococcus pyogenes, or group A streptococcus (GAS), is an important human pathogen responsible for a wide spectrum of infections ranging from simple, noninvasive pharyngitis to invasive, life-threatening infections, including necrotizing soft tissue infections (NSTIs) and streptococcal toxic shock syndrome (STSS). Among these, the combination of necrotizing fasciitis (NF; a dominant subgroup of NSTI) plus STSS represents the most severe form of invasive GAS infections (1–6). GAS produces potent immunotoxins, referred to as superantigens (SAgs), that cross-link host HLA class II molecules expressed on antigen-presenting cells (APCs) and T cell receptors (TCRs) carrying specific Vβ chains (TCRVβ) expressed on T cells. The binding of SAg to TCR is Vβ specific and results in the polyclonal activation of T cells and in the proliferation and release of proinflammatory mediators that lead to increased morbidity and mortality (7–9). Previously, we reported that polymorphisms of HLA-II alleles strongly influence the outcomes and severity of NF/STSS (10). The biological relevance of HLA-II allelic variations against STSS was further confirmed in vivo by using transgenic (tg) mice carrying human HLA-II alleles associated with either protection (DQB1*06 [DQ6]) or neutral risk (DRB1*04/DQB1*0302 [DR4/DQ8]) and in vitro by evaluating responses to GAS SAg (11, 12).
T regulatory cells (Tregs), a subset of CD4+ T cells that constitutively express CD25 and the transcription factor FoxP3, are critical for the suppression of immune responses to a variety of microbial antigens. They limit inflammatory responses by employing various mechanisms (13, 14). While CD25 is considered a putative marker for the identification of FoxP3+ Tregs, this receptor is also highly expressed on activated CD4+ T cells, thus making it difficult to adequately determine whether activated CD4+ CD25+ cells expressing Foxp3 are functionally suppressive. However, studies have shown that the generation of CD4+ CD25+ Foxp3+ Tregs induced by exposure to SAg contributes to immunosuppression mediated either by cell contact (15) or by secretion of suppressor cytokines such as interleukin-10 (IL-10) and transforming growth factor β1 (TGF-β1) (16, 17). TGF-β, the critical cytokine associated with the conversion of naive T cells into FoxP3-expressing cells, has a suppressor function in vitro and a protective function in vivo (18–20). TGF-β is synthesized as pro-TGF-β, which is then processed by furin proprotein convertase to form a latent complex noncovalently associated with the propeptide latency-associated peptide (LAP) (20). LAP is expressed on the surface of activated Tregs, where it is anchored to the membrane through glycoprotein A repetitions predominant (GARP/LRRC32) and confers a suppressive phenotype for FoxP3-expressing Tregs (21–23).
It is not clear whether variations in HLA-II alleles that present SAgs to T cells play a role in the induction of Tregs during GAS-mediated NSTI. Tregs comprise heterogeneous subsets with distinct phenotypic and functional characteristics, so we postulated that identification of these diverse Treg subsets would be critical to understanding the mechanisms underlying NSTI outcomes and severity. In the present study, we phenotypically characterized GARP-, LAP-, and FoxP3-expressing Treg subsets in vivo after subcutaneous GAS infections as well as in SAg SmeZ-stimulated splenocytes in vitro in transgenic mice carrying human HLA-II alleles associated with either protection or neutral risk for combined NF/STSS. Using in vivo and in vitro approaches, we demonstrated that, compared to the protective allele, there is a significant attenuation of FoxP3- and GARP-expressing Tregs in vivo and that this attenuation was SmeZ concentration dependent in vitro in the neutral-risk allele. Further, our in vitro studies showed that presentation of SmeZ by the neutral-risk allele is associated with a significant increase in T cell proliferative responses, expression of effector cytokines gamma interferon (IFN-γ) and IL-2, and upregulation of CD4+ CD25+ TCRVβ11+ T cells and mRNA expression of the Th1 transcription factor Tbx21. Taken together, our data suggest that HLA-II alleles associated with neutral risk potentially drive effector T cell responses and Th1 differentiation while attenuating the induction of Tregs associated with suppressive function in GAS-mediated NSTI.
RESULTS
An HLA-II tg mouse model of NSTI for Treg analysis.
In order to understand the mechanisms and disease signatures underlying NSTI, we used a forward-genetics approach in an advanced recombinant inbred BxD mouse model specifically optimized for NSTI (24). To elucidate the role of HLA-II alleles and their influence on T-effector and Treg polarization during NSTI, we used a subcutaneous skin infection model of GAS-mediated NSTI in HLA-II transgenic mice that mimic lesions and inflammatory responses observed in humans. We initially planned to use HLA-II tg mice expressing HLA-II DRB1*15/DQB1*06 (DR15/DQ6) alleles associated with protection for combined NF/STSS and to compare their responses to those of mice expressing DR4/DQ8 alleles associated with neutral risk for combined NF/STSS, as identified in our epidemiological study (10). However, the mice expressing DR15/DQ6 alleles were not readily available. Since our previous studies showed that the proliferative responses of splenocytes from mice expressing either DR15 or DQ6 alleles were similar (11), the present study was conducted using transgenic mice expressing the DR15 allele, and the responses were compared with those seen with mice carrying DR4/DQ8 alleles. All mice were subcutaneously infected with 1 × 108 to 10 × 108 CFU of GAS 2006, a clinical isolate from an NSTI patient. At 72 h postinfection, GAS was not detectable in the blood of DR15 or DR4/DQ8 mice. However, GAS persisted in the skin at the site of infection and showed systemic dissemination into the spleen. Our data suggest a trend toward a difference between DR15 and DR4/DQ8 mice in bacterial burden, but the results did not reach statistical significance in either the skin (P = 0.11) or the spleen (P = 0.88) (Fig. 1). Subcutaneous infections in control C57BL/6J (B6) and DBA2/J (D2) mice that did not express human HLA-II alleles showed similar levels of bacterial burden in the skin and spleen (Fig. 1). Spleens from GAS-infected or uninfected control mice were collected for analysis of Tregs by flow cytometry.
FIG 1.
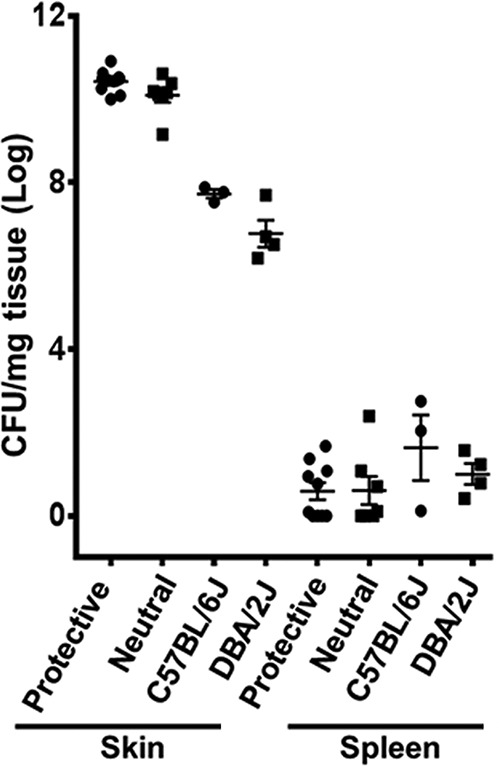
No significant differences in bacterial loads and dissemination were observed between HLA-II DR15 and DR4/DQ8 mice. A total of 7 to 10 mice expressing HLA-II DR15 (protective) or DR4/DQ8 (neutral risk) were infected subcutaneously with 1 × 108 to 10 × 108 CFU/mouse of M1T1 GAS isolate 2006. C57BL/6J (B6) and DBA/2J (D2) mice that were not transgenic for HLA-II were used as controls. At 72 h postinfection, skin and spleen were recovered for enumeration of bacterial load. Each mouse is represented by a symbol. Data presented represent log10-transformed bacterial loads, and error bars represent the SEM.
The frequency of activated CD4+ CD25+ cells was increased in vivo during GAS infection and in vitro in response to SAg stimulation.
The function of IL-2α receptor CD25 is critical for T-cell proliferation, and its surface expression is upregulated in activated T cells (25). We examined the effects of GAS dissemination into the spleen and in vitro SmeZ stimulation of splenocytes on the expression of CD25 in CD4+ T cells. We used SmeZ because it is the most potent SAg produced by GAS and because SmeZ binds to the beta chain of the HLA-DR allele (26). As shown in Fig. 2A, in vivo, there was an increased frequency of activated CD4+ CD25+ T cells in splenocytes isolated from DR4/DQ8 mice compared with DR15 mice, and yet the difference was not statistically significant (P = 0.11). However, compared with uninfected controls, the induction of CD4+ CD25+ T cells was significant in DR4/DQ8 mice (P ≤ 0.05) but not in DR15 mice (Fig. 2A). Significantly higher frequencies of CD25+ T cells were observed in vitro when SmeZ was presented by DR4/DQ8 than when it was presented by DR15. This was evident at lower concentrations (1 and 0.1 ng/ml) but not at the highest concentration (10 ng/ml) (Fig. 2B). Because CD25 upregulation is a key event in the regulation of T-cell proliferative responses, we measured in vitro proliferative responses to different concentrations of SmeZ. As shown in Fig. 2C, SmeZ induced significantly higher T-cell proliferative responses when it was presented by DR4/DQ8 (P ≤ 0.0005, P ≤ 0.005, and P ≤ 0.05 when the SmeZ concentrations were 10, 1, and 0.1 ng/ml, respectively). Similarly to SmeZ, the partially purified native mixture of SAgs induced significantly higher proliferative responses (P ≤ 0.05) in splenocytes expressing DR4/DQ8 alleles than in those expressing the protective DR15 allele (Fig. 2D). These differences were specific to the SmeZ/native mix of SAg because proliferative responses to the polyclonal mitogen concanavalin A (ConA) did not differ between splenocytes that expressed DR4/DQ8 and those that expressed the protective DR15 allele (Fig. 2E).
FIG 2.

The increased frequency of activated CD4+ CD25+ cells during GAS infections in vivo and in response to SmeZ stimulations in vitro mirrored SAg-specific T cell proliferation responses. (A and B) GAS infections or SmeZ stimulations induced expansion of CD4+ CD25+ cells in vivo (A) and in vitro (B). Splenocytes were isolated from mice that either were left uninfected (Uninf) (n = 2, each from mice expressing the protective or neutral-risk [Neutral] allele) or were infected (n = 7 to 10) subcutaneously with 1 × 108 to 10 × 108 CFU/mouse of M1T1 GAS isolate 2006 for 72 h. For in vitro stimulations, splenocytes (4 × 106 to 5 × 106/well) from HLA-II tg mice expressing the protective or neutral-risk allele that were left unstimulated (control) or stimulated with 10, 1, or 0.1 ng/ml of SmeZ (Z10, Z1, Z 0.1, respectively) were cultured for 72 h at 37°C in an atmosphere of 5% CO2. Splenocytes were stained with anti-CD3, anti-CD4, and anti-CD25 and analyzed by flow cytometry. The bar graph shows the mean levels of expression of CD25 within viable CD4+ T cells. Error bars represent the SEM. (C to E) HLA-II allelic variations affect in vitro proliferative responses to SmeZ (C) and a native mix of SAg (D). Splenocytes (2 × 106/well) from HLA-II tg mice expressing the protective or neutral-risk allele were left unstimulated or had been stimulated with recombinant SmeZ at a concentration of 10 ng/ml (Z10), 1 ng/ml (Z1), or 0.1 ng/ml (Z0.1) (C) or either a mixture of native M1T1 SAgs (dilution ratio, 1:100) (D) or ConA (1 μg/ml) (E). Cells were cultured for 72 h at 37°C in an atmosphere of 5% CO2. Proliferation was measured by analysis of [3H]thymidine incorporation 72 h after stimulation, and the levels of incorporation are represented as bar graphs (the corresponding y axis is on the left). In each panel, the mean values (representing counts per minute) are presented as white bars (protective) or black bars (neutral risk), and error bars represent the SEM. Experiments were repeated at least three times. *, P ≤ 0.05; **, P ≤ 0.005; ***, P ≤ 0.0005.
HLA-II allelic variations play a dominant role in modulating FoxP3 expression in vivo and in vitro.
The concentration of antigen or mitogenic stimulus is critical in regulating the induction of FoxP3 expression in Tregs both in vitro and in vivo (27–29). Our in vivo data showed that while the frequency of CD4+ CD25+ T cells was unchanged, Foxp3 expression within the CD4+ CD25+ T cells was significantly lower in the neutral-risk DR4/DQ8 alleles than in the protective DR15 allele (P ≤ 0.05, Fig. 3A). In vitro, we observed that when 10 ng/ml of SmeZ was presented by either the DR15 or DR4/DQ8 alleles, induction of FoxP3 expression in activated CD4+ CD25+ T cells was diminished compared with the levels of spontaneous expression seen in unstimulated cells (Fig. 3B). Presentation of SmeZ (10 ng/ml) by the protective DR15 allele induced more FoxP3 expression than presentation of SmeZ by DR4/DQ8. However, the differences were not significant. Strikingly, FoxP3 expression was most efficiently induced in CD4+ CD25+ T cells upon stimulation with lower concentrations of SmeZ (1 and 0.1 ng/ml) when presented by the protective DR15 allele. In contrast, FoxP3 induction was significantly diminished in response to all three concentrations of SmeZ when presented by the neutral-risk DR4/DQ8 alleles (P ≤ 0.05, Fig. 3B). Together, our data strongly indicate that the protective DR15 allele promoted significantly greater FoxP3 induction than the neutral-risk DR4/DQ8 alleles in vivo and in a concentration-dependent manner in vitro.
FIG 3.

Variations in HLA-II alleles played a dominant role in regulating the expression of FoxP3+ cells within populations of activated CD4+ CD25+ cells in vivo and in vitro. (A) Splenocytes were isolated from mice that were either left uninfected (n = 2, each from mice expressing protective or neutral-risk allele) or infected subcutaneously (n = 7 to 10) with 1 × 108 to 10 × 108 CFU/mouse of M1T1 GAS isolate 2006 for 72 h. (B) For in vitro stimulations, splenocytes (4 × 106 to 5 × 106/well) from HLA-II tg mice expressing the protective or neutral-risk allele were either left unstimulated (control) or stimulated with 10, 1, or 0.1 ng/ml of SmeZ and cultured for 72 h at 37°C in an atmosphere of 5% CO2. Cells were stained with antibodies to surface CD3, CD4, and CD25, as well as intracellular FoxP3, and levels were measured by flow cytometry gated on viable CD3+/CD4+ and CD25+/FoxP3 cells. Each mouse is represented by a symbol. Error bars represent the SEM. In vitro data are shown as bar graphs and indicate the mean frequencies of FoxP3-expressing cells within activated CD4+ CD25+ cells. Error bars represent the SEM from the mean results of at least three biological replicates. *, P ≤ 0.05.
Surface GARP expression indicative of the presence of Tregs with suppressive function was significantly downregulated in vivo and in vitro in the DR4/DQ8 allele.
Since the mere induction of Foxp3 expression does not necessarily confer suppressive function, we expanded our analysis of FoxP3+ Tregs to include measurements of surface GARP and LAP expression. Probst-Kepper et al. identified GARP on the basis of gene expression analysis of Tregs and T-helper cells following TCR stimulation and reported that GARP expression was specifically induced in CD4+ CD25+ FoxP3+ Tregs. Thus, GARP was considered a Treg-specific activation marker and functionally suppressive (30). Our results show that the changes in the frequencies of FoxP3-expressing CD4+ CD25+ T cells were accompanied by similar changes in GARP expression. In vivo, there was a significant reduction in the GARP+ LAP− population (P ≤ 0.05, Fig. 4A) in DR4/DQ8 alleles compared with the results seen with the DR15 allele, which mirrored the FoxP3 expression profile (Fig. 3A). In vitro, 10 ng/ml of SmeZ downregulated GARP expression when presented by either DR15 or DR4/DQ8 alleles to levels similar to those seen with FoxP3 expression. Interestingly, GARP expression was restored in response to a lower concentration of SmeZ (1 ng/ml) with the DR15 allele (P ≤ 0.005) but not with DR4/DQ8 (Fig. 4B). Representative flow cytometry data are shown as contour plots in Fig. 4C (in vivo) and Fig. 4D (in vitro).
FIG 4.

The protective allele resulted in significant induction of GARP+ LAP− Tregs both in vivo and in vitro. (A) A total of 5 to 7 mice expressing either the HLA-II protective or neutral-risk allele were infected subcutaneously with 1 × 108 to 10 × 108 CFU/mouse of M1T1 GAS isolate 2006 for 72 h. Splenocytes were isolated and stained with antibodies to surface CD3, CD4, CD25, GARP, and LAP, and levels were measured by flow cytometry. Each mouse is represented by a symbol. Error bars represent the SEM. (B) Splenocytes from HLA-II tg mice expressing either the protective or neutral-risk allele were either left unstimulated or stimulated with 10 or 1 ng/ml of SmeZ and cultured for 72 h at 37°C and 5% CO2. After 72 h, the cells were washed and stained with antibodies against surface CD3, CD4, CD25, GARP, and LAP. Their expression levels were measured by flow cytometry. Results are shown as bar graphs and indicate the mean frequencies of GARP+ LAP− cells within activated CD4+ CD25+ T cells gated on viable cells. Error bars represent the SEM from the means of results of at least three biological replicates. (C and D) Representative flow cytometry data are presented as a contour plot of GARP+ LAP− cells in vivo (C) and in vitro (D). *, P ≤ 0.05; **, P ≤ 0.005.
FoxP3-negative surface LAP-expressing Tregs were significantly upregulated in vivo and in vitro in the DR4/DQ8 alleles associated with neutral risk.
LAP is expressed on activated FoxP3-expressing Tregs that exert their suppressive function in a TGF-β-dependent manner. However, small subsets of CD4+ LAP+ cells that do not express FoxP3 have also been reported, and interestingly, those cells still exerted suppressive ability (31, 32). In the present study, we observed a significant induction of CD4+ CD25+ LAP+ T cells that lacked FoxP3 expression during GAS-mediated NSTI by the neutral-risk allele compared with the protective allele (P ≤ 0.05; Fig. 5A). Similarly, in vitro SmeZ stimulation at all three concentrations significantly induced FoxP3− LAP+ cells when presented by the neutral-risk allele compared with the protective allele (P ≤ 0.05; Fig. 5B). Representative contour plots are shown in Fig. 5C (in vivo) and Fig. 5D (in vitro). These findings prompt further investigations to determine the contribution of this novel subset of Tregs during GAS-mediated NSTI.
FIG 5.

The neutral-risk allele resulted in significant induction of FoxP3-negative LAP-expressing Tregs in vivo and in vitro. (A) A total of 7 to 10 mice expressing either the HLA-II protective or neutral-risk allele were infected subcutaneously with 1 × 108 to 10 × 108 CFU/mouse of M1T1 GAS isolate 2006 for 72 h. Splenocytes were isolated and stained with antibodies against surface CD3, CD4, CD25, GARP, and LAP, as well as intracellular FoxP3, and levels were measured by flow cytometry. Data shown represent the frequencies of FoxP3− LAP+ cells within activated CD4+ CD25+ T cells gated on viable cells. Each mouse is represented by a symbol. Error bars represent the SEM. (B) Splenocytes from HLA-II tg mice expressing either the protective or neutral-risk allele were either left unstimulated or stimulated with 10, 1, and 0.1 ng/ml of SmeZ. Cells were cultured for 72 h at 37°C and 5% CO2. After 72 h, the cells were washed and incubated with antibodies to surface CD3, CD4, CD25, GARP, and LAP, as well as intracellular FoxP3, and subjected to flow cytometry. Results are shown as bar graphs and indicate the mean frequencies of FoxP3− LAP+ cells within activated CD4+ CD25+ T cells gated on viable cells. Error bars represent the SEM from the means of results of at least three biological replicates. (C and D) Representative flow cytometry data as a contour plot of FoxP3− LAP+ cells in vivo (C) and in vitro (D). *, P ≤ 0.05.
In vitro presentation of SmeZ by neutral-risk alleles DR4/DQ8 promoted the expansion of TCRVβ11+ T cells within the activated CD4+ CD25+ population.
Unlike conventional antigens, SAgs activate a large proportion (10% to 30%) of T cells by simultaneously cross-linking major histocompatibility complex class II (MHC-II) expressed on APCs and TCR carrying specific Vβ domains (26, 33). The binding of SAgs to MHC-II is affected by HLA-II allelic variations that profoundly modulate the magnitude of T cell responses. SmeZ binds the polymorphic HLA-DRβ chain, and in order to determine the effect of SmeZ presentation by the protective allele versus the neutral-risk allele on TCRVβ responses, we determined the frequency of CD4+ CD25+ TCRVβ11+ T cells, since expression of TCRVβ11 is specific for SmeZ in mice (11). We observed that presentation of SmeZ (at 10 and 1 ng/ml) by the neutral-risk allele induced a greater frequency of CD4+ CD25+ TCRVβ11+ T cells than presentation of SmeZ by the protective allele (Fig. 6). Our data also revealed a unique subset of CD4+ CD25+ TCRVβ11+ FoxP3+ T cells in the neutral-risk allele. However, compared with the unstimulated control, there was no significant increase in the frequency of CD4+ CD25+ TCRVβ11+ FoxP3+ T cells when SmeZ (at 10 and 1 ng/ml) was presented by the neutral-risk allele compared with presentation of SmeZ by the protective allele (Fig. 6).
FIG 6.

Presentation of SmeZ by the neutral-risk allele promoted the induction of TCRVβ11+ cells within activated CD4+ CD25+ cells. Splenocytes (4 × 106 to 5 × 106 per well) from HLA-II tg mice expressing either the protective or neutral-risk HLA-II allele were left unstimulated (control) or stimulated with 10 ng/ml of SmeZ (SmeZ 10) or 1 ng/ml of SmeZ (SmeZ 1) (n = 3 mice per group). Cells were cultured for 72 h at 37°C in an atmosphere of 5% CO2. Cultured cells were isolated and stained with surface antibodies against CD3, CD4, CD25, and TCRVβ11, as well as intracellular FoxP3, and levels were measured by flow cytometry gated on viable CD3+ CD4+/CD25+/TCRVβ11+ FoxP3+ cells. Data are shown as a contour plot of one representative mouse expressing either the protective or neutral-risk HLA-II allele.
Presentation of SmeZ by the neutral-risk allele resulted in significant production of IFN-γ and IL-2 and a trend toward upregulation of Tbx21 mRNA in vitro.
In this study, we demonstrated that in vitro SmeZ presentation by the neutral-risk allele resulted in increased expression of the activation marker CD25, SmeZ-specific T-cell proliferative responses, and expansion of CD4+ CD25+ TCRVβ11+ T cells, with a significantly lower frequency of CD4+ CD25+/FoxP3+ T cells. The decreased expression of FoxP3 has been related to its ability to produce effector cytokines such as IFN-γ and IL-2 in addition to IL-4, IL-17, and tumor necrosis factor alpha (TNF-α) (34). Using a bead-based immunoassay (BioLegend), we assessed the expression of a panel of Th1/Th2 cytokines in cell-free supernatants from SmeZ-stimulated cultures. Our results show that while there were no differences in the levels of IL-4, IL-5, IL-6, IL-10, IL-13, and TNF-α, there was significant induction of IFN-γ (P < 0.05; SmeZ at 10 and 1 ng/ml) (Fig. 7A) and IL-2 (P < 0.05; SmeZ at 1 and 0.1 ng/ml) (Fig. 7B) by the neutral-risk allele compared with the protective allele. It is well established that when naive CD4+ T cells encounter antigens in the presence of IFN-γ, coordinated signaling is activated through the TCR and signal transducer and activator of transcription 1, resulting in increased expression of Tbx21 (a key regulator for lineage commitment in CD4+ Th1), which in turn induces the expression of IFN-γ by upregulating IL-12Rβ2 expression (35). Both IFN-γ and IL-12 are important cytokines that guide the development of their master transcription factor, Tbx21. Although it seems redundant, the sequential induction of IFN-γ is a powerful mechanism that amplifies effector differentiation initiation by Tbx21, stabilization, and maintenance of the Th1 phenotype. We assessed Tbx21 mRNA levels in SmeZ-stimulated (1 ng/ml) splenocytes by quantitative real-time PCR (RT-qPCR). As shown in Fig. 7C, the mRNA expression levels of Tbx21 transcripts showed a considerable trend toward significance in the neutral-risk allele compared with the protective allele, but the results did not reach statistical significance (P = 0.0571). Similarly, although there were no significant differences in Foxp3 mRNA levels when SmeZ was presented by either the protective or the neutral-risk allele, analysis of the Tbx21/FoxP3 transcription factor ratio revealed a trend toward significance (P = 0.0571) when SmeZ was presented by the neutral-risk allele (data not shown).
FIG 7.

SmeZ presentation by the neutral-risk allele significantly increased expression of the proinflammatory cytokines IFN-γ and IL-2 and upregulated Th1 transcription factor Tbx21 mRNA. (A and B) Cell-free culture supernatants from stimulated splenocytes were analyzed for IFN-γ production (A) and IL-2 production (B). (C) mRNA expression levels of the Th1 master transcription factor Tbx21 were determined by quantitative real-time PCR in splenocytes stimulated with 1 ng/ml of SmeZ. Results are shown as bar graphs and indicate the mean levels of IFN-γ or IL-2. Tbx21 mRNA was normalized against GAPDH expression. Error bars represent the SEM from the means of results from at least three biological replicates. *, P ≤ 0.05.
DISCUSSION
Our data lay the foundation for the idea of an as-yet-unidentified role for HLA-II allelic variations in regulating effector T cell responses and Th1 differentiation versus induction of Tregs in GAS-mediated NSTI and in response to SAg stimulations in vitro. We show that, compared with the protective allele, there was a significant attenuation of FoxP3- and GARP-expressing Tregs during GAS-mediated NSTI and that the attenuation in response to SmeZ stimulations was concentration dependent in the neutral-risk allele in vitro. Further, presentation of SmeZ by the neutral-risk allele was associated with a significant increase in T cell proliferative responses and expression of the effector cytokines IFN-γ and IL-2, as well as upregulation of CD4+ CD25+ TCRVβ11+ T cells and mRNA expression of the Th1 transcription factor Tbx21.
We observed a loss of FoxP3 induction by DR15 or DR4/DQ8 alleles at a high SmeZ concentration (10 ng/ml). High antigen concentrations have been reported to downregulate the induction and maintenance of FoxP3 expression, which is modulated by the strength of both TCR stimulation and costimulatory signals (28). One proposed mechanism is represented by the fact that high antigen concentrations that trigger TCR stimuli can antagonize the differentiation of naive T cells into Tregs and stimulate the expression of IFN-γ, IL-17A, IL-17F, or IL-9 (28). Our in vivo and in vitro data suggest that the neutral-risk allele induces a higher frequency of CD4+ CD25high FoxP3low cells, which are perhaps effector Tregs with the potential to secrete inflammatory cytokines, than the protective DR15 allele. Indeed, in vitro SmeZ presentation by the neutral-risk allele induced IFN-γ and IL-2 effector cytokines and Tbx21 mRNA compared with presentation by the protective allele. Tbx21 is the transcription factor for Th1 cells and plays a critical role in regulating IFN-γ production. We are currently investigating whether Th1 differentiation signals and induction of Tbx21 are similarly driven by neutral-risk alleles during GAS-mediated NSTI.
In this study, we evaluated two additional surface markers, GARP and LAP, on FoxP3-expressing Tregs that are critical to understanding the nature of Treg-mediated suppression of inflammatory responses associated with GAS-mediated NSTI. GARP regulates the bioavailability of active TGF-β, and its expression has been reported to correlate with the expression of both FoxP3 and LAP (36). While the levels of the GARP+ LAP− subsets were significantly increased in the protective allele both in vivo and in vitro, in contrast, we also observed a significant increase in the frequency of FoxP3− LAP+ cells (and, similarly, GARP− LAP+ cells; data not shown) in the neutral-risk allele both in vivo and in vitro. Oida and Weiner reported that overexpression of TGF-β1 in murine CD4+ CD25− cells induced LAP expression that was independent of Foxp3 (31) and GARP (37) where LAP was anchored through GRP78 (a molecular chaperone belonging to the Hsp70 family). The role of the GARP/FoxP3-independent CD4+ CD25+ LAP+ subset in GAS-mediated NSTI needs to be assessed in future studies.
The immunological synapse resulting from the combination of costimulatory signals and those induced by the interactions of the MHC-II–SAg–TCR trimolecular complex orchestrates T-cell activation and proliferation (38, 39). It is well established that TCR signaling is required for the induction and maintenance of FoxP3 expression, although excessive levels of TCR stimuli can downregulate Foxp3 expression (34, 40). We did not measure the frequency of CD4+ CD25+ TCRVβ11+ T cells during in vivo GAS infections. However, our in vitro data suggest an increased frequency of CD4+ CD25+ TCRVβ11+ T cells when SmeZ is presented by the neutral-risk allele. This result is expected and likely due to the differences in the binding affinities or stimulus strengths (or both) between the HLA-II–SmeZ complex and T cells bearing TCRVβ11. Our data also reveal that expression of a subset of CD4+ CD25+/TCRVβ11+ FoxP3+ T cells was more pronounced when SmeZ was presented by the neutral-risk allele than when it was presented by the protective allele. We speculate that the CD4+ CD25+/TCRVβ11+ FoxP3+ T cells seen in the neutral-risk allele might represent activation-induced FoxP3-expressing effector Tregs and might not have suppressive functions.
Our in vivo and in vitro data demonstrate for the first time that protective HLA class II alleles promote the induction of GARP and FoxP3-expressing Tregs whereas neutral-risk alleles influence effector T cells and the Tbx21-expressing Th1 phenotype. However, our study had the following limitations: (i) we did not isolate the GARP+ LAP+ Tregs and test their suppressive ability in vitro or in vivo; (ii) we did not induce GARP expression as a means of promoting FoxP3-expressing cells with suppressive function; and (iii) we did not characterize the TGF-β/Th17 cytokines/transcription factors in vitro or in vivo. Our future studies will address these gaps and show how Treg versus Th1 responses influence the cytokine storm and consequently the clinical outcomes of GAS-mediated NSTI.
MATERIALS AND METHODS
Ethics statement.
Mice were bred and maintained in accordance with the guidelines established by the University of North Dakota Institutional Animal Care and Use Committee. All experimental protocols were approved by the University of North Dakota Institutional Animal Care and Use Committee.
HLA-II transgenic (HLA-II tg) mice.
To generate HLA-II tg mice expressing the protective DR15 allele, the desired HLA-II amplicon was cloned into the EcoRI site of the pDOI-5 mammalian expression vector. This site is downstream of the mouse H2 I-Eα promoter and the rabbit β-globin intron (which served as an enhancer) and upstream of the β-globin 3′ untranslated region and polyadenylation signal. Clones with inserts in the sense direction (5′ to 3′) were confirmed by sequencing for subsequent HLA-DRα and HLA-II DRβ assembly. The SalI-XhoI-BglI fragment excised from the pDOI-5 clone that included the H2 I-Eα promoter/rabbit β-globin/HLA-II DRα or β gene was assembled into one construct in the pSP73 vector. DNA was extracted from positive clones after confirmation by restriction digestion and sequencing and then microinjected into ova of C57BL/6 mice. Transgenic founders were transported to the Jackson Laboratory for mating to B6 (IAbIEb)null mice to generate progeny that were completely deficient in mouse MHC class II but expressed the desired HLA-II transgenes. HLA-II tg, MHC class II-deficient (H-2 Ab0) mice that expressed the human DRB1*04/DQB1*0302 (DR4/DQ8) haplotype (associated with neutral-risk outcomes for combined NF/STSS) were generated in the laboratory of C. S. David (Mayo Clinic, Rochester, MN) (11). We confirmed the presence of the appropriate HLA-II transgenes by PCR-based genotyping using the following allele-specific oligonucleotide primers: DR15 sense 5′-TCC TGT GGC AGC CTA AGA G-3′ and DR15 antisense 5′-TCG CCG CTG CAC TGT GAA G-3′; DQ8 sense 5′-AGG ATT TGG TGT ACC AGT TTA AGG GCA T-3′ and DQ8 antisense 5′-TGC AAG GTC GTG CGG AGC TCC AA-3′; and DR4 sense 5′-GTT TCT TGG AGC AGG TTA AAC A-3′ and DR4 antisense 5′-CTG CAC TGT GAA GCT CTC AC-3′ (11). We confirmed the expression of HLA-II DR/DQ molecules in mouse whole blood by flow cytometry using an LSR-II flow cytometer (Becton, Dickinson) after staining whole blood with allophycocyanin-labeled anti-HLA-DR (clone L243) (eBioscience/Tonbo Bio) and a phycoerythrin (PE)-labeled monoclonal antibody (MAb) specific to HLA-DQ (clone DQB1) (BioLegend).
GAS bacteria and in vivo infections.
We used the clinical isolate M1 GAS 2006 for the in vivo studies. GAS 2006 was grown under static conditions in Todd-Hewitt broth supplemented with 1.5% yeast extract for 17 h at 37°C (11, 24, 41). The bacteria were harvested by centrifugation, washed three times with sterile endotoxin-free Dulbecco's phosphate-buffered saline (DPBS), and resuspended in DPBS. Age- and gender-matched 16-to-24-week-old mice expressing either HLA-II DR15 or DR4/DQ8 alleles were subcutaneously infected with 0.1 ml of GAS suspension (optical density at 600 nm [OD600] adjusted to yield ∼1 × 108 to 10 × 108 CFU/0.1 ml). Inocula were determined by plating on sheep blood agar. Mice were monitored daily, and skin lesion areas were measured using digital calipers. At 72 h postinfection, the mice were euthanized by CO2 inhalation. Blood was drawn through cardiac puncture for bacteremia estimations, and the necrotic skin tissue and spleen were recovered under sterile conditions. A portion of the recovered skin/spleen was homogenized using a motorized homogenizer (Omni International, Marietta, GA). For enumeration of bacterial load and dissemination, 10-fold dilutions of the homogenates were prepared in DPBS and plated on sheep blood agar plates. The remaining portion of the spleen was used to isolate splenocytes for Treg immunophenotyping.
M1T1 GAS recombinant SmeZ1 SAg and preparation of partially purified native, secreted streptococcal SAgs.
We generated recombinant SmeZ1 (SmeZ) by expressing it as a histidine-fusion protein in Escherichia coli and subsequently purifying it as described previously (11). Purified recombinant SmeZ1 was treated with polymyxin B agarose (Boehringer Mannheim, Indianapolis, IN) to remove any contaminating endotoxin. We used an isolate of clonal M1T1 strain 5448 that had been passaged in animals to prepare a partially purified mixture of the native secreted streptococcal SAgs as described previously (12).
In vitro splenocyte proliferation assays.
Splenocyte proliferation assays were carried out as described previously (11). Briefly, spleens were aseptically removed from the HLA-II tg mice expressing either the protective DR15 allele or the DR4/DQ8 alleles associated with neutral-risk outcomes of NF/STSS. Single-cell suspensions were prepared, and the cells were treated with NH4Cl red blood cell (RBC) lysis buffer, washed three times in Hanks’ balanced salt solution (HBSS), and resuspended to a concentration of 2 × 106 cells/ml in RPMI 1640 medium with 1% (vol/vol) heat-inactivated and polymyxin B-treated fetal bovine serum (FBS), 4 mM l-glutamine, 25 mM HEPES, 50 U/ml penicillin, 50 µg/ml streptomycin, and 50 µM 2-mercaptoethanol (2-ME) (RPMI 1640 complete medium). The cells were seeded at a concentration of 2 × 105 per well into U-bottom, 96-well microtiter plates (Costar) and stimulated with optimal concentrations/dilutions of (i) SmeZ (10, 1, or 0.1 ng/ml), (ii) a 1:100 dilution of partially purified native, secreted streptococcal SAgs, or (iii) concanavalin A (ConA; 1 µg/ml). After incubation for 72 h at 37°C in an atmosphere of 5% CO2, the cultures were pulsed for the final 6 h with 1 µCi [3H]thymidine (Perkin Elmer, Waltham, MA) (specific activity = 6.7 Ci/mM) per well, harvested onto glass fiber filters, and washed. [3H]thymidine uptake was measured by counting using a beta counter (Perkin Elmer). All [3H]thymidine measurements were performed in triplicate, and the data are presented as mean counts per minute ± standard errors of the means (SEM). Each experiment was repeated at least three times. Parallel cultures were set up for Treg immunophenotyping by flow cytometry and RNA isolation and cDNA synthesis for quantitative real-time PCR.
Flow cytometry and Treg immunophenotyping.
Splenocytes were isolated from 7 to 10 mice expressing either HLA-II DR15 or DR4/DQ8 and infected subcutaneously with 1 × 108 to 10 × 108 CFU in 100 μl of M1T1 GAS isolate 2006 for 72 h. The cells were then stained with appropriate Abs. Antibodies and permeabilization kits were obtained from BioLegend, eBioscience, and Tonbo Biotechnology Corporation. Unstained, single stained, and fluorescence-minus-one (FMO) controls were used to set gating parameters, determine specificity, and set up compensation panels for acquisition to avoid spectral overlaps. At least 50,000 cells were acquired in the live gate for further analysis. Expression of surface markers and intracellular cytokines or FoxP3 was evaluated by staining with the following panel: BV510-Live/Dead fixable viability dye for live/dead cell staining, BV450-conjugated anti-mouse CD3 (clone 145-2C11), PECy7-conjugated anti-mouse CD4 (clone RM4-5), and PE/fluorescein isothiocyanate (PE/FITC)-conjugated anti-mouse CD25 (clone PC61). PE-conjugated anti-mouse GARP (clone F011-5) and allophycocyanin-conjugated anti-mouse LAP (TW7-16B4) were used as highly specific markers of activated Tregs. FoxP3 staining was carried out using a PECy5.5-conjugated anti-mouse FoxP3 (clone FJK16S) antibody following the manufacturer’s instructions (eBioscience). Briefly, after live/dead cell staining, the cells were incubated with respective antibodies against surface markers for 45 min at room temperature or left overnight at 4°C. Cells were washed twice with fluorescence-activated cell sorter (FACS) buffer (PBS–2% FBS) followed by permeabilization/fixation for 15 min at room temperature in the dark. Cells were washed with permeabilization buffer, and nonspecific binding sites were blocked by incubation with normal rat serum (dilution ratio, 1:300) for 10 min at room temperature in the dark. These cells were subsequently incubated with an anti-FoxP3 antibody for 2 h at room temperature. Cells were washed twice with permeabilization buffer and finally resuspended in FACS buffer containing 5 mM EDTA. Cells were acquired using an LSR II flow cytometer (BD Biosciences), and data were analyzed using FloJo software (FloJO, Treestar, Ashland, OR).
Quantitative real-time PCR.
RNA from SAg-stimulated splenocytes was isolated using an RNeasy minikit (Qiagen) following standard manufacturer protocols. RNA concentrations were analyzed using a NanoDrop spectrophotometer (ND-1000; Thermo Fisher). A total of 0.5 to 1 µg of RNA was used for cDNA synthesis using oligo(dT) and random hexamer primer mix (Promega), 10 mM deoxynucleoside triphosphate (dNTP), and Superscript III reverse transcriptase (Invitrogen) with RiboLock RNase inhibitor (Thermo Scientific) in a 10-µl final volume. RT-qPCR was performed in 10-µl volumes in triplicate on a CFX Connect real-time PCR detection system (Bio-Rad) using 2× SYBR green master mix (Roche). The primers used for Tbx21 were 5′-TTT CCA AGA GAC CCA GTT CAT TG-3′ (sense) and 5′-ATG CGT ACA TGG ACT CAA AGT T-3′ (antisense). At least three mice per group were assayed, each in triplicate. Relative Tbx21 gene expression levels were calculated using the comparative threshold cycle (2−ΔΔCq) method (42) normalized against GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA levels.
Cytokine assays.
Cell-free culture supernatants from SAg-stimulated splenocytes were assayed using a LEGENDplex mouse Th1/Th2 panel (8-plex) (BioLegend). This kit allows the simultaneous detection of eight mouse Th1/Th2 cytokines: IL-2, IL-4, IL-5, IL-6, IL-10, IL-13, IFN-γ, and TNF-α. Assays were carried out according to the instructions of the manufacturers. Unstimulated cultures were used as controls, and cytokine levels were detected by flow cytometry using BD-FACSymphony A3 (BD Biosciences).
Statistical analysis.
Results are presented as mean values ± standard errors of the means (SEM). Comparisons among three groups were made using one-way analysis of variance (ANOVA), and the differences between two groups were assessed with the nonparametric Mann-Whitney U test (GraphPad Prism version 7.00 for Windows; GraphPad Software, La Jolla, CA, USA) (*, P ≤ 0.05; **, P ≤ 0.005; ***, P ≤ 0.0005). Significance was achieved when the P value was ≤0.05.
ACKNOWLEDGMENTS
We thank Chella David for HLA-II DRB1*04/DQB1*0302 (DR4/DQ8) mice. We acknowledge Ellen Olson for help with infections and Tyler Staskivige for help in animal husbandry. We are grateful to K. Beiswenger for manuscript editing.
This work was supported by the University of North Dakota (UND) Faculty Seed Money awarded to S.N. and grants from the European Union (FP7/2012-2017) under grant agreement 305340 (M.K.). The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award P20GM113123. The flow cytometry reported here was performed in the North Dakota Flow Cytometry Cell Sorting Core supported by Institutional Development Awards (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grants P20GM103442 and P20GM113123.
REFERENCES
- 1.Stevens DL, Tanner MH, Winship J, Swarts R, Ries KM, Schlievert PM, Kaplan E. 1989. Severe group A streptococcal infections associated with a toxic shock-like syndrome and scarlet fever toxin A. N Engl J Med 321:1–7. doi: 10.1056/NEJM198907063210101. [DOI] [PubMed] [Google Scholar]
- 2.Donaldson PM, Naylor B, Lowe JW, Gouldesbrough DR. 1993. Rapidly fatal necrotising fasciitis caused by Streptococcus pyogenes. J Clin Pathol 46:617–620. doi: 10.1136/jcp.46.7.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kazmi SU, Kansal R, Aziz RK, Hooshdaran M, Norrby-Teglund A, Low DE, Halim AB, Kotb M. 2001. Reciprocal, temporal expression of speA and speB by invasive M1T1 group A streptococcal isolates in vivo. Infect Immun 69:4988–4995. doi: 10.1128/IAI.69.8.4988-4995.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kansal RG, Aziz RK, Kotb M. 2005. Modulation of expression of superantigens by human transferrin and lactoferrin: a novel mechanism in host-Streptococcus interactions. J Infect Dis 191:2121–2129. doi: 10.1086/430386. [DOI] [PubMed] [Google Scholar]
- 5.Aziz RK, Kansal R, Abdeltawab NF, Rowe SL, Su Y, Carrigan D, Nooh MM, Attia RR, Brannen C, Gardner LA, Lu L, Williams RW, Kotb M. 2007. Susceptibility to severe streptococcal sepsis: use of a large set of isogenic mouse lines to study genetic and environmental factors. Genes Immun 8:404–415. doi: 10.1038/sj.gene.6364402. [DOI] [PubMed] [Google Scholar]
- 6.Kansal RG, Datta V, Aziz RK, Abdeltawab NF, Rowe S, Kotb M. 2010. Dissection of the molecular basis for hypervirulence of an in vivo-selected phenotype of the widely disseminated M1T1 strain of group A Streptococcus bacteria. J Infect Dis 201:855–865. doi: 10.1086/651019. [DOI] [PubMed] [Google Scholar]
- 7.Kappler J, Kotzin B, Herron L, Gelfand EW, Bigler RD, Boylston A, Carrel S, Posnett DN, Choi Y, Marrack P. 1989. V beta-specific stimulation of human T cells by staphylococcal toxins. Science 244:811–813. doi: 10.1126/science.2524876. [DOI] [PubMed] [Google Scholar]
- 8.Kotb M. 1998. Superantigens of gram-positive bacteria: structure-function analyses and their implications for biological activity. Curr Opin Microbiol 1:56–65. doi: 10.1016/S1369-5274(98)80143-4. [DOI] [PubMed] [Google Scholar]
- 9.Norrby-Teglund A, Thulin P, Gan BS, Kotb M, McGeer A, Andersson J, Low DE. 2001. Evidence for superantigen involvement in severe group A streptococcal tissue infections. J Infect Dis 184:853–860. doi: 10.1086/323443. [DOI] [PubMed] [Google Scholar]
- 10.Kotb M, Norrby-Teglund A, McGeer A, El-Sherbini H, Tevik Dorak M, Khurshid A, Green K, Peeples J, Wade J, Thomson G, Schwartz B, Low DE. 2002. An immunogenetic and molecular basis for differences in outcomes of invasive group A streptococcal infections. Nat Med 8:1398–1404. doi: 10.1038/nm1202-800. [DOI] [PubMed] [Google Scholar]
- 11.Nooh MM, El-Gengehi N, Kansal R, David CS, Kotb M. 2007. HLA transgenic mice provide evidence for a direct and dominant role of HLA class II variation in modulating the severity of streptococcal sepsis. J Immunol 178:3076–3083. doi: 10.4049/jimmunol.178.5.3076. [DOI] [PubMed] [Google Scholar]
- 12.Nooh MM, Nookala S, Kansal R, Kotb M. 2011. Individual genetic variations directly effect polarization of cytokine responses to superantigens associated with streptococcal sepsis: implications for customized patient care. J Immunol 186:3156–3163. doi: 10.4049/jimmunol.1002057. [DOI] [PubMed] [Google Scholar]
- 13.Hori S, Nomura T, Sakaguchi S. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science 299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 14.Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, Treuting PM, Rudensky AY. 2015. A distinct function of regulatory T cells in tissue protection. Cell 162:1078–1089. doi: 10.1016/j.cell.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura K, Kitani A, Strober W. 2001. Cell contact-dependent immunosuppression by Cd4(+) Cd25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor β. J Exp Med 194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seo KK, Lee SU, Park YH, Davis WC, Fox LK, Bohach GA. 2007. Long-term staphylococcal enterotoxin C1 exposure induces soluble factor-mediated immunosuppression by bovine CD4+ and CD8+ T cells. Infect Immun 75:260–269. doi: 10.1128/IAI.01358-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor AL, Llewelyn MJ. 2010. Superantigen-induced proliferation of human CD4+CD25- T cells is followed by a switch to a functional regulatory phenotype. J Immunol 185:6591–6598. doi: 10.4049/jimmunol.1002416. [DOI] [PubMed] [Google Scholar]
- 18.Fantini MC, Becker C, Tubbe I, Nikolaev A, Lehr HA, Galle P, Neurath MF. 2006. Transforming growth factor beta induced FoxP3+ regulatory T cells suppress Th1 mediated experimental colitis. Gut 55:671–680. doi: 10.1136/gut.2005.072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. 2005. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol 6:1219–1227. doi: 10.1038/ni1265. [DOI] [PubMed] [Google Scholar]
- 20.Tran DQ. 2012. TGF-beta: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol 4:29–37. doi: 10.1093/jmcb/mjr033. [DOI] [PubMed] [Google Scholar]
- 21.Stockis J, Colau D, Coulie PG, Lucas S. 2009. Membrane protein GARP is a receptor for latent TGF-beta on the surface of activated human Treg. Eur J Immunol 39:3315–3322. doi: 10.1002/eji.200939684. [DOI] [PubMed] [Google Scholar]
- 22.Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. 2009. GARP (LRRC32) is essential for the surface expression of latent TGF- on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A 106:13445–13450. doi: 10.1073/pnas.0901944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA. 2011. Latent TGF-β structure and activation. Nature 474:343–351. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chella Krishnan K, Mukundan S, Alagarsamy J, Hur J, Nookala S, Siemens N, Svensson M, Hyldegaard O, Norrby-Teglund A, Kotb M. 2016. Genetic architecture of group A streptococcal necrotizing soft tissue infections in the mouse. PLoS Pathog 12:e1005732. doi: 10.1371/journal.ppat.1005732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fazekas De St Groth B, Smith AL, Higgins CA. 2004. T cell activation: in vivo veritas. Immunol Cell Biol 82:260–268. doi: 10.1111/j.0818-9641.2004.01243.x. [DOI] [PubMed] [Google Scholar]
- 26.Proft T, Fraser JD. 2007. Streptococcal superantigens. Chem Immunol Allergy 93:1–23. doi: 10.1159/000100851. [DOI] [PubMed] [Google Scholar]
- 27.Shevach EM, Tran DQ, Davidson TS, Andersson J. 2008. The critical contribution of TGF-β to the induction of Foxp3 expression and regulatory T cell function. Eur J Immunol 38:915–917. doi: 10.1002/eji.200738111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molinero LL, Miller ML, Evaristo C, Alegre M-L. 2011. High TCR stimuli prevent induced regulatory T cell differentiation in a NF-κB-dependent manner. J Immunol 186:4609–4617. doi: 10.4049/jimmunol.1002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gabryšová L, Christensen JR, Wu X, Kissenpfennig A, Malissen B, O’Garra A. 2011. Integrated T-cell receptor and costimulatory signals determine TGF-β-dependent differentiation and maintenance of Foxp3+ regulatory T cells. Eur J Immunol 41:1242–1248. doi: 10.1002/eji.201041073. [DOI] [PubMed] [Google Scholar]
- 30.Probst-Kepper M, Geffers R, Kröger A, Viegas N, Erck C, Hecht HJ, Lünsdorf H, Roubin R, Moharregh-Khiabani D, Wagner K, Ocklenburg F, Jeron A, Garritsen H, Arstila TP, Kekäläinen E, Balling R, Hauser H, Buer J, Weiss S. 2009. GARP: a key receptor controlling FOXP3 in human regulatory T cells. J Cell Mol Med 13:3343–3357. doi: 10.1111/j.1582-4934.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oida T, Weiner HL. 2010. TGF-β induces surface LAP expression on murine CD4 T cells independent of Foxp3 induction. PLoS One 5:e0015523. doi: 10.1371/journal.pone.0015523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gandhi R, Farez MF, Wang Y, Kozoriz D, Quintana FJ, Weiner HL. 2010. Cutting edge: human latency-associated peptide+ T cells: a novel regulatory T cell subset. J Immunol 184:4620–4624. doi: 10.4049/jimmunol.0903329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herman A, Kappler JW, Marrack P, Pullen AM. 1991. Superantigens: mechanism of T-cell stimulation and role in immune responses. Annu Rev Immunol 9:745–772. doi: 10.1146/annurev.iy.09.040191.003525. [DOI] [PubMed] [Google Scholar]
- 34.Williams LM, Rudensky AY. 2007. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol 8:277–284. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- 35.Petermann F, Korn T. 2011. Cytokines and effector T cell subsets causing autoimmune CNS disease. FEBS Lett 585:3747–3757. doi: 10.1016/j.febslet.2011.03.064. [DOI] [PubMed] [Google Scholar]
- 36.Wang R, Zhu J, Dong X, Shi M, Lu C, Springer TA. 2012. GARP regulates the bioavailability and activation of TGF. Mol Biol Cell 23:1129–1139. doi: 10.1091/mbc.e11-12-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oida T, Weiner HL. 2010. Overexpression of TGF-β1 gene induces cell surface localized glucose-regulated protein 78-associated latency-associated peptide/TGF-β. J Immunol 185:3529–3535. doi: 10.4049/jimmunol.0904121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saline M, Rödström KEJ, Fischer G, Orekhov VY, Karlsson BG, Lindkvist-Petersson K. 2010. The structure of superantigen complexed with TCR and MHC reveals novel insights into superantigenic T cell activation. Nat Commun 1:119. doi: 10.1038/ncomms1117. [DOI] [PubMed] [Google Scholar]
- 39.Kasper KJ, Zeppa JJ, Wakabayashi AT, Xu SX, Mazzuca DM, Welch I, Baroja ML, Kotb M, Cairns E, Cleary PP, Haeryfar SMM, McCormick JK. 2014. Bacterial superantigens promote acute nasopharyngeal infection by Streptococcus pyogenes in a human MHC class II-dependent manner. PLoS Pathog 10:e1004155. doi: 10.1371/journal.ppat.1004155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohkura N, Sakaguchi S. 2010. Regulatory T cells: roles of T cell receptor for their development and function. Semin Immunopathol 32:95–106. doi: 10.1007/s00281-010-0200-5. [DOI] [PubMed] [Google Scholar]
- 41.Krishnan KC, Mukundan S, Alagarsamy J, Laturnus D, Kotb M. 2016. Host genetic variations and sex differences potentiate predisposition, severity, and outcomes of group A Streptococcus-mediated necrotizing soft tissue infections. Infect Immun 84:416–424. doi: 10.1128/IAI.01191-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]