
Figure 2: Proposed scheme for mechanisms and pathological consequences of age-related oxidative stress in vascular endothelial cells.
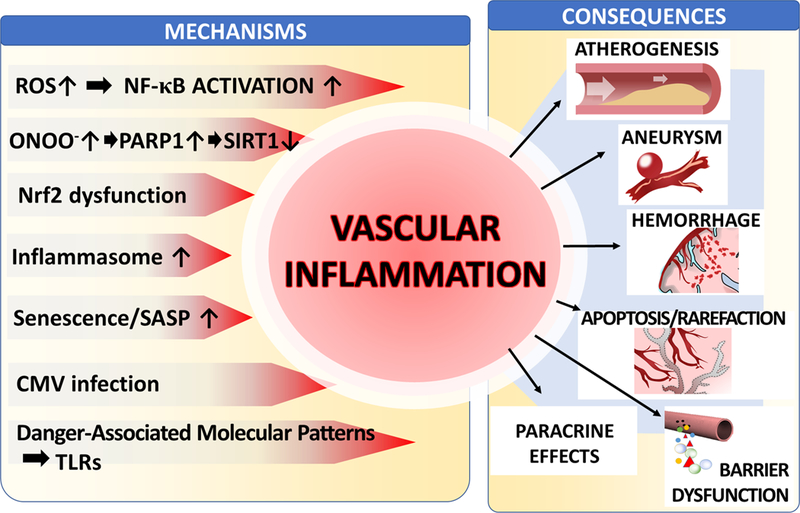
The model predicts that in aged endothelial cells dysfunctional mitochondria and NAD(P)H oxidases are critical sources of increased ROS production. Increased levels of O2.- generated by the electron transport chain are dismutated to H2O2, which can penetrate the mitochondrial membrane increasing cytoplasmic H2O2 levels. Increased oxidative stress is exacerbated by age-related impairment of Nrf2-dependent homeostatic antioxidant defense mechanisms. H2O2 plays important signaling roles, including activation of NF-κB, which contribute to age-associated low grade chronic vascular inflammation. Increased levels of O2.- generated by NAD(P)H oxidases (stimulated by elevated TNFα levels and/or by the activated local renin-angiotensin system [RAS] in the vascular wall) decrease the bioavailability of NO by forming ONOO-. Increased nitrative stress lead to PARP-1 activation, which promotes vascular inflammation and contributes to cellular energetic dysfunction by consuming NAD+ , compromising sirtuin-mediated anti-aging pathways. Impaired bioavailability of NO promotes vasodilator dysfunction and compromises endothelial viability. In addition, increased vascular oxidative stress in aging also induces MMP activation, promoting the pathogenesis of intracerebral hemorrhages, aneurysm formation and blood brain barrier disruption.