

Abstract
In utero gene editing has the potential to prenatally treat genetic diseases that result in significant morbidity and mortality before or shortly after birth. We assessed the viral vector-mediated delivery of clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated 9 (CRISPR-Cas9) or base editor 3 (BE3) in utero, seeking therapeutic modification of Pcsk9 or Hpd in wild-type mice or the murine model of hereditary tyrosinemia type 1 (HT1), respectively. We observed long-term postnatal persistence of edited cells in both models, with reduction of plasma PCSK9 and cholesterol levels following in utero Pcsk9 targeting and rescue of the lethal phenotype of HT1 following in utero Hpd targeting. The results of this proof-of-concept work demonstrate the possibility to efficiently perform gene editing before birth, pointing to a potential new therapeutic approach for select congenital genetic disorders.
The developing fetus has several properties that would make it favorable for therapeutic gene editing. It is immunologically immature; studies of in utero gene therapy demonstrated tolerance to the transgene product and lack of an immune response to the vector following prenatal viral vector delivery1,2. This contrasts with the development of antibody and T cell-mediated responses following postnatal gene therapy1–4. The small fetal size allows high levels of vectors to be given per fetal weight. Cells of multiple organs are highly proliferative and accessible for efficient vector transduction during fetal development5. Finally, in utero editing offers the potential to target genes before disease onset, critical for diseases with high prenatal or perinatal morbidity and mortality.
We sought to establish the feasibility of in utero gene editing with Streptococcus pyogenes Cas9 (SpCas9) genome editing or BE3 base editing6. We undertook standard genome editing with adeno-associated viral (AAV) vectors in R26mTmG/+ mice, which constitutively express red fluorescence in each cell until deletion of a loxP-flanked cassette switches the fluorescence to green. We injected AAV9 encoding Cre recombinase (AAV9.Cre; positive control) or two AAV9 vectors (AAV9.SpCas9.mTmG) encoding SpCas9 and a loxP-targeting guide RNA (gRNA) into embryonic day 16 (E16) R26mTmG/+ fetuses via the vitelline vein (Figure 1a, Supplementary Video 1), providing first-pass effect to liver. On day of life 1 (DOL1), we observed editing predominantly in liver and heart (Supplementary Figure 1).
Figure 1.
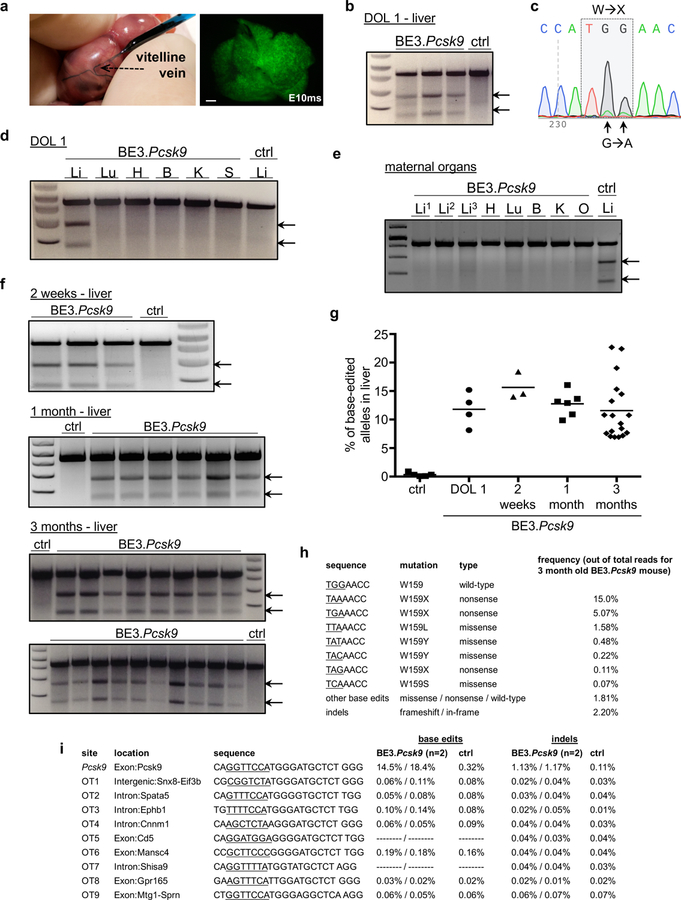
In utero base editing of the Pcsk9 gene. (a) Vitelline vein injection of E16 fetus; DOL1 liver, Ad.GFP injection (fluorescence stereomicroscopy, GFP filter). Scale bar = 1mm. (b-i) E16 Balb/c fetuses were injected with Ad.BE3.Pcsk9 or Ad.BE3.Null (ctrl). Genomic DNA from organs of injected fetuses (b-d; f-i) or of dams of injected fetuses (e) were assessed for Pcsk9 on-target (b-h) and off-target (i) editing by Surveyor assays (b, d-f), Sanger sequencing (c), and NGS (g-i). Maternal organ analysis (e) occurred 1 week after fetal injection, 3 separate liver samples per mother, ctrl Li = liver DNA from injected fetus. (b, N=3 BE3.Pcsk9, 1 ctrl; c, representative of 4 mice replicates; d, representative of 3 mice replicates; e, representative of 2 mice replicates; f, 2 weeks: N=3 BE3.Pcsk9, 1 ctrl, 1 month: N=6 BE3.Pcsk9, 1 ctrl, 3 months: N=18 BE3.Pcsk9, 2 ctrl) (g) NGS, liver DNA in control mice (N=5) and at DOL1 (N=4), 2 weeks (N=3), 1 month (N=6), and 3 months of age (N=18) following in utero Ad.BE3.Pcsk9 injection; measure of centre = mean. (h) Frequencies of base-edited and indel-bearing alleles, liver DNA, 3-month-old prenatal recipient of Ad.BE3.Pcsk9. Underlined bases indicate the target codon. (i) Base editing and indel rates for the on-target and top 9 predicted off-target sites, NGS, liver DNA at 2 weeks of age from 2 prenatal Ad.BE3.Pcsk9 recipients (results separated by dashes) and a control mouse. Underlined bases indicate the base editing window based on distance from the PAM. In cases in which no base editing rates are shown, there were no C bases within the window. DOL, day of life; BE3, base editor 3; PCSK9, proprotein convertase subtilisin/kexin type 9; ctrl, control; exp; Li, liver; Lu, lung; H, heart; B, brain; K, kidney; S, spleen; arrows, Surveyor cleavage products.
The ultimate objective of this work was to use BE3 for in utero base editing of hepatocytes. BE3 can make site-specific CT or GA changes in coding sequences without double-strand DNA breaks, making it potentially safer than standard CRISPR-Cas9 genome editing7. The large size of SpCas9-based BE3 (~5.1 kb) preempts its delivery via AAV (capacity ~4.7 kb). We thus turned to adenoviral (Ad) vectors for the subsequent series of proof-of-concept experiments, fully recognizing the limitations of Ad vectors for clinical translation due to adverse host immune responses that might lead to systemic toxicity including production of pro-inflammatory cytokines, thrombocytopenia, coagulopathy, and liver damage8. We injected E16 wild-type fetuses with an Ad vector expressing GFP (Ad.GFP) via the vitelline vein and observed robust hepatocyte transduction at DOL1, with decreased levels at 3 months (Figure 1a, Supplementary Figure 2). We then injected Ad vectors encoding either SpCas9 and a loxP-targeting gRNA (Ad.SpCas9.mTmG) or Cre recombinase (Ad.Cre) into E16 R26mTmG/+ fetuses and observed editing in various organs on DOL1, with the most robust editing seen in liver and heart (Supplementary Figure 1).
Loss-of-function mutations in PCSK9 reduce cholesterol levels and coronary heart disease risk without serious adverse consequences9. Previous studies demonstrated CRISPR-Cas9-mediated nonhomologous end-joining (NHEJ) or base editing to disrupt PCSK9 orthologs in mouse10–13 and human hepatocytes14 in vivo postnatally. We evaluated if in utero base editing of murine Pcsk9 reduces postnatal plasma PCSK9 and cholesterol levels. We used an Ad vector containing BE3 and a gRNA targeting Pcsk9 codon W159 (Ad.BE3.Pcsk9) that in a previous adult mouse study resulted in conversion to stop codons12. Following injection of E16 Balb/c fetuses, Ad.BE3.Pcsk9 resulted in base editing in liver apparent on DOL1 without evidence of editing in other organs (Figure 1b-d). Analysis of DNA from organs of mothers of injected fetuses showed no significant Pcsk9 editing [Figure 1e; next-generation sequencing (NGS) on-target Pcsk9 base-edited alleles: 0.06–0.14%, N=2 mothers; negative control: 0.06–0.3%].
Assessed by Surveyor assays and NGS of the target site, the proportion of Pcsk9 base-edited alleles in livers of in utero Ad.BE3.Pcsk9-injected mice was stable at 10–15% between DOL1 and 3 months (Figure 1f,g). The indel rate was low (~2%) (Figure 1h), contrasting with >40% indel rates seen in previous postnatal studies using NHEJ to disrupt Pcsk910,11. NGS analysis of 9 top predicted off-target sites in liver DNA from two 2-week-old Ad.BE3.Pcsk9-injected mice showed no evidence of editing (Figure 1i).
Compared with a control Ad vector, in utero Ad.BE3.Pcsk9 resulted in decreased postnatal levels of PCSK9 protein and total cholesterol at 1 and 3 months, with no differences in alanine aminotransferase levels and grossly normal liver histology (Figure 2a-l). Mice treated postnatally at 5 weeks of age with Ad.BE3.Pcsk9 had substantial editing at early post-injection timepoints (5 days, 1 month) but attenuated editing at later timepoints (2 and 3 months) (Figure 2m,n). In contrast, prenatal Ad.BE3.Pcsk9 recipients had stable editing over time, with significantly higher editing rates than postnatal recipients at 3 months despite having similar rates at 5 days (Figure 2n). Previous studies of gene therapy documented immune responses to both the Ad vector and the transgene product following postnatal delivery that were diminished with prenatal delivery15,16. We assessed if there was a different immune response to the Ad vector and SpCas9-based BE3 transgene product following prenatal versus postnatal Ad.BE3.Pcsk9 delivery, which might explain the difference in editing stability. Serum anti-Ad and anti-SpCas9 antibodies were higher in postnatal compared to prenatal recipients and naïve controls at 1 and 3 months post-injection, and equal or lower in prenatal recipients compared to naïve controls (Figure 2o,p).
Figure 2.
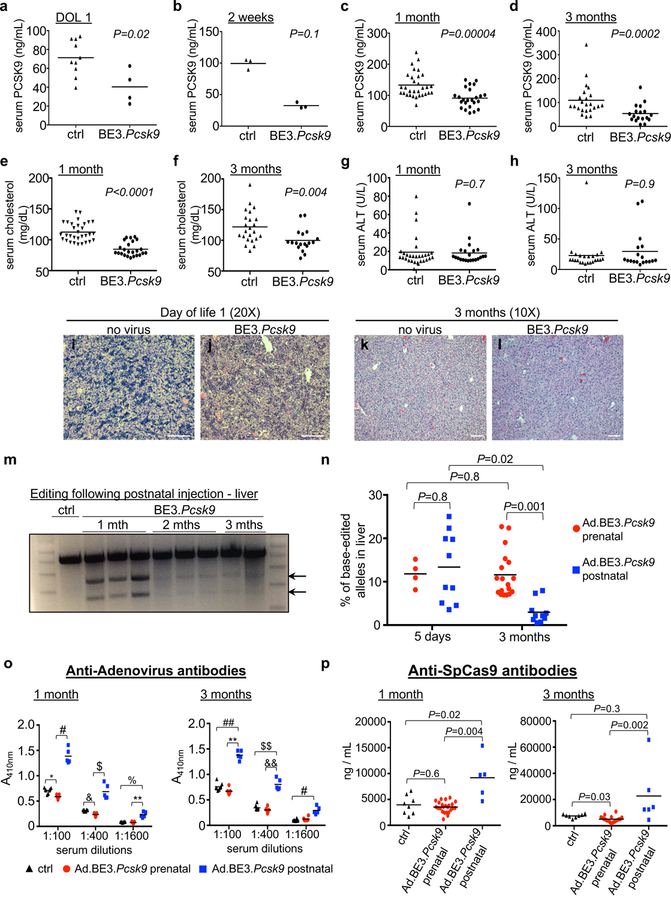
Functional effects of in utero Pcsk9 base editing and comparison to postnatal editing. (a-l) E16 Balb/c fetuses were injected with Ad.BE3.Pcsk9 or Ad.BE3.Null (ctrl). (a-d) Plasma PCSK9 protein levels at DOL1 (ctrl, N=10; BE3.Pcsk9, N=4), 2 weeks (ctrl, N=3; BE3.Pcsk9, N=3), 1 month (ctrl, N=31; BE3.Pcsk9, N=24), and 3 months (ctrl, N=23; BE3.Pcsk9, N=18). (e,f) Plasma cholesterol levels at 1 (ctrl, N=31; BE3.Pcsk9, N=24) and 3 months (ctrl, N=23; BE3.Pcsk9, N=18). (g,h) Plasma ALT levels at 1 (ctrl, N=30; BE3.Pcsk9, N=22) and 3 months (ctrl, N=23; BE3.Pcsk9, N=18). (i-l) Liver histology (hematoxylin and eosin staining) from non-injected and Ad.BE3.Pcsk9 injected fetuses. Representative of 3 mice replicates per group per time point. Scale bar = 100μm (m) Ad.BE3.Pcsk9 was injected into 5-week-old B6 mice. Surveyor assays, liver genomic DNA at 1 month (N=3), 2 months (N=3), and 3 months (N=2) post-injection. (n) Ad.BE3.Pcsk9 was injected into E16 Balb/c fetuses and 5-week-old Balb/c mice. Percentage base-edited Pcsk9 on-target alleles, NGS, liver genomic DNA at 5 days and 3 months post-injection (prenatal 5 days-N=4, 3 months-N=18; postnatal 5 days-N=10, 3 months-N=10). (o,p) Serum from mice injected at E16 or 5 weeks of age with Ad.BE3.Psck9 was assessed at 1 and 3 months post injection for antibodies to adenovirus (o) and SpCas9 (p) (anti-Ad: prenatal 1 month-N=11, 3 months-N=6; postnatal 1 month-N=5, 3 months-N=5; anti-SpCas9: prenatal 1 month-N=24, 3 months-N=18; postnatal 1 month-N=5, 3 months-N=6); Control non-injected, age-matched Balb/c mice (N=8). DOL, day of life; BE3, base editor 3; PCSK9, proprotein convertase subtilisin/kexin type 9; ALT, alanine aminotransferase; arrows, Surveyor cleavage products; * P=0.01; # P=0.001; $ P=0.00008; & P=0.006; % P=0.03; ** P=0.0007; ## P=0.04; $ $ P=0.02; && P=0.002. Statistical analysis performed with two-tailed Mann-Whitney U test (a-d,g,h), two-tailed Student’s t test (e, t=7.8, df=53; f, t=3.2; df=39) and Kruskal-Wallis test (n-p). Measure of centre = mean (a-h, n-p).
Having demonstrated in utero base editing of Pcsk9, we next sought to target a gene for which the in utero approach would be more relevant. HT1 results from a mutated Fah gene blocking the tyrosine catabolic pathway.17 Inhibiting the upstream HPD enzyme in this pathway with the drug NTBC prevents the accumulation of toxic metabolites and rescues the lethal liver failure (Supplementary Figure 3). We sought to introduce a nonsense mutation in the Hpd gene in utero to permanently knock out gene function. We screened 8 gRNAs in vitro and observed the most editing at codon Q352 (Supplementary Figure 3). In utero base editing of Hpd in E16 wild-type fetuses with an Ad vector encoding BE3 and the Q352 gRNA (Ad.BE3.Hpd) resulted in a mean editing rate of ~15% in liver at 2 weeks of age; analysis of other organs showed no editing (Supplementary Figure 3).
Fah–/– mice, a model of HT1, experience neonatal lethality and can be rescued with NTBC delivered via the mother’s breast milk. Previous studies have demonstrated amelioration of HT1 in this model with postnatal gene editing via homology-direct repair or NHEJ18–20. We mated Fah–/– adult mice on NTBC and administered Ad.BE3.Hpd to E16 fetuses, and on DOL1 we placed the recipients with foster mothers not on NTBC (Figure 3a). Base editing in liver was substantially higher in recipient mice analyzed at 1 month and 3 months than at DOL1 (37% and 40% versus 14%) (Figure 3b, c), likely due to the survival advantage and subsequent expansion of edited cells21–23. In addition to the desired CT nonsense mutation at the target site, there were much lower rates of alternative missense mutations and indels (Figure 3d). We observed no evidence of editing in other organs including gonads at 1 month (Figure 3e); sperm from two 3-month-old recipient mice with liver Hpd editing rates of 46–57% demonstrated no significant editing by Surveyor assays (Figure 3e) or NGS (0.2–0.8%; controls: 0.03–0.6%). We observed BE3 expression in heart (Supplementary Figure 3) and speculate the Hpd locus was inaccessible to BE3 in cardiomyocytes, where Hpd is not expressed24. NGS analysis of 10 top-predicted off-target sites in liver DNA from 1-month-old recipient mice showed no evidence of editing (Figure 3f).
Figure 3.
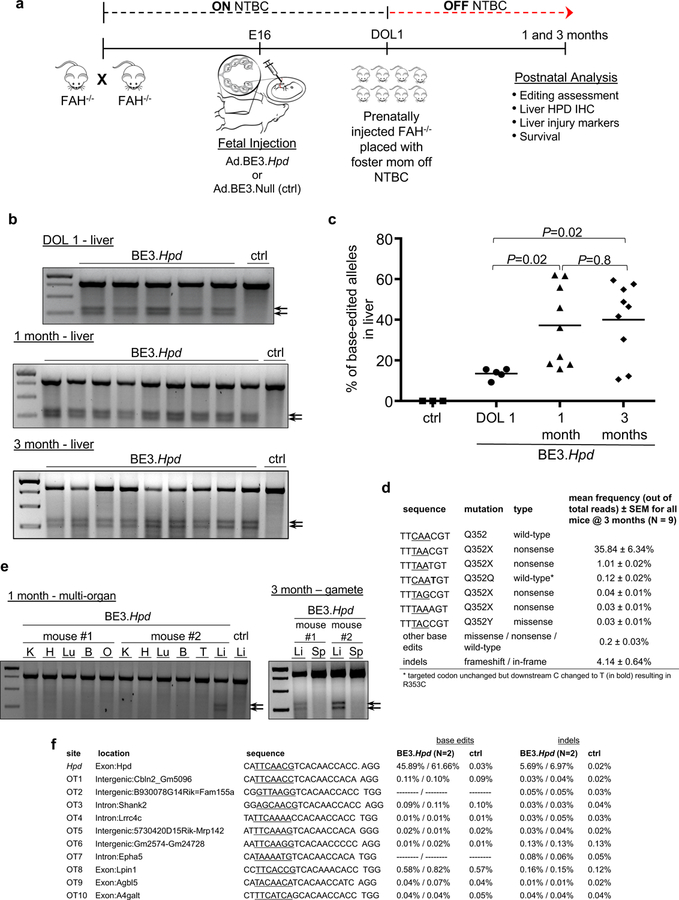
In utero base editing of Hpd in the Fah–/– mouse model. (a) Experimental scheme. (b) Surveyor assays to assess Hpd base editing, liver genomic DNA at DOL1, 1 month, and 3 months of age (Ad.BE3.Hpd DOL1, N=5; 1 month, N=9; 3 months, N=9) or Ad.BE3.Null (ctrl, N=1). (c) The percentage of base-edited Hpd on-target alleles was assessed by NGS of liver genomic DNA in fetal recipients of Ad.BE3.Null (ctrl, N=3 at DOL1 to 2 weeks of age) and Ad.BE3.Hpd at DOL1 (N=5), 1 month (N=9), and 3 months of age (N=9). Measure of centre = mean. (d) Frequencies of base-edited and indel-bearing alleles were assessed at 1 month of age via NGS of liver genomic DNA of prenatal Ad.BE3.Hpd recipients (N=9). Underlined bases indicate the target codon. (e) Genomic DNA isolated from other organs at 1 month of age (two independent mice as shown) and sperm at 3 months of age (2 independent mice as shown) was assessed by Surveyor assays for Hpd editing following prenatal Ad.BE3.Hpd injection. – ctrl = liver DNA from Ad.BE3.Null-injected fetus. (f) NGS analysis of the Hpd on-target site and the top 10 predicted off-target sites in liver genomic DNA harvested at 1 month of age from 2 Ad.BE3.Hpd recipients and 1 Ad.BE3.Null (ctrl) recipient. BE3, base editor; HPD, hydroxyphenylpyruvate dioxygenase; NTBC, 2-(2-nitro-4-trifluoro-methylbenzyol)-1,3 cyclohexanedione, FAH, fumarylacetoacetate hydrolase; IHC, immunohistochemistry; DOL, day of life; K, kidney; H, heart; Lu, lung; B, brain; O, ovary; T, testis; L, liver; arrows, Surveyor cleavage products. Statistical analysis performed with Kruskal-Wallis test.
In utero Ad.BE3.Hpd treatment rescued the lethal phenotype in Fah–/– mice following withdrawal of NTBC at birth. In contrast to recipients of the control Ad.BE3.Null vector, all of which lost weight prior to death and did not survive beyond 21 days, Ad.BE3.Hpd recipients demonstrated appropriate weight gain with 89% survival at 3 months (Figure 4a,b). Notably, weights of Ad.BE3.Hpd-injected mice exceeded those of non-injected Fah–/– mice maintained on NTBC. Additionally, liver function of Ad.BE3.Hpd-injected Fah–/– mice and non-injected, NTBC-treated Fah–/– mice was similar at 1 and 3 months and significantly improved compared to Ad.BE3.Null recipients prior to their death (Figure 4c-e, Supplementary Figure 4). Improved survival and liver function in Ad.BE3.Hpd recipients correlated with a substantial reduction in HPD+ cells on immunohistochemistry at DOL1 (Supplementary Figure 4) and one month (Figure 4f,g). Liver histology in 1-month-old Ad.BE3.Hpd recipients revealed no significant inflammation or abnormality; in Ad.BE3.Null-injected mice, variability in nuclear size and apoptotic hepatocytes consistent with liver injury were evident (Supplementary Figure 4).
Figure 4.
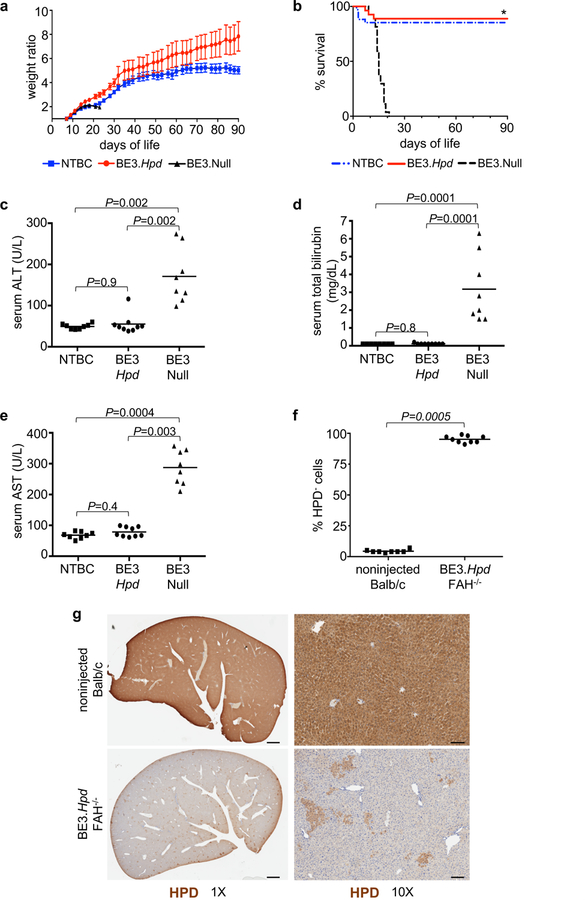
In utero Hpd base editing improves liver function and rescues the lethal phenotype of Fah–/– mice. (a, b) Fah–/– mice injected prenatally with Ad.BE3.Hpd (N=26) or Ad.BE3.Null (N=27) and taken off NTBC at DOL1 and non-injected Fah–/– mice maintained on NTBC (N=33) were serially weighed and followed for survival. Weight ratio represented as mean ± standard error of mean. Survival statistical analysis performed with log-rank test; * P=8×10−11 for BE3.Hpd vs. BE3.Null. (c-e) Plasma ALT, total bilirubin, and AST levels were assessed at 1 month of age (or just prior to death in Ad.BE3.Null-injected mice) in Fah–/– mice injected prenatally with Ad.BE3.Hpd (N=9) or Ad.BE3.Null (N=8) and taken off NTBC at DOL1 and non-injected Fah–/– mice maintained on NTBC (N=8). (f, g) Livers of Fah–/– mice prenatally injected with Ad.BE3.Hpd and taken off NTBC at DOL1 (N=9) and non-injected Balb/c mice (N=8) were assessed for HPD staining at 1 month of age. ~100,000 to 300,000 hepatocytes were assessed per sample and the percentages of HPD-negative cells determined. Scale bar = 1mm (g, left panels) and 100μm (g, right panels). Measure of centre = mean (a, c-f). ΒΕ3, base editor; HPD, hydroxyphenylpyruvate dioxygenase; NTBC, 2-(2-nitro-4-trifluoro-methylbenzyol)-1,3 cyclohexanedione; FAH, fumarylacetoacetate hydrolase; ALT, alanine aminotransferase; AST, aspartate aminotransferase. Statistical analysis performed with Kruskal-Wallis test (c-e) and two-tailed Mann-Whitney U test (f).
In summary, we established the feasibility of in utero CRISPR-mediated therapeutic editing of metabolic genes. We used Ad vectors for these initial proof-of-concept studies, recognizing that safer alternative methods such as lipid nanoparticles13,25 will need to be explored and optimized in order for translation to the clinic to occur. This notwithstanding, our work highlights the potential of in utero base editing to target a gene—either by disruption, as done here with Hpd, or potentially by directly correcting disease-causing mutations—for the purpose of treating a congenital genetic disorder that can be diagnosed early in pregnancy26. Although HT1 served as a proof-of-concept disease model to investigate in utero base editing, this approach holds greater potential for diseases that have no effective treatment for the majority of patients and result in profound morbidity and mortality shortly after birth.
ONLINE METHODS
Screening and selection of guide RNAs
gRNAs targeting the mouse Pcsk9 and Hpd genes were screened in vitro in Neuro-2a cells (N2a) for base editing activity via Surveyor assays or Sanger sequencing as previously described10–12. Specifically, the protospacer and protospacer adjacent motif (PAM) (5’-CAGGTTCCATGGGATGCTCT|GGG-3’) previously demonstrated to target the mouse Pcsk9 gene at W159 were used in the current study12. Screening and selection of the gRNAs targeting the mouse Hpd gene involved a similar protocol. pCMV-BE3 was a gift from David Liu (Addgene plasmid #73021). The mouse Hpd sequence was visually inspected, and codons that could be potentially base-edited into nonsense codons were identified. gRNAs were selected if the BE3 PAM sequence (NGG) was 13–17 nucleotides distal to the target cytosine base(s). If base editing resulted in a nonsense codon in an appreciable number of alleles (as indicated by the height of the alternative base peak on Sanger sequencing) the gRNA was designated (https://crispor.tefor.net)27. The protospacer and PAM sequences screened and corresponding target codon are listed in Supplementary Table 1. For the R26mTmG/+ mouse model, gRNAs targeting the loxP sites flanking the mT gene were selected based on their predicted high on-target efficiency and low off-target effects as determined by the online tool CRISPOR27. The protospacer and PAM used to target the loxP sites in the current studies was 5’-ATTATACGAAGTTATATTAA|GGG-3’.
Generation of adenovirus vectors
The BE3-encoding gene and synthetic polyadenylation sequence from pCMV-BE3, the CAG reporter from pCas9_GFP (Addgene plasmid #44719), and the U6 promoter-driven gRNA cassette from pGuide (Addgene plasmid #64711) with the protospacer sequence 5’-CAGGTTCCATGGGATGCTCT-3’ (for Pcsk9 studies), the protospacer sequence 5’-CATTCAACGTCACAACCACC-3’ (for the Hpd studies), or the protospacer sequence 5’-GGTGCTAGCCTTGCGTTCCG-3’ (control studies: irrelevant protospacer not matching any sequence in the mouse genome) were cloned into pDUAL-Basic expression vector. For R26mTmG/+ experiments, which used SpCas9 and not the BE3, the mTmG protospacer (5’-ATTATACGAAGTTATATTAA-3’) was cloned into plasmid pX330-U6-Chimeric_BB-CBh-hSpCas9 (a gift from Feng Zhang; Addgene plasmid # 42230). Vector Biolabs (Malvern, PA) used these constructs to generate recombinant adenovirus type 5 particles. Premade adenovirus type 5 particles containing the GFP transgene or Cre recombinase under a CMV promoter were obtained from Vector Biolabs. Ad viral vectors are referred to as Ad.BE3.Pcsk9, Ad.BE3.Hpd, Ad.BE3.Null, Ad.SpCas9.mTmG, Ad.GFP, and Ad.Cre, and the titers are indicated in Supplementary Table 2.
Generation of AAV vectors
AAV9 serotype vectors containing SpCas9 and the gRNA targeting the mTmG protospacer (as above) were generated by Vector Biolabs. This dual-AAV vector system in which two vectors are used to deliver SpCas9 and mTmG gRNA is referred to as AAV9.SpCas9.mTmG. An AAV9 serotype vector containing Cre recombinase (AAV9.Cre) was obtained from the University of Pennsylvania Vector Core.
Animals
Balb/c, C57BL/6J (called B6), B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J (called R26mTmG/+; stock #007676), and Fah1RTyrc/RJ (called FAH; stock #018129) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). FAH mice were provided as heterozygotes and subsequently bred to homozygosity (Fah–/–) in our animal facility and maintained on nitisinone (NTBC, CAS# 104206–65-7, Yecuris) in their drinking water at a concentration of 16.5 mg/L. NTBC was removed from experimental animals and continued on control animals as indicated. Animals were housed in the Laboratory Animal Facility of the Abramson Research Center and the Colket Translational Research Building at The Children’s Hospital of Philadelphia (CHOP). The experimental protocols were approved by the Institutional Animal Care and Use Committee at CHOP and followed guidelines set forth in the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals.
Genotyping
FAH mice were genotyped to confirm the Fah–/– genotype. At weaning or the time of sacrifice, 2-mm tail snips were placed in 100 μL of 1× Lysis buffer (50× Lysis buffer: 1.25M NaOH, 10 mM EDTA) and incubated at 95°C for 1 hour. 100 μL of Neutralization buffer (50× Neutralization buffer: 2M tris-HCl) was then added and samples were vortexed. Extracted DNA was amplified using primers Fah-F (5’-TCTCCCCCGCACTTAGTTTCC-3’) and Fah-R (5’-GGACTCAGATGCTGGGCTGATG-3’), and PCR products were digested with the BsrBI restriction enzyme (#R0102, New England BioLabs) according to the manufacturer’s instructions. Digested samples were run on ethidium bromide-stained 1.2% agarose gels for analysis. The mutated Fah allele lacks the BsrBI restriction enzyme site. The genotype was also confirmed with Sanger sequencing.
In utero and postnatal mouse injections
Intravenous in utero injections were performed as previously described28 (Supplementary Video 1). Fetuses of time-dated Balb/c, Fah–/–, and R26mTmG/mTmG × B6 (to generate R26mTmG/+ fetuses) mice were injected at gestational day (E) 16. Under isoflurane anesthesia and after providing local anesthetic (0.25% bupivacaine subcutaneously), a midline laparotomy was made and the uterine horn exposed. The vitelline vein, which runs along the uterine wall and enters the portal circulation resulting in first-pass effect to liver and systemic delivery via the ductus venosus, was identified under a dissecting microscope and 10 μL of virus (1×108 to 1×109 viral particles) was injected per fetus using a 100-μm beveled glass micropipette. A successful injection was confirmed by temporary clearance of the blood from the vein and absence of extravasation of the injectate. The uterus was then returned to the abdominal cavity and the laparotomy incision was closed in a single layer with 4–0 Vicryl suture.
Viral injections into adult mice (Balb/c and B6 mice for Pcsk9 studies) were performed via the retroorbital vein under isoflurane anesthesia. A total volume of 200 μL was injected such that ~4×109 viral particles were injected per mouse.
Screening Balb/c and R26mTmG/+ animal studies
E16 Balb/c fetuses were injected via the vitelline vein with 1×108 Ad.GFP particles. Livers of injected mice were assessed on DOL1 and 3 months of age by fluorescence stereomicroscopy and immunohistochemistry for GFP expression. Similarly, brain, heart, lung, and kidney of injected mice were assessed for GFP expression on DOL1.
E16 R26mTmG/+ fetuses were injected via the vitelline vein with 1×109 Ad.SpCas9.mTmG, 1×108 Ad.Cre, or 1×1011 AAV9.SpCas9.mTmG particles. Injected mice were sacrificed on DOL1 (5 days post injection) and liver, heart, lung, and brain were isolated. A portion of each organ was used to extract genomic DNA using the DNeasy Blood and Tissue Kit (QIAGEN) according to the manufacturer’s instructions, and the remainder of the organ was used to assess GFP and TdTomato expression by immunohistochemistry. Genomic DNA was assessed for editing by Sanger sequencing and by PCR analysis using primers (Supplementary Table 3) flanking the protospacer sequences within the loxP sites such that successful editing resulted in amplification of a 545-bp band while unsuccessful editing yielded a 2951-bp band.
Pcsk9 mouse studies
Prenatal experiments:
E16 Balb/c fetuses were injected with 4×108 Ad.BE3.Pcsk9 or 6×108 Ad.BE3.Null particles via the vitelline vein. Injected mice were subsequently sacrificed at DOL1, 2 weeks, 1 month, and 3 months of age. Plasma was collected at the time of sacrifice from all mice and at 1 month for mice sacrificed at 3 months of age. Prior to sacrifice and/or plasma isolation, mice were fasted for 4 hours. At the time of sacrifice, genomic DNA was isolated from a portion of liver, heart, lung, brain, kidney, spleen and ovary for analysis of gene editing by Sanger sequencing, Surveyor assays, and next-generation sequencing (NGS, see on-target and off-target mutagenesis analyses below). The remaining liver was fixed in 10% buffered formalin for hematoxylin and eosin staining for histologic analysis. Plasma was collected for analysis of PCSK9 protein, total cholesterol, and alanine transaminase (ALT) levels. Whole blood was obtained by retro-orbital bleed and spun at 10,000 rpm for 2 minutes. Plasma PCSK9 protein and total cholesterol levels were measured using the Mouse Proprotein Convertase9/PCSK9 Quantikine ELISA Kit (R&D Systems) and the Infinity Cholesterol Reagent (Thermo Fisher Scientific), respectively, according to the manufacturer’s instructions. Blood ALT levels were measured using the infinity ALT (GPT) Liquid Stable Reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Genomic DNA from maternal organs of dams whose fetuses underwent prenatal Ad.BE3.Pcsk9 injection were assessed by Surveyor assays and NGS for on-target Pcsk9 editing 1 week after injection. Specifically, DNA from heart, lung, brain, kidney, ovary, and three distinct liver samples was assessed per mother.
Postnatal experiments:
5-week-old Balb/c or B6 mice were injected with 4×109 Ad.BE3.Pcsk9 particles via the retro-orbital vein. Liver was harvested at 5 days and 3 months post injection, and genomic DNA was extracted for analysis of editing by Surveyor assays and NGS. In the subset of mice sacrificed at 3 months post injection, serum was isolated at 1 and 3 months post injection for analysis of anti-SpCas9 and anti-Ad antibody levels (see below).
Fah–/– mouse studies
An initial in vivo screening study of the Ad.BE3.Hpd viral vector was performed in time-dated Balb/c fetuses. E16 Balb/c fetuses were injected with 5×108 Ad.BE3.Hpd particles, and injected fetuses were sacrificed at 2 weeks of age. Liver was harvested for immunohistochemical analysis of HPD staining, and genomic DNA was isolated from liver, heart, brain, and lung for analysis of gene editing by Sanger sequencing, Surveyor assays, and NGS. For experiments in Fah–/– mice, Fah–/– mice maintained on NTBC were time-dated, and E16 fetuses were injected with 5×108 Ad.BE3.Hpd or 6×108 Ad.BE3.Null particles. An additional control group consisting of non-injected Fah–/– mice maintained on NTBC was included. Injected pups were fostered on DOL1 with Balb/c dams that were not maintained on NTBC, thus removing NTBC from the breast milk received by injected pups. All mice were weighed every other day beginning on DOL7 and survival was monitored daily. Liver genomic DNA was assessed for gene editing by Surveyor assays and NGS at DOL1, 1 month, and 3 months of age in recipients of Ad.BE3.Hpd and at ~DOL20 in recipients of Ad.BE3.Null (the time of clinical deterioration). In addition, genomic DNA isolated at 1 month of age from kidney, heart, lung, brain, and gonads of Ad.BE3.Hpd-injected mice was assessed for gene editing by Surveyor assays. Furthermore, genomic DNA from the sperm of Ad.BE3.Hpd-injected mice was isolated at 3 months of age for on-target editing analysis by NGS. Prior to sacrifice at 1 month of age for Ad.BE3.Hpd-injected Fah–/– mice and control Fah–/– mice on NTBC and just prior to death in the Ad.BE3.Null-injected Fah–/– mice, liver function was assessed by serum biochemical analysis. Specifically, blood was collected by retroorbital bleed, maintained on ice, and immediately centrifuged at 14,000 rpm for 15 minutes at 4°C. Total bilirubin, aspartate transaminase (AST), and ALT were measured in fresh serum samples using the Vitros 350 Chemistry Analyzer.
Histology
At the time of tissue collection, mice were euthanized by decapitation (DOL1 mice) or CO2 inhalation (all other mice). Organs were harvested and fixed in 10% buffered formalin or 4% paraformaldehyde. After serial dehydration in ascending concentrations of ethanol and xylene, organs were paraffin-embedded and sectioned. Haematoxylin and eosin staining was performed for morphologic analysis. Immunohistochemistry was performed to determine the expression of GFP, TdTomato, and the SpCas9-based BE3 using the following antibodies for immunofluorescence images: GFP (goat, Abcam, ab6673, 1:100), RFP (rabbit, Rockland, 600–401-379, 1:250), and SpCas9 (mouse, Cell Signaling Technology, 14697, 1:50). For GFP analysis by immunoperoxidase staining, slides were initially incubated with anti-GFP (rabbit, Thermo Fisher, A11122, 1:400) overnight at 4°C, washed, and then incubated at room temperature for 30 minutes with the HRP polymer in the SuperPicture Polymer Detection Kit (Invitrogen, #878963) and subsequently developed with the DAB Peroxidase (HRP) Substrate Kit (Vector Labs, #SK-4100). For HPD analysis, HPD (rabbit, St. John’s Laboratory, STJ28588, 1:1000) staining was performed on a Bond Max automated staining system (Leica Biosystems). The Bond Refine polymer staining kit (Leica Biosystems) was used. The standard protocol was followed with the exception that primary antibody incubation was extended to 1 hour at room temperature and the post-primary step was excluded. Antigen retrieval was performed with Epitope Retrieval Solution 2 BOND (Leica Biosystems) for 20 minutes. Stained slides were digitally scanned at 20× magnification on an Aperio CS-O slide scanner (Leica Biosystems).
Liver HPD quantification
Liver HPD expression was quantified by immunohistochemistry in prenatal recipients of Ad.BE3.Hpd and non-injected, age-matched Balb/c control mice. Following HPD staining and digital slide scanning, whole slide image analysis using Aperio ImageScope (Leica) was used to count the total number of cells and the number of HPD-negative cells in each section as determined through thresholding and area measurement. Between 100,000 and 300,000 cells were counted for each mouse liver sample.
Image analysis
For all histologic analyses except the quantification of HPD expression, images were taken on a Nikon Eclipse 80i fluorescence microscope. Images for confocal microscopy were taken on a Zeiss LSM 710 microscope. Fluorescence stereomicroscopy (MZ16FA; Leica, Heerburg, Switzerland) was also used to visualize GFP expression following Ad.GFP and Ad.SpCas9.mTmG injection.
Anti-SpCas9 and anti-Ad serum antibody analysis
The levels of anti-SpCas9 antibodies and presence of anti-Ad antibodies in the serum of mice injected with Ad.BE3.Pcsk9 prenatally were compared to those injected with Ad.BE3.Pcsk9 postnatally and naïve, non-injected 1-and 3-month-old Balb/c controls. Serum was isolated at 1 and 3 months after injection, and antibody levels were determined by ELISA as previously described1,29. Briefly, 96 well Nunc MaxiSorp Plates (Thermo Fisher Scientific) were coated with SpCas9 protein (PNA Bio #CP01) at 0.5 μg/well or heat-inactivated (30 minutes at 90°C) Ad viral particles (5×109 particles/well) in 1× coating buffer diluted from Coating Solution Concentrate Kit (KPL) and placed at 4°C overnight. Plates were washed with 1× Wash buffer and blocked with 1% BSA Blocking Solution (KPL) at room temperature for 1 hour. For the anti-SpCas9 studies, serum was diluted 1000-fold with 1% BSA Dilutent Solution (KPL) and added to wells for 1 hour at room temperature with shaking. The mouse monoclonal anti-SpCas9 antibody (Epigentek; clone 7A9, #A-9000–100) was serially diluted in 1% BSA Dilutent Solution and used as a standard to quantify anti-SpCas9 IgG1 levels. For the anti-Ad studies, serum was assessed at three different dilutions secondary to the lack of a standard mouse anti-Ad antibody for quantification. Thus, serum was diluted 1:100, 1:400, and 1:1600 with 1% BSA Dilutent Solution and added to wells for 1 hour at room temperature with shaking. After the 1-hour incubation, wells were washed, and 100 μL of HRP-labeled mouse IgGκ binding protein (Santa Cruz Biotechnology #sc-516102) was added to each well for an additional 1 hour at room temperature. Wells were subsequently washed 4 times and incubated with 100 μL of ABTS ELISA HRP Substrate (KPL). The SpectraMax M5 plate reader (Molecular Devices) with SoftMax Pro 6.3 software was used to measure Optical density at 410 nm.
On-target and off-target sequence analysis
On-target editing of the Hpd and Pcsk9 genes was assessed by Surveyor nuclease assays (CEL-I nuclease assays) as previously described12. Briefly, genomic DNA from the indicated organs (liver, heart, lung, brain, spleen, kidney, or gonads) was isolated using the DNeasy Blood and Tissue Kit (QIAGEN) as per the manufacturer’s instructions. For sperm DNA isolation, sperm were isolated from within the epididymis and vas deferens, suspended in warm PBS, centrifuged (14,500 rpm for 10 minutes), and the supernatant discarded. Isolation of DNA from the sperm pellet was then performed with the DNeasy Kit according to the manufacturer’s instructions with an additional incubation step (70°C for 10 minutes) prior to addition of Buffer AL. PCR amplicons (see Supplementary Table 3 for the primers used for Surveyor assays) were purified using the QIAquick PCR Purification Kit (QIAGEN), analyzed using the Surveyor Mutation Detection Kit (Integrated DNA Technologies) according to the manufacturer’s instructions, and run on ethidium bromide-stained 2.5% agarose gels. PCR amplicons of on-and off-target predicted sites for Pcsk9 and Hpd were also subjected to NGS at the Massachusetts General Hospital CCIB DNA Core (CRISPR Sequencing Service; (https://dnacore.mgh.harvard.edu/new-cgi-bin/site/pages/crispr_sequencing_main.jsp). Off-target sites were predicted using CRISPOR (https://crispor.tefor.net)27, and the top sites as ranked by the mitOfftargetScore were also subjected to NGS. Supplementary Tables 4 and 5 list the predicted Pcsk9 and Hpd off-target sites and the PCR primers used for on-target and off-target NGS analysis.
We determined on-target and off-target base editing proportions and indel proportions as previously described12 using data from next-generation sequencing performed at the Massachusetts General Hospital Center for Computational & Integrative Biology DNA Core. In brief, the Core typically obtained at least 50,000 paired-end reads for each PCR amplicon at each target site for each sample. We used custom scripts to map the processed sequencing reads, using the expected PCR amplicon sequence as the reference and discarding the reads that were not successfully mapped. To determine the base editing proportions, since the expected window for base editing spans from positions 4 to 8 in the protospacer, we used a window corresponding to positions 3 to 9 in the protospacer for each mapped read. In accordance with previously published analyses30 we discarded a read if the 4 bases proximal to the window (positions –1 to 2) and 4 bases distal to the window (positions 10 to 13) did not perfectly match the reference. In each of the remaining reads (denominator), we assessed each of the cytosine bases within the window in the reference for a change to another base; any read with at least one such change was tallied as a base-edited read (numerator).
Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR)
Organ samples stored in RNAlater TissueProtect Tubes (#76154, QIAGEN) were used for RNA preparation with the RNeasy Mini Kit (#74104, QIAGEN), according to the manufacturers’ instructions. Reverse transcription was performed after removal of contaminating genomic DNA, using SuperScript™ IV VILO™ Master Mix with ezDNase™ Enzyme (#11766050, Thermo Fisher Scientific).
Gene expression was measured using the following TaqMan Gene Expression Assay along with TaqMan Gene Expression Master Mix (Thermo Fisher Scientific): Mouse GAPD (GAPDH) Endogenous Control (VIC/MGB probe, primer limited) (#4352339E), for Gapdh as the reference gene. For the BE3 gene, the primers F: 5’-AAGCGCATACAACAAGCACA-3’ and R: 5’-GAATCAGTGTCGCGTCTAGC-3’ were used with SYBR Green (Thermo Fisher Scientific). Each 10 μL qRT-PCR reaction contained 3 μL cDNA (diluted 1:5 with water) and was performed in technical triplicate. Reactions were carried out on the QuantStudio 7 Flex System (Thermo Fisher Scientific). Relative expression levels were quantified by the 2–ΔΔCt method.
Statistics
A two-tailed Student’s t test was used for experiments involving the comparison of two groups in which data was normally distributed as determined by the D’Agostino and Pearson omnibus test of normality. The Mann-Whitney U test was used for experiments involving the comparison of two groups in which data was not normally distributed. The Kruskal-Wallis rank sum test for multiple independent samples using the Dunn method with adjustment of the P-value according to the false discovery rate procedure of Benjamini-Hochberg was used for experiments involving the comparison of more than 2 experimental groups. Survival statistics were assessed with the log-rank test. Unless otherwise indicated, data are represented as the mean ± SEM or the mean with individual values.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by grants T32-HL007843 (A.C.W.), R01-HL118744 and R01-HL126875 (K.M.) from the United States National Institute of Health (NIH); grant UL1-TR001878 from the National Center for Advancing Translational Sciences of the NIH and the Institute for Translational Medicine and Therapeutics at the University of Pennsylvania (UPenn) (W.H.P. and K.M.); grant GE-16–001-IU from the UPenn Orphan Disease Center (W.H.P.); the Winkelman Family Fund in Cardiovascular Innovation (K.M.); and generous family gifts to The Children’s Hospital of Philadelphia (CHOP). We thank A. Weilerstein and L. Ma for their help with animal care, the Translational Core Laboratory at CHOP for assistance with liver function tests, and Ms. A. Radu, the Pathology Core at CHOP and the Histology Core at the Cardiovascular Institute at UPenn for their assistance with histology.
Footnotes
ACCESSION CODES
DNA sequencing data has been deposited on the NCBI Sequence Read Archive SRP155635.
Data availability
The data that support the findings of this study are available within the paper and its supplementary information files. DNA sequencing data has been deposited on the NCBI Sequence Read Archive SRP155635.
Code availability
The custom code used to calculate the percent editing of on-target and off-target sites as determined by NGS has previously been validated12,31 and is available from the corresponding authors upon reasonable request.
Reporting Summary
Additional information on experimental design and reagents can be found in the Life Sciences Reporting Summary accompanying this manuscript.
COMPETING FINANCIAL INTERESTS
The authors have no conflicts of interest to declare.
REFERENCES
- 1.Davey MG et al. Induction of immune tolerance to foreign protein via adeno-associated viral vector gene transfer in mid-gestation fetal sheep. PLoS One 12, e0171132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sabatino DE et al. Persistent expression of hF.IX After tolerance induction by in utero or neonatal administration of AAV-1-F.IX in hemophilia B mice. Mol. Ther 15, 1677–1685 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Mingozzi F et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med 13, 419–422 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Moss RB et al. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum. Gene Ther 18, 726–732 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Endo M et al. The developmental stage determines the distribution and duration of gene expression after early intra-amniotic gene transfer using lentiviral vectors. Gene Ther 17, 61–71 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim D et al. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat. Biotechnol 35, 475–480 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Ahi YS, Bangari DS & Mittal SK Adenoviral vector immunity: its implications and circumvention strategies. Curr. Gene Ther 11, 307–320 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen JC, Boerwinkle E, Mosley TH Jr. & Hobbs HH Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med 354, 1264–1272 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Ding Q et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res 115, 488–492 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ran FA et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chadwick AC, Wang X & Musunuru K In vivo base editing of PCSK9 (proprotein convertase subtilisin/kexin type 9) as a therapeutic alternative to genome editing. Arterioscler. Thromb. Vasc. Biol 37, 1741–1747 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yin H et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat. Biotechnol 35, 1179–1187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X et al. CRISPR-Cas9 targeting of PCSK9 in human hepatocytes in vivo. Arterioscler. Thromb. Vasc. Biol 36, 783–786 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lipshutz GS, Flebbe-Rehwaldt L & Gaensler KM Reexpression following readministration of an adenoviral vector in adult mice after initial in utero adenoviral administration. Mol. Ther 2, 374–380 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Waddington SN et al. In utero gene transfer of human factor IX to fetal mice can induce postnatal tolerance of the exogenous clotting factor. Blood 101, 1359–1366 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Morrow G & Tanguay RM Biochemical and clinical aspects of hereditary tyrosinemia type 1. Adv. Exp. Med. Biol 959, 9–21 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Yin H et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol 32, 551–553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yin H et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol 34, 328–333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pankowicz FP et al. Reprogramming metabolic pathways in vivo with CRISPR/Cas9 genome editing to treat hereditary tyrosinaemia. Nat. Commun 7, 12642 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aponte JL et al. Point mutations in the murine fumarylacetoacetate hydrolase gene: animal models for the human genetic disorder hereditary tyrosinemia type 1. Proc. Natl. Acad. Sci. U. S. A 98, 641–645 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Azuma H et al. Robust expansion of human hepatocytes in Fah−/−/Rag2−/−/Il2rg−/−mice. Nat. Biotechnol 25, 903–910 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paulk NK et al. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology 51, 1200–1208 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daer RM, Cutts JP, Brafman DA & Haynes KA The impact of chromatin dynamics on Cas9-mediated genome editing in human cells. ACS Synth. Biol 6, 428–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finn JD et al. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep 22, 2227–2235 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Rafati M, Mohamadhashem F, Hoseini A, Ramandi SD & Ghaffari SR Prenatal diagnosis of tyrosinemia type 1 using next generation sequencing. Fetal Pediatr. Pathol 35, 282–285 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Haeussler M et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17, 148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boelig MM et al. The intravenous route of injection optimizes engraftment and survival in the murine model of in utero hematopoietic cell transplantation. Biol. Blood Marrow Transplant 22, 991–999 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Wang D et al. Adenovirus-mediated somatic genome editing of Pten by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum. Gene Ther 26, 432–442 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim YB et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 35, 371–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol 34, 334–338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.