

Abstract
Centriole assembly initiates when Polo-like kinase 4 (Plk4) interacts with a centriole “targeting-factor.” In Drosophila, Asterless/Asl (Cep152 in humans) fulfills the targeting role. Interestingly, Asl also regulates Plk4 levels. The N-terminus of Asl (Asl-A; amino acids 1-374) binds Plk4 and promotes Plk4 self-destruction, although it is unclear how this is achieved. Moreover, Plk4 phosphorylates the Cep152 N-terminus, but the functional consequence is unknown. Here, we show that Plk4 phosphorylates Asl and mapped 13 phospho-residues in Asl-A. Nonphosphorylatable alanine (13A) and phosphomimetic (13PM) mutants did not alter Asl function, presumably because of the dominant role of the Asl C-terminus in Plk4 stabilization and centriolar targeting. To address how Asl-A phosphorylation specifically affects Plk4 regulation, we generated Asl-A fragment phospho-mutants and expressed them in cultured Drosophila cells. Asl-A-13A stimulated kinase activity by relieving Plk4 autoinhibition. In contrast, Asl-A-13PM inhibited Plk4 activity by a novel mechanism involving autophosphorylation of Plk4’s kinase domain. Thus, Asl-A’s phosphorylation state determines which of Asl-A’s two opposing effects are exerted on Plk4. Initially, nonphosphorylated Asl binds Plk4 and stimulates its kinase activity, but after Asl is phosphorylated, a negative-feedback mechanism suppresses Plk4 activity. This dual regulatory effect by Asl-A may limit Plk4 to bursts of activity that modulate centriole duplication.
INTRODUCTION
Centrioles give rise to several physiologically important organelles including cilia, flagella, and centrosomes, and so centriole copy number must be strictly controlled in cells (Fu et al., 2015; Lattao et al., 2017). In the case of dividing cells, centriole overduplication (or “amplification”) interferes with proper spindle morphology (or orientation) and chromosome attachment, which can compromise mitotic fidelity and lead to oncogenic transformation (Ganem et al., 2009; Silkworth et al., 2009; Coelho et al., 2015; Serçin et al., 2016; Levine et al., 2017). Centriole assembly is controlled by the conserved Ser/Thr kinase Polo-like kinase 4 (Plk4; Avidor-Reiss and Gopalakrishnan, 2013; Arquint and Nigg, 2016) whose activity is tightly regulated, like other Polo family members (Zitouni et al., 2014), because an increase in Plk4 protein level (and thus activity) causes rampant centriole amplification (Kleylein-Sohn et al., 2007; Peel et al., 2007; Rodrigues-Martins et al., 2007).
Currently, Plk4 regulation is believed to occur primarily through two mechanisms: autoinhibition and self-destruction. Newly translated Plk4 is autoinhibited by its Linker 1 domain (L1), which is thought to inhibit trans-autophosphorylation of the activation loop within the nearby kinase domain, thereby suppressing kinase activity (Klebba et al., 2015a; Lopes et al., 2015). Relief of autoinhibition occurs through interactions between Plk4’s Polo box 3 domain (PB3) and SCL/TAL1 Interrupting Locus (STIL), the human homologue of the conserved SAS-5/Ana2/STIL family of centriole proteins (Arquint et al., 2015; Moyer et al., 2015). Plk4 autoinhibition can also be relieved by autophosphorylation of L1 (Klebba et al., 2015a). During most of the cell cycle, Plk4 acts as a suicide kinase, homodimerizing (perhaps in a concentration-dependent process) through its tandem PB1-PB2 domains and then trans-autophosphorylating its downstream regulatory element (DRE), inducing the recruitment of SCFSlimb ubiquitin ligase (Figure 1A; Cunha-Ferreira et al., 2009, 2013; Rogers et al., 2009; Guderian et al., 2010; Holland et al., 2010; Slevin et al., 2012; Klebba et al., 2013, 2015a; Lopes et al., 2015). Polyubiquitination of PB1 mediates Plk4 proteolysis, which is critical in suppressing centriole amplification (Klebba et al., 2015a).
FIGURE 1:

Plk4 phosphorylates the N-terminal domain of Asl. (A, B) Linear maps of Drosophila Plk4 and Asterless (Asl) showing functional and structural domains. CC, coiled-coil; DRE, downstream regulatory element (contains the SCFSlimb-binding motif); PB, Polo boxes; L1 and L2, linkers. The three regions of Asl are indicated with circled letters. Both Asl-A and Asl-C bind Plk4. (C) In vitro kinase assay of purified recombinant Plk4 (consisting of kinase domain + DRE), GST-tagged Asl regions, and γP32-ATP. Plk4 phosphorylates itself and Asl-A but not Asl-B, Asl-C, or the GST control. Coomassie-stained SDS–PAGE gels and corresponding autoradiographs (or phosphorimage) are shown. (D) Linear map of Asl-A showing phosphorylated residues identified by MS/MS from samples of purified Asl-A incubated with Plk4 in vitro (top) or immunoprecipitated Asl from lysates of S2 cells coexpressing Plk4 (bottom). (E) Lysates of S2 cells coexpressing Asl-A-V5 and either GFP, kinase-dead (KD) Plk4-GFP, or active nondegradable (ND) Plk4-GFP were resolved by 2D electrophoresis and the Western blot probed with anti-V5 antibody. Resolution in the horizontal axis is by isoelectric point. Note that expressed Asl-A exists as multiple differently charged species and that coexpression of active Plk4 causes an acidic shift for the majority of Asl-A.
In addition to STIL, the conserved coiled-coil protein Asterless (Asl) acts as a Plk4 “regulator” and, notably, first interacts with Plk4 before STIL/Ana2 is recruited to the nascent procentriole (Avidor-Reiss and Gopalakrishnan, 2013). Intriguingly, both overexpression and depletion of Asl stabilize cellular Plk4 levels (Klebba et al., 2015b). This is likely because Asl contains two spatially distinct Plk4-binding domains within its Asl-A and Asl-C regions (Figure 1B; Cizmecioglu et al., 2010; Dzhindzhev et al., 2010; Hatch et al., 2010; Klebba et al., 2015b), and, curiously, these regions display opposing effects on Plk4 levels during the cell cycle. Asl stabilizes Plk4 during mitosis when levels of the kinase peak; this activity is primarily attributed to the C-terminal Asl-C region, which is sufficient to suppress Plk4 turnover and to target the kinase to centrioles (Klebba et al., 2015b). In contrast, the N-terminal Asl-A region induces Plk4 homodimerization and autophosphorylation in interphase cells (Klebba et al., 2015b). Because Asl-A stimulates kinase activity (and thus Plk4 self-destruction), Asl-A functions as a Plk4 activator, though the mechanism of activation is unknown. Furthermore, previous studies have identified the N-terminus of Cep152 (the human homologue of Asl) as a Plk4 substrate (Hatch et al., 2010). However, the phosphoresidues in Cep152 have not been mapped, and the functional significance of Cep152 phosphorylation is unknown.
In this study, we show that phosphorylation of Asl by Plk4 is conserved in Drosophila and map 13 phosphosites in the N-terminal Asl-A region. We examine the functional significance of these phosphorylation events by replacing endogenous Asl with phosphomutant Asl-A variants in cells, and testing their impact on Plk4 binding, turnover, and activity, and ultimately, centriole duplication. Additionally, we use biochemical approaches to test whether phosphorylation of Asl-A can impact Plk4 kinase activity directly. Overall, our study reveals important insights into the complex mechanisms of Plk4 regulation, and how these mechanisms directly affect centriole assembly.
RESULTS
The Asterless N-terminus (Asl-A) is phosphorylated by Plk4 in vitro and in cells
To determine whether phosphorylation of Asl by Plk4 occurs in flies, we performed in vitro kinase assays by incubating a minimal Drosophila Plk4 construct containing the kinase domain and DRE (amino acids 1–317) with purified GST-tagged Asl regions (Asl-A, B, and C) and γ32P-ATP. In addition to labeling itself, Plk4 phosphorylated Asl-A but not Asl-B, Asl-C, or control GST (Figure 1C), consistent with a previous in vitro study (Dzhindzhev et al., 2014). To identify the phosphorylated residues, we performed tandem mass spectrometry (MS/MS) on Asl-A mixed with Plk4 and ATP, as well as a control sample containing Asl-A and ATP. We identified seven phosphorylated residues in Asl-A incubated with Plk4, none of which was phosphorylated in control Asl-A (Supplemental Table 1). In parallel, we used MS/MS analysis of full-length (FL) Asl-GFP immunoprecipitated (IPed) from lysates of fly S2 cells coexpressing catalytically active Plk4 (or kinase-dead [KD]-Plk4 as a control) to identify Asl-A residues phosphorylated in cells. Within the Asl-A region, we identified 10 phosphorylated residues when purified from cells coexpressing active Plk4 (Supplemental Table 1); total coverage of the Asl-A region was 83%. No phosphorylated peptides from the Asl-A region were recovered from cells coexpressing control KD-Plk4; although the coverage for this control was low (53%), coverage included all of the residues identified from the in vivo active-Plk4 sample. In total, we identified 13 phosphorylated residues that primarily cluster at the N- and C-termini of the Asl-A region (Figure 1D).
Asl phosphorylation in Drosophila cells was also examined with two-dimensional (2D) immunoblot analysis of lysates from S2 cells coexpressing Asl-A-V5 with either negative-control green fluorescent protein (GFP), inactive Plk4-KD-GFP, or an active nondegradable (ND) Plk4-GFP mutant. Asl-A appeared as multiple discrete spots when coexpressed with GFP or with Plk4-KD (Figure 1E, arrowheads), suggesting that it is phosphorylated to different extents when expressed in cells. Strikingly, Asl-A shifted to a more negatively charged (i.e., more acidic) species when coexpressed with active ND-Plk4, suggesting that Asl-A phosphorylation increases in the presence of active Plk4 in cells (Figure 1E). These data further support the conclusion that Plk4 phosphorylates the N-terminus of Asl, and that phosphorylation of the Asl/Cep152 N-termini by Plk4 is a conserved event in Drosophila and humans.
Within the FL Asl protein, the presence of Asl-C masks potential phenotypes arising from Asl-A phosphorylation
Some Asl-A phosphopeptides recovered from the MS/MS analyses contained multiple, closely spaced Ser/Thr residues. For example, the recovered phosphopeptides included some derived from the C-terminus of Asl-A, which contains a tight cluster of three Thr residues: T337, T338, and T339. Even though two or more of these residues were identified as phosphosites in both the in vitro and in vivo samples, the contiguity of the residues usually prevented the phosphosites from being identified with high confidence (i.e., with phosphate localization probability ≥95%; Supplemental Table 1). Given the proximity of these residues and their potential to be phosphorylated, we combined the phosphosites found in the in vitro and in vivo MS/MS data and included them in the pool of potential Plk4-targeted phosphosites in Asl-A. This approach was used to avoid overlooking functionally important sites. Thus, we made nonphosphorylatable and phosphomimetic (PM) Asl-GFP constructs by mutating all 13 identified residues to alanines (13A) or aspartates/glutamates (13PM) within the FL protein (Figure 2A).
FIGURE 2:

Asl-A phosphomutants are largely α-helical and self-oligomerize. (A) Abbreviated primary sequence of Asl-A indicating the 13 hydroxyl residues phosphorylated by Plk4 (bold) and mutated to nonphosphorylatable alanine (13A) or phosphomimetic aspartate/glutamate (13PM). Gray highlight indicates regions of predicted coiled-coil. (B) In the absence of endogenous Asl, expressed Asl-A-13A restores Plk4 self-destruction, whereas Asl-A-13PM has no effect on Plk4 protein levels. S2 cells were treated with Asl-C dsRNA for 6 d to deplete endogenous Asl without affecting transgenic Asl-A. On day 4, cells were transfected with inducible Plk4-GFP alone or with the indicated Asl-A-V5 construct and then induced to express the next day for 24 h. Immunoblots of lysates were probed with anti-GFP, V5, Asl, and α-tubulin (loading control). The graph shows the relative amounts of Plk4-GFP as determined by densitometry of the anti-GFP immunoblots, normalized to α-tubulin, and plotted relative to control (Cntrl). Error bars, SEM. n = 4 experiments. In all figures, asterisks indicate significance and error bars show SEM. (C) Asl-A mutants exhibit no significant differences of secondary structure. Circular dichroism analysis of Asl-A mutants suggests that Asl-A retains its α-helical structure even after phosphomimetic and nonphospho mutations are introduced. (D) Asl-A-WT, 13A, and 13PM constructs analyzed using SEC-MALS. Elution time from the Superdex 200 column is indicated on the x-axis in minutes. Normalized refractive index is indicated on the left y-axis (gray trace) and molecular mass is indicated on the right y-axis (black trace, kDa). Asl-A-WT elutes just after 20 min with an experimentally determined average molecular mass of 85.2 kDa, indicative of a stable dimeric state (calculated formula weights for Asl-A oligomeric states are monomer: 41.5 kDa; dimer: 83.0 kDa; trimer: 123.5 kDa; tetramer: 166.0 kDa; theoretical oligomeric masses are indicated by horizontal dashed lines). Control runs using bovine serum albumin (BSA; unpublished data) revealed that the globular BSA dimer (132 kDa) eluted at 25 min, suggesting that the Asl-A dimer, which has a slightly smaller molecular mass but elutes earlier, likely adopts an extended conformation. Asl-A-13A and 13PM constructs elute earlier than the Asl-A-WT protein and show higher experimentally determined molecular masses (110.9 and 100.6 kDa, respectively), suggesting that these mutant dimers may transiently self-associate into higher molecular weight complexes during the course of the gel filtration run. The vertical dashed line indicates the position of the elution peak for Asl-A-WT. (E) Asl-A phosphomutants retain the ability to homodimerize. S2 cells were RNAi-treated for 6 d to deplete Asl. On day 4, cells were cotransfected with the indicated GFP-tagged and V5-tagged Asl-A constructs and then induced to express the next day for 24 h. Samples were prepared by anti-GFP immunoprecipitation from cell lysates. Immunoblots were probed for GFP, V5, and α-tubulin.
Next, we examined the subcellular localizations of the wild-type (WT) and mutant Asl constructs and their effects on centriole duplication by counting centrioles in transiently transfected S2 cells immunostained for pericentrin-like protein (PLP), a centriole marker (Martinez-Campos et al., 2004). Because Asl oligomerizes (Klebba et al., 2015b; Galletta et al., 2016), we first eliminated the possible influence of endogenous Asl on the localization of transgenic Asl by targeting the Asl untranslated region with RNAi. After depletion of endogenous Asl, all of the GFP-Asl variants still localized to centrioles (Supplemental Figure 1A). Next, measurement of the centriole numbers revealed that RNAi-induced Asl depletion caused centriole loss in control cells, but expression of GFP-Asl proteins not only rescued centriole duplication but induced centriole amplification regardless of its pseudophosphorylation state (Supplemental Figure 1B). Moreover, Plk4 co-IPed with both FL Asl mutants (Supplemental Figure 1C). These results seemingly suggest that the phosphorylation state of the N-terminal Asl region does not regulate centriole assembly or Plk4 binding. However, the use of FL Asl in these assays may fail to reveal Asl-A activities because these Asl constructs contain the centriole-targeting Asl-C region that, by itself, contains a Plk4-binding domain (as does the Asl-A region) and is sufficient to induce centriole amplification when overexpressed (Klebba et al., 2015b). Therefore, the use of FL Asl in these assays risks masking phosphorylation-dependent changes of Asl-A function because the potentially dominant Asl-C region is always present.
Replacement of endogenous Asl with Asl-A phosphorylation mutants modulates Plk4 protein levels in a manner that requires kinase activity
To analyze the biological functions of Asl-A phosphorylation apart from the potentially dominant effects of Asl-C, we generated truncation constructs that express only Asl-A and transfected these into cells depleted of endogenous Asl. Notably, Asl depletion blocks centriole duplication (Dzhindzhev et al., 2010; Klebba et al., 2015b) and was not rescued by overexpression of WT or phosphomutant Asl-A proteins (unpublished data), likely because they lack the Asl-C region. Although most of these cells contained no centrioles, we could identify cells in transfected proliferating cultures that contained remnant centrioles with which to assess Asl-A localization. Unlike the FL Asl mutants that localized to centrioles, phospho-Asl-A mutant proteins were cytoplasmic and weakly localized to centrioles (Supplemental Figure S1D), as we previously reported for the Asl-A-WT fragment (Klebba et al., 2015b).
Because Asl-A uniquely facilitates Plk4 homodimerization and trans-autophosphorylation in S2 cells (Klebba et al., 2015b), we next examined the effects of 13A and 13PM Asl-A mutants on coexpressed Plk4-GFP protein levels (Figure 2B). As previously reported (Klebba et al., 2015b), Asl depletion without replacement significantly increased Plk4 levels (Figure 2B; compare lanes 1 and 2); without Asl, Plk4 accumulates because proteolysis is diminished. Notably, we observed a sevenfold increase in Plk4 levels compared with a twofold increase in the previous study (Klebba et al., 2015b), possibly due to a more efficient Asl knockdown in these experiments. Curiously, replacement with Asl-A-WT only partially reduced the Plk4 level (lane 3), whereas replacement with Asl-A-13A completely restored Plk4 to its low level (lane 4). Strikingly, the Plk4 level after replacement with Asl-A-13PM was not significantly different than the level in the endogenous Asl-depleted control (lane 5 vs. lane 2). When this experiment was repeated in S2 cells expressing KD Plk4, levels were not significantly altered by any Asl-A variant (Supplemental Figure 1E), indicating that Asl-A modulation of Plk4 levels is dependent on Plk4 kinase activity. Thus, the phosphorylation state of Asl-A regulates Plk4 levels and, because Asl-mediated regulation requires Plk4 catalytic activity, our findings suggest that nonphosphorylated Asl-A promotes Plk4 self-destruction. In contrast, Plk4 is stable in the presence of phosphorylated Asl-A. Because the cells used in these experiments were depleted of endogenous Asl and mostly lacked centrioles, it is likely that Asl-A modulation of Plk4 levels occurs in the cytoplasm and not on centrioles. Moreover, coexpression of Plk4 and Asl-A (WT or phosphomutants) did not induce the formation of cytoplasmic aggregates (Supplemental Figure 2A), a potential Plk4-stabilizing effect that we previously observed when Plk4 was coexpressed with either FL Asl or the Asl-C fragment (Klebba et al., 2015b).
The Asl-A phosphorylation state does not influence its structure, oligomerization, or ability to bind Plk4
The Asl-A phosphorylation state could regulate Plk4 self-destruction in different ways. We first explored the possibility that the introduction of 13A and 13PM mutations into Asl could disrupt the structure of the proteins and thus compromise their ability to regulate Plk4 levels. Circular dichroism (CD) analysis of purified Asl-A mutant proteins revealed that the largely alpha-helical structure of Asl-A was unaffected by the changing phosphostate of the mutants (Figure 2C). These results suggest that the 13 phosphomutations do not alter Asl-A structure.
Asl-A is known to form a homodimer that binds to two Plk4 molecules and likely facilitates Plk4 homodimerization and autophosphorylation (Klebba et al., 2015b). Possibly, Asl-A phosphorylation disrupts its ability to dimerize and, consequently, Plk4 homodimers fail to form. To test this, we used size exclusion chromatography with multiangle light scattering (SEC-MALS) to examine the oligomeric state of purified Asl-A phosphomutants. Our analysis revealed that Asl-A-WT, 13A, and 13PM oligomerize, based on measurements of their molecular mass (Figure 2D). In addition, we used IP from S2 cell lysates depleted of endogenous Asl to examine whether self-association of Asl-A is influenced by phosphorylation. Both Asl-A-13A and 13PM were able to co-IP themselves (Figure 2E), suggesting that Asl-A phosphorylation state does not affect its oligomerization.
Another possibility is that Asl-A phosphorylation blocks Plk4 binding, potentially explaining why Plk4 fails to self-destruct when coexpressed with Asl-A-13PM (Figure 2B). To test this, we performed GST-pulldown assays using purified Asl-A-His6 proteins and GST-PB1-PB2 (the Asl-binding domain in Plk4; Cizmecioglu et al., 2010; Dzhindzhev et al., 2010; Hatch et al., 2010). No differences in Plk4 binding by the Asl-A phosphomutants in vitro were observed (Supplemental Figure 2B). We also examined PB1-PB2-GFP binding to Asl-A proteins in S2 cells by using anti-GFP–coated resin to purify transgenic PB1-PB2-GFP from endogenous Asl-depleted S2 cell lysates. Again, the expressed Asl-A constructs did not differ in their association with PB1-PB2 (Supplemental Figure 2C). (The Asl-A mutants did not bind a Plk4 truncation mutant lacking PB1-PB2 [Plk4-1-381; Supplemental Figure 2D].) Finally, quantitative co-IP experiments using FL Plk4, instead of the PB1-PB2 fragment, also showed no differences in the binding of Asl-A-WT or phosphomutants (Figure 3, A and B). Thus, the phosphorylation state of Asl-A does not affect Plk4 binding.
FIGURE 3:

The phosphorylation state of Asl-A controls Plk4 activity. (A) Asl-A-13A enhances Slimb binding to Plk4, whereas Asl-A-13PM diminishes this interaction. Samples of anti-GFP immunoprecipitates were prepared from lysates of S2 cells treated as described in Figure 2B. Immunoblots were probed for GFP, V5, Slimb, and α-tubulin. (B, C) Graphs show relative amounts of Asl-A-V5 (B) or Slimb (C) bound to Plk4-GFP. Values were measured by densitometry of anti-V5 or anti-Slimb immunoblots of IP samples, normalized to the respective Plk4-GFP band intensity, and plotted relative to WT control. n = 3 experiments. ns, not significantly different. (D) Asl-A mutants modulate Plk4 autophosphorylation state. Samples of anti-GFP immunoprecipitates were prepared from lysates of S2 cells treated as described in Figure 2B except, in this experiment, cells were cotransfected with inducible Plk4-myc. Immunoblots were probed for GFP, myc, V5, and α-tubulin. Dashed lines mark Plk4-myc with different electrophoretic mobilities, indicating changes in phosphorylation state. (E) Asl-A-13PM suppresses Plk4-dependent Ana2 phosphorylation. Anti-GFP immunoprecipitates were prepared from lysates of S2 cells expressing GFP-Ana2, nondegradable Plk4-ND-myc, and the indicated Asl-V5 construct. Samples of anti-GFP immunoprecipitates were prepared from lysates of S2 cells treated as described in Figure 2B. Immunoblots were probed with anti-GFP to detect total GFP-Ana2 levels and with anti-pS318 to detect phospho-Ana2.
Phospho-null Asl-A (13A) stimulates Plk4 kinase activity, whereas phosphomimetic Asl-A (13PM) inhibits Plk4 catalytic activity
Because phosphorylation of Asl-A does not control its oligomerization or Plk4 binding, we next asked whether Asl-A’s phosphorylation state directly regulates Plk4 kinase activity. If this regulation exists, then our previous results predict that Asl-A-13A should activate Plk4 and Asl-A-13PM should inhibit. To test this, we first examined Plk4 mobility on SDS–PAGE as a readout of Plk4 autophosphorylation activity. Catalytically active Plk4 extensively autophosphorylates and normally migrates as a multiband smear; in contrast, Plk4 collapses to a single fast-migrating band if mutated to KD (Klebba et al., 2013) or when expressed in Asl-depleted cells (Figure 3D, lane 1; Klebba et al., 2015b). (These electrophoretic shifts in mobility were most obvious when Plk4 was immunoprecipitated from cell lysates.) In cells depleted of endogenous Asl, coexpression of Plk4 with Asl-A-WT partially restored Plk4 activity, producing multiple bands that included a species with intermediate mobility (lane 2). However, when coexpressed with Asl-A-13A, Plk4 shifted to a low-mobility, hyperphosphorylated form (lane 3). In contrast, coexpression with Asl-A-13PM had little to no effect on Plk4 mobility, appearing most similar to Plk4 from cells with no Asl-A replacement (lane 4). As an additional readout of kinase activity, we quantified the amount of Slimb that co-IPed with Plk4 in Asl-depleted cells because Slimb binds only autophosphorylated Plk4 (Guderian et al., 2010; Holland et al., 2010; Cunha-Ferreira et al., 2013; Klebba et al., 2013). Compared to cells coexpressing Asl-A-WT, Plk4 bound significantly more Slimb (threefold) when expressed with Asl-A-13A but bound less Slimb when expressed with Asl-A-13PM (Figure 3, A and C). Results from both experimental approaches support the conclusion that Plk4 activity is highest in cells expressing the phospho-null Asl-A-13A mutant and least with phosphomimetic Asl-A-13PM.
In addition to Plk4 itself, we examined phosphorylation of another Plk4 substrate. To do this, we used a phosphospecific antibody generated against phosphorylated S318 (pS318) in Ana2 (Supplemental Figure 2E; McLamarrah et al., 2018), a conserved residue phosphorylated by Plk4 (Dzhindzhev et al., 2014; Ohta et al., 2014; Kratz et al., 2015; Moyer et al., 2015). Western blots of GFP-Ana2 purified by IP from cell lysates were probed with the anti-pS318 antibody. This antibody recognized GFP-Ana2 when coexpressed with ND Plk4 (Figure 3E, lane 2). Surprisingly, coexpression of Asl-A-WT with ND-Plk4 decreased Ana2 phosphorylation, as indicated by the relatively diminished pS318 band observed by Western blot (lane 3). Possibly, Asl-A-WT is a preferred Plk4 substrate in cells, resulting in decreased Ana2 phosphorylation. In comparison, Asl-A-13A coexpression increased the detected pS318 (lane 4), whereas the pS318 level in cells coexpressing Asl-A-13PM was nearly undetectable, similar to the level in control cells lacking active ND-Plk4 (lanes 1 and 5).
We next performed in vitro kinase assays to address whether the phosphostate of Asl-A can modulate Plk4 catalytic activity directly. Purified FL Plk4 was mixed with each Asl-A phosphomutant and γ32P-ATP, and Plk4 kinase activity was evaluated by measuring its autophosphorylation over time (Figure 4A). For each condition, the extent of Plk4 autophosphorylation was compared with the autophosphorylation of control (Plk4 alone) at the first time point (5 min). We found that Plk4 alone is relatively inactive, presumably because it is autoinhibited (Figure 4A, lanes 1–3; Klebba et al., 2015a). In contrast, the phospho-null mutant Asl-A-13A significantly stimulated Plk4 activity (Figure 4, A, lanes 7–9, B, and C). Unlike the 13A mutant, Asl-A-13PM did not activate Plk4 (Figure 4, A, lanes 10–12, B, and C), perhaps because the phosphomimetic mutant cannot relieve Plk4 autoinhibition. However, we were surprised to observe that Asl-A-13PM failed to completely inhibit Plk4 kinase activity because our previous experiments (above) support the conclusion that Plk4 activity is suppressed by the 13PM mutant in cells. Likewise, the addition of Asl-A-WT had no significant effect on Plk4 autophosphorylation compared with the control (Figure 4, A, lanes 4–6, B, and C), possibly because Asl-A-WT relieves Plk4 autoinhibition only until Asl-A is phosphorylated and then resembles Asl-A-13PM. If Asl-A-WT phosphorylation is rapid in vitro, Plk4 autoinhibition could quickly resume and prevent substantial autophosphorylation. These in vitro results support our observations that Asl-A-13A directly activates Plk4 in cells. The lack of inhibition by Asl-A-13PM in vitro was unexpected.
FIGURE 4:

Nonphospho Asl-A (13A) stimulates Plk4 activity in vitro. (A) Purified Plk4-Flag-His6 was mixed with equimolar amounts of Asl-A and incubated with γ32P-ATP. Reactions were sampled at 5, 30, and 60 min, and the samples resolved by SDS–PAGE. Top, Coomassie-stained gel; bottom, corresponding autoradiogram. Arrows indicate Plk4 bands presumably differing by phosphorylation state; the asterisk marks a contaminating band from an added reagent (a protease inhibitor) that migrates between the Plk4 bands. (B) Graphical representation of Plk4 autophosphorylation over time. y-Axis values are measurements of radioactivity in the Plk4 bands for each sample, expressed as the fold increase of a sample measurement over the Plk4 radioactivity present in the control (Plk4-only) at the initial (5 min) time point. n = 3 except Asl-A-13PM where n = 2. SEMs smaller than the radii of the graphing points are not plotted. (C) Asl-A-13A significantly increases Plk4 activity. The average values for Plk4 phosphorylation at the last time point (60 min) from the in vitro kinase assays shown in B are graphed. ns, not significant.
Phosphomimetic Asl-A (13PM) inhibits centriole duplication
If Asl-A-13PM inhibits Plk4 activity in cells, it follows that its expression should block centriole duplication. Therefore, we overexpressed Asl-A-GFP proteins in S2 cells (containing endogenous Asl) for 3 d, immunolabeled centrioles with anti-PLP antibody, and then measured centriole numbers (Figure 5A). Expression of Asl-A-WT or 13A did not significantly alter centriole numbers compared with GFP-expressing control cells. However, Asl-A-13PM expression significantly increased the number of cells with less than two centrioles. Although Asl-A-13A stimulates Plk4 kinase activity, overexpression of Asl-A-13A by itself was not sufficient to cause significant centriole amplification (Figure 5A). Thus, Asl-A-13PM acts as a dominant negative and inhibits centriole duplication in cells. In contrast, overexpression of Asl-C has the opposite effect and induced centriole amplification (Figure 5A), as we previously reported (Klebba et al., 2015b). Therefore, we asked whether co-overexpression of Asl-C could mask the dominant-negative effect of Asl-A-13PM. We found that overexpression of Asl-C with Asl-A-13PM also showed centriole amplification, demonstrating that the presence Asl-C, either in cis (Supplemental Figure 1B) or trans (Figure 5A), masks the effects of an Asl-A variant.
FIGURE 5:

Asl-A phosphomutants control kinase activity by manipulating Plk4 autoinhibition. (A) Asl-A-13PM expression causes centriole loss. The indicated Asl-GFP constructs were transfected into S2 cells and then induced the next day to express for 72 h. Cells were immunostained for PLP to mark centrioles. Significant centriole loss (less than two centrioles) occurs in cells expressing Asl-A-13PM. In contrast, centrioles are amplified (more than two centrioles) in cells expressing Asl-C, and centrioles are similarly amplified in cells coexpressing Asl-C and Asl-A-13PM. n = 3 experiments per construct (total 300 cells/construct). (B) Coexpressed Asl-A-13PM not only blocks the centriole amplification induced by Plk4 expression but causes significant centriole loss. Plk4-GFP/Asl-A-V5 dual-gene expression plasmids were transfected into S2 cells, and then were induced the next day to express for 72 h. Cells were immunostained for PLP to mark centrioles, and centriole numbers measured. n = 3 experiments per construct (total 300 cells/construct). (C) Regulation of Plk4 by Asl-A mutants occurs independently of Ana2. S2 cells were codepleted of endogenous Asl and Ana2 for 7 d. On day 5, Plk4-GFP and the indicated Asl-A-V5 constructs were cotransfected into cells and expression was induced the next day. Immunoblots of cell lysates were probed for Ana2, GFP, V5, and α-tubulin. (D) Asl-A-WT and Asl-A-13A can activate an autoinhibited Plk4-ΔPB3 mutant. S2 cells were transfected with Plk4-ΔPB3-GFP/Asl-A-V5 dual-gene expression plasmids, and samples were prepared as in B. n = 3 experiments per construct (total 300 cells/construct). (E) A Plk4 mutant incapable of autoinhibition is inhibited by Asl-A-13PM. S2 cells were transfected with Plk4-ΔL1-GFP/Asl-A-V5 dual-gene expression plasmids, and samples were prepared as in B. n = 3 experiments per construct (total 300 cells/construct).
To test whether the effect of Asl-A-13PM on centrioles is likely due to Plk4 inhibition, we generated “dual-gene” vectors that express inducible Asl-A and Plk4 from the same plasmid, thereby ensuring that transfected cells contain both proteins (Figure 5B). In this approach, Asl-A mutants were expressed at higher than endogenous Asl levels but similar to one another (Supplemental Figure 3A). Plk4 overexpression by itself caused significant centriole amplification compared with control. However, coexpression of Plk4 with Asl-A-13PM, but not WT or 13A, not only blocked amplification but caused significant centriole loss (Figure 5B). Thus, expression of Asl-A-13PM inhibits both centriole duplication and Plk4-induced centriole amplification.
Asl-A stimulates Plk4 kinase activity by relieving L1-mediated autoinhibition
To understand how the phosphorylation state of Asl-A regulates Plk4 kinase activity, we next examined whether Asl-A influences Plk4 autoinhibition. Plk4 initially exists in an autoinhibited conformation mediated by Linker 1 (L1; Klebba et al., 2015a). Current models suggest that L1 interacts with the activation loop of the kinase domain and blocks its trans-autophosphorylation (Arquint et al., 2015; Klebba et al., 2015a; Moyer et al., 2015). Relief of autoinhibition occurs when PB3 binds the conserved centriole protein STIL, which repositions L1 so that it no longer obstructs activation loop phosphorylation (Arquint et al., 2015; Moyer et al., 2015). To test whether Asl-A requires Ana2 (the fly homologue of STIL; Goshima et al., 2007) to regulate Plk4 activity, we replaced endogenous Asl with Asl-A proteins and examined the electrophoretic mobility of Plk4 IPed from lysates of Ana2-depleted cells (Figure 5C). Notably, Ana2 depletion had no observable effect on the ability of Asl-A-WT or the phosphomutants to modulate Plk4 autophosphorylation. Thus, if Asl-A controls Plk4 autoinhibition, then it does so in an Ana2-independent manner.
Previously, we showed that Plk4 lacking PB3 (ΔPB3) is catalytically inactive because it cannot relieve autoinhibition, and its expression in cultured cells causes centriole loss (Klebba et al., 2015a). Unlike STIL/Ana2, Asl binds PB1-PB2 (Cizmecioglu et al., 2010; Dzhindzhev et al., 2010; Hatch et al., 2010), but not PB3. Therefore, we reasoned that expression of Asl-A might relieve autoinhibition in the Plk4-ΔPB3 mutant. To test this, we redesigned our dual-gene plasmid to coexpress Plk4-ΔPB3 and Asl-A proteins, and measured centriole numbers in transfected cells (Figure 5D). As expected, cells expressing only Plk4-ΔPB3 displayed increased centriole loss (less than two centrioles). Remarkably, coexpression of Plk4-ΔPB3 with Asl-A-WT or Asl-A-13A induced centriole amplification, whereas centrioles were lost after coexpression with Asl-A-13PM. The simplest interpretation of these results is that Asl-A can directly relieve Plk4 autoinhibition and activate the kinase, bypassing the need for PB3 to do so. However, this is not the case when Asl-A is phosphorylated.
Plk4 autophosphorylation of its kinase domain promotes Asl-A-13PM–mediated inhibition
The ability of Asl-A-13A to activate Plk4-ΔPB3 could be explained if the 13A mutant repositions the inhibitory L1 domain so that it no longer interferes with kinase activity, whereas 13PM cannot. Therefore, we generated a dual-gene plasmid to express Plk4 lacking L1 (ΔL1, which cannot autoinhibit) alone or with the Asl-A mutants, and then measured centriole numbers in cells, with the expectation that all Asl-A proteins would induce centriole amplification (Figure 5E). We found that expression of Plk4-ΔL1 alone or with Asl-A-WT or Asl-A-13A resulted in a significant increase in cells with more than two centrioles. Surprisingly, coexpression with Asl-A-13PM still produced significant centriole loss. These results suggest that Asl-A-13PM suppresses catalytic activity in an L1-independent manner.
How does phosphomimetic Asl-A-13PM inhibit Plk4 kinase activity if not through L1-mediated autoinhibition? Similarities with Plk1 regulation might hold a clue. Like Plk4, Plk1 is autoinhibited by its L1 (Xu et al., 2013), but Plk1 has an additional mechanism of autoinhibition: L1-independent autoinhibition occurs when its two Polo boxes bind the kinase domain, which rigidifies the hinge region of the kinase domain and inhibits catalytic activity. This autoinhibition is relieved by phosphorylation of a hinge residue in Plk1 that disrupts the interaction (Jang et al., 2002; Lowery et al., 2005; Xu et al., 2013). Similarly, Asl-A-13PM may bind Plk4’s kinase domain in a unique conformation that could, for example, block the active site.
We previously identified a residue in the fly Plk4 kinase domain (S228) that is autophosphorylated both in vitro and in vivo (Klebba et al., 2015a), but the functional consequence of this modification is unknown. The crystal structure of the human Plk4 kinase domain places this residue within an exposed loop near the kinase domain’s C-terminus (which we refer to as the “C-loop”; Figure 6A; Wong et al., 2015). Although the corresponding residue is not conserved in humans (A226 in human Plk4), in vitro autophosphorylation of a nearby C-loop residue (S232) in human Plk4 has been reported (Figure 6A; Sillibourne et al., 2010). Thus, we explored the possibility that autophosphorylation of S228 contributes to Asl-A-13PM’s inhibitory mechanism.
FIGURE 6:
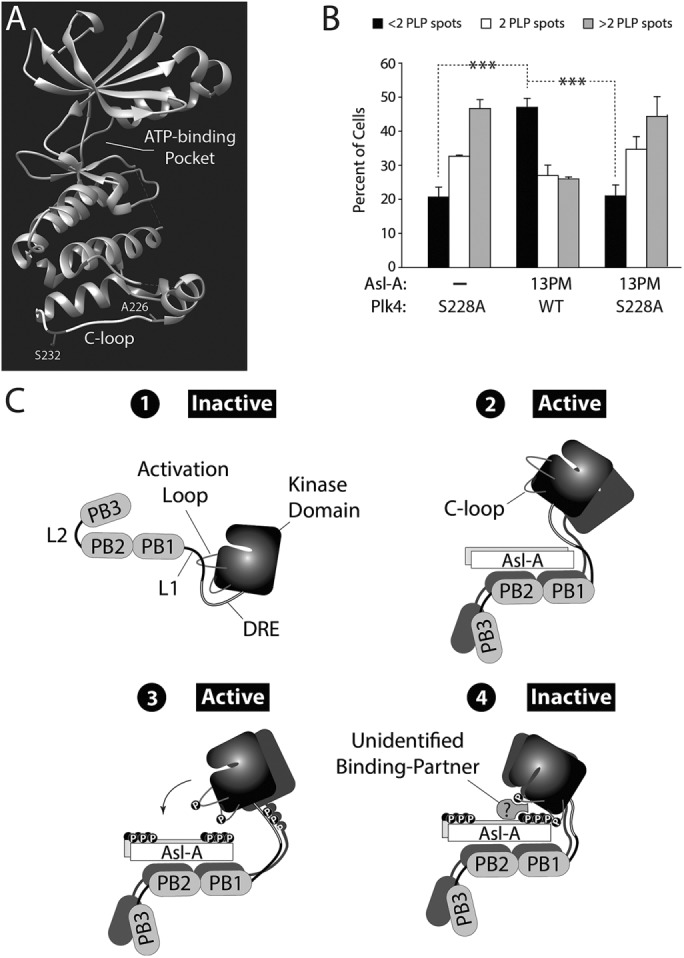
Plk4 phosphorylates its kinase domain and Asl-A, generating a state that inhibits kinase activity. (A) Atomic structure of the human Plk4 kinase domain (Wong et al., 2015). The residue, A226, is the human equivalent of Drosophila S228 within the C-loop. Human Plk4 autophosphorylates C-loop residue S232 in vitro (Sillibourne et al., 2010). (B) Asl-A-13PM does not inhibit Plk4-S228A from amplifying centrioles. Plk4-GFP/Asl-A-V5 dual-gene expression plasmids were transfected into cells and the next day were induced to express for 72 h. Centrioles were visualized with PLP immunostaining, and the number of centrioles per cell was counted. n = 3 experiments per construct (total 300 cells/construct). (C) Model: the phosphorylation state of Asl-A regulates Plk4 kinase activity by a multistep process of stimulation followed by negative feedback. 1, Initially, Plk4 is autoinhibited by its L1 domain, which masks its activation loop. 2, Nonphosphorylated Asl-A binds Plk4 and relieves autoinhibition to activate the kinase. 3, Homodimerized Plk4 phosphorylates itself (including its activation and C-loops) and, importantly, Asl-A. 4, Together, phospho-Asl-A and the phospho-C-loop recruit an unknown factor(s) to inactivate Plk4, perhaps by generating a complex that obstructs or distorts the active site.
First, we generated nonphosphorylatable S228A and phosphomimetic S228D Plk4 and determined that these S228 substitutions do not compromise kinase activity because, unlike KD-Plk4, their expression promotes centriole amplification and maintains low Plk4 protein levels in cells, similar to WT-Plk4 (Supplemental Figure 3, B and C). If phosphorylation of S228 does contribute to the inhibitory activity of Asl-A-13PM, then coexpression of Plk4-S228A should prevent this. Therefore, we altered our dual-gene plasmid to coexpress Asl-A with Plk4-S228A and measured centriole numbers in transfected cells (Figure 6B). As before, coexpression of Asl-A-13PM prevented WT-Plk4 from amplifying centrioles and caused significant centriole loss. Strikingly, however, when coexpressed with Plk4-S228A, Asl-A-13PM no longer suppressed centriole assembly and amplification ensued. Thus, our findings suggest that, in order to generate an inhibitory conformation with Asl-A, Plk4 phosphorylates not only Asl-A but its kinase domain as well.
One possible model for this inhibition is that Asl-A-13PM directly binds the kinase domain of Plk4 (containing phospho-S228) and inhibits its enzymatic activity (e.g., competitively or allosterically). Possible interactions between Asl-A mutants and mutant Plk4 fragments containing only the kinase domain and DRE (amino acids 1–317) were tested using co-IPs from lysates of cells expressing the constructs, but no such interactions were detected (Supplemental Figure 3D), demonstrating the requirement of PB1-PB2 for stable Asl-A/Plk4 binding. Additionally, we performed co-IPs to see whether the S228D mutation in FL Plk4 would result in a significant increase in Asl-A-13PM binding. Perhaps not surprisingly, we did not observe an increase in binding (Supplemental Figure 3E), presumably due to the stable interaction between Asl-A-13PM and Plk4 PB1-PB2 (Supplemental Figure S2, B and C). Taken together, our findings suggest that in order for Asl-A-13PM to inhibit Plk4 kinase activity, it requires the presence of an unidentified factor.
DISCUSSION
Plk4 kinase activity is required for centriole duplication (Holland et al., 2010), and a number of Plk4-mediated phosphorylation events that promote centriole assembly have been described (Kitagawa et al., 2009; Hatch et al., 2010; Sillibourne et al., 2010; Puklowski et al., 2011; Bahtz et al., 2012; Dzhindzhev et al., 2014; Ohta et al., 2014; Kratz et al., 2015; Galletta et al., 2016; Lee et al., 2017; McLamarrah et al., 2018). Of these, the most conserved targets include autophosphorylation of its activation loop (required for full kinase activation) and the STAN domain in STIL/Ana2 (promotes binding of the cartwheel protein, Sas6; Dzhindzhev et al., 2014; Ohta et al., 2014; Klebba et al., 2015a; Kratz et al., 2015; Lopes et al., 2015; Moyer et al., 2015). Plk4 activity also suppresses centriole duplication by down-regulating itself via autophosphorylation of its DRE (induces ubiquitin-mediated proteolysis; Guderian et al., 2010; Holland et al., 2010; Cunha-Ferreira et al., 2013; Klebba et al., 2013).
In this study, we have identified another conserved Plk4 target, namely, the Asl N-terminus; using mass spectrometry, 13 phosphoresidues of Asl-A were identified that primarily cluster at both ends of the Asl-A region. Overexpression of mutants containing phospho-null or phosphomimetic substitutions in all of these sites within the FL Asl protein had no effect on centriole duplication, Plk4 binding, or centriole localization in cultured cells. We attribute this to the presence of the Asl-C region because Asl-C contains a second Plk4-binding site as well as the centriole-targeting domain, and, most importantly, overexpression of Asl-C alone is sufficient to induce centriole amplification (Klebba et al., 2015b). The conclusion that the presence of the Asl-C region can obscure the biological effects of Asl-A phosphorylation is further supported by our finding that coexpression of Asl-A-13PM does not significantly alter centriole amplification induced by Asl-C overexpression (Figure 5A). Thus, Asl is a multifunction protein whose activities are parsed between its domains and, interestingly, the effects of the Asl-C region on centriole assembly are apparently dominant over the Asl-A region. Understanding how these two domains coordinate their functions to regulate centriole duplication is an important goal.
To determine how Asl-A modification may affect Plk4 regulation, we employed an approach that involved depleting endogenous Asl (via RNAi) and replacing it with an overexpressed Asl-A fragment. Though CRISPR/Cas9 genome editing could be used to express a mutated Asl gene at endogenous levels and has been used in Drosophila S2 cells (Bassett et al., 2014; Böttcher et al., 2014), the technique is not without its challenges because S2 cells possess a stable aneuploid genome with approximately two X chromosomes and two major autosomes, where each autosome is present in four copies (Zhang et al., 2010). Although we note that our results must be appraised carefully because of potential difficulties arising from overexpression, our replacement experimental strategy allowed us to coexpress a variety of Asl and Plk4 mutant constructs and use techniques (such as immunoprecipitation) to rapidly probe a new complex mechanism of Plk4 regulation. We also performed in vitro kinase assays and discovered that nonphospho Asl-A activates Plk4, whereas WT and phosphomimetic Asl-A do not, suggesting that the observed in vivo phenotypes are not simply artifacts of protein overexpression. Under these conditions, Asl-A-WT functions like Asl-A-13PM presumably because it is rapidly phosphorylated in vitro and then (like 13PM) can no longer relieve Plk4 autoinhibition, restoring Plk4 to its low basal rate of autophosphorylation. Surprisingly, in contrast to our in vivo observations, Asl-A-13PM did not inhibit Plk4 activity compared with Plk4 alone. Thus, we propose that phospho Asl-A requires an additional, unidentified factor to inhibit Plk4, and that the factor is probably not Ana2 (Figure 5C).
This study revealed a functional significance of Asl phosphorylation by Plk4, and we summarize our major findings in the following model that diagrams a new mechanism regulating Plk4 kinase activity (Figure 6C). We propose that Asl-A binds PB1-PB2 in Plk4 and initially relieves autoinhibition of inactive Plk4 (stage 1) by repositioning L1 in a PB3-independent manner (stage 2). In turn, Plk4 trans-autophosphorylates its activation loop and residue S228 of its C-loop, in addition to extensively phosphorylating the N- and C-termini of Asl-A (stage 3). Phosphorylation of Asl-A and Plk4’s C-loop result in Plk4 inactivation, perhaps after the recruitment of an unidentified protein to form an inhibitory complex (stage 4). Our model of this novel inhibitory mechanism resembles the “product inhibition” commonly observed in metabolic pathways (Frieden and Walter, 1963), because Plk4 phosphorylates Asl-A to generate a modified product that then suppresses kinase activity. Kinase inhibition could occur in a variety of ways; stage 4 of our model depicts only one possibility, where the active site is obstructed and competitively inhibited. Future studies of the molecular structures of the activating and inhibitory complexes would be very informative.
Asl-mediated activation of Plk4 followed immediately by negative feedback to inhibit the kinase could function to ensure that Plk4 kinase activity is limited to brief bursts when encountering Asl. How then could Asl-A–mediated activation/inactivation of Plk4 participate in the centriole duplication cycle? We propose that the Asl-A mechanism described here likely occurs in the cytoplasm, facilitating Plk4 destruction to suppress rampant centriole amplification in interphase cells. Specifically, Plk4 activity would be confined to just the Asl-Plk4 complex, ensuring that 1) autophosphorylation of the DRE triggers Plk4 ubiquitination and 2) phosphorylated Asl-A, perhaps with an unidentified binding partner, holds the kinase in an inactivate state to prevent unwanted centriole assembly while Plk4 awaits degradation. If true, then Asl is the primary facilitator of interphase Plk4 degradation in Drosophila cells. Supporting this idea is the finding that Asl RNAi increases Plk4 protein levels in interphase cells (Klebba et al., 2015b). Moreover, the stabilized Plk4 in Asl-depleted cells migrates as a fast-migrating single polypeptide on SDS–PAGE (similar to KD Plk4; Klebba et al., 2015a), suggesting that, without Asl, Plk4 accumulates in an autoinhibited, nonphosphorylated state. Interestingly, a similar phenotype is observed in human cells depleted of STIL (Moyer et al., 2015), suggesting that STIL replaced Asl/Cep152 as the predominant interphase Plk4 activator during human evolution.
Although Asl-C overexpression by itself induces centriole amplification, we predict that under physiologic conditions the Asl-A and Asl-C domains coordinate their respective Plk4 regulatory functions to ensure centriole duplication occurs only once per cell cycle. During mitotic exit when procentriole assembly is initiated (Dzhindzhev et al., 2014), the Asl-C domain likely stabilizes Plk4 at the centriole (Klebba et al., 2015b). Plk4 activation/deactivation mediated by Asl-A then could ensure that Plk4 activity is restricted both spatially and temporally at the parent centriole surface. Like the Asl-A region, STIL stimulates Plk4 kinase activity, probably by also relieving autoinhibition (Arquint et al., 2015; Ohta et al., 2018). Thus, Asl is the second Plk4 activator identified to date. Notably, Plk4 is predicted to be regulated by Asl before Ana2/STIL based on the hierarchal recruitment of the two regulators to procentrioles (Fu et al., 2016). In this regard, Asl-A–mediated activation of Plk4 may play a role in STIL recruitment, as Plk4 kinase activity is required to maintain STIL on centrioles (Moyer et al., 2015). In this scenario, Asl could direct Plk4 activity to an initial set of essential centriole substrates before handing off Plk4 to STIL/Ana2, which, after binding and stimulating Plk4, facilitates Sas6 recruitment (Dzhindzhev et al., 2014; Ohta et al., 2014; Kratz et al., 2015; Moyer et al., 2015). Additionally, a recent study has shown that Plk4 controls the rate of centriole growth during early fly embryogenesis, and that oscillations in Plk4 activity during different cell cycle phases are likely required to establish proper centriole length (Aydogan et al., 2018). Our finding that Asl-A can both stimulate and inhibit Plk4 activity suggests that Asl may be a primary regulator of these oscillations in Plk4 activity, thus ensuring that centrioles reach their proper size during each cell cycle. Future studies are necessary to determine when this new mechanism of Plk4 regulation occurs during the centrosome cycle and whether additional Plk4 “activators” exist, as they may be necessary to stimulate additional centriole assembly steps.
MATERIALS AND METHODS
Drosophila cell culture
Drosophila S2 cell culture was performed as previously described (Rogers and Rogers, 2008). S2 cells (Life Technologies; catalogue no. R69007) were cultured at room temperature (20–25°C) in Sf900II SFM medium (Life Technologies; catalogue no. 10902104).
Double-stranded RNA (dsRNA) interference
dsRNA interference (RNAi) was performed as previously described (Rogers and Rogers, 2008). Briefly, cells were cultured in six-well plates at 50–90% confluency in 1 ml of medium. Cells were treated with 5–10 μg dsRNA every day for 5–7 d. Control dsRNA was synthesized from noncoding DNA in a pET28a vector template (Clontech) using the primers 5′-ATCAG GCGCTCTTCCGC and 5′-GTTCGTGCACACAGCCC. (All primers used for dsRNA synthesis begin with the T7 promoter sequence 5′-TAATACGACTCACTATAGGG, followed by template-specific sequence.) DNA template for Asl dsRNA (which targeted coding sequence not present in the Asl-A coding sequence) was generated using the primers 5′-CGTCTGATCC ATCGCCC-3′ and 5′-CATCGCCTCTTCGTGGG-3′. The DNA template for Ana2 dsRNA was generated using the primers 5′-TAATACGACTCACGCTCTGGTATCCC-3′ and 5′-TAATAC GACTCACGTTGCTCCTCGGG-3′ to amplify a region of coding sequence from Ana2 cDNA. Immunoblots confirmed the depletion of endogenous Asl or Ana2 (Figures 2B and 5C, respectively).
Immunofluorescence microscopy
S2 cells were fixed and processed as previously described (Rogers and Rogers, 2008) by spreading cells on concanavalin A–coated glass bottom dishes and fixing with 10% formaldehyde for 10 min. Primary antibodies were diluted to concentrations ranging from 1 to 20 μg/ml and included rabbit anti-PLP (our laboratory) and guinea pig anti-Asl (our laboratory). Secondary antibodies (conjugated with Cy5 or Rhodamine red-X [Jackson ImmunoResearch Laboratories]) were used at a dilution of 1:1500. To stain DNA, Hoechst 33342 (Life Technologies; catalogue no. H3570) was used at a final concentration of 3.2 μM. Cells were mounted in phosphate-buffered saline (PBS) containing 0.1 M n-propyl galate, 90% (by volume) glycerol. Specimens were imaged using a DeltaVision Core system (Applied Precision) equipped with an Olympus IX71 microscope, a 100× objective (NA 1.4) and a cooled charge-coupled device camera (CoolSNAP HQ2; Photometrics). Images were acquired with softWoRx v1.2 software (Applied Science).
Immunoblotting
S2 cell extracts were produced by lysing cells in cold PBS containing 0.1% Triton X-100. Laemmli sample buffer was then added and the samples were boiled for 5 min. Samples of equal total protein were resolved by SDS–PAGE, blotted, probed with primary and secondary antibodies, and scanned on an Odyssey imager (Li-Cor Biosciences). Care was taken to avoid saturating the scans of blots. Antibodies used for Western blotting include guinea pig anti-Asl (our laboratory), rat anti-Asl (our laboratory), rabbit anti-Ana2 (our laboratory), mouse anti-GFP monoclonal JL-8 (Clontech; catalogue no. 632380), mouse anti-V5 monoclonal (Life Technologies; catalogue no. R960-25), mouse anti-myc (Cell Signaling Technology; catalogue no. 2276S), and mouse anti-a tubulin (Sigma-Aldrich; catalogue no. T9026) at dilutions ranging from 1:1000 to 1:3000. IRDye 800CW secondary antibodies (Li-Cor Biosciences) were prepared according to manufacturer’s instructions and used at 1:3000 dilution.
To generate anti-phosphospecific S318 Ana2 antibody, rat polyclonal antibodies were raised against the phosphopeptide: AKPNTEK{pSer}MVMNELAC. A nonphosphopeptide with the sequence AKPNTEKSMVMNELAC was also generated (Pocono Rabbit Farm, PA). Antibodies were affinity purified from antisera using phosphopeptide coupled to Affi-Gel. The anti-phosphopeptide solution was concentrated using 10k Ultrafree concentrators (Millipore). Antibodies were used at a 1:500 dilution.
For 2D gel electrophoresis, S2 cell extracts were produced by lysing cells in sample rehydration buffer (8 M urea, 33 mM CHAPS, 0.5% Zoom carrier ampholytes [Novex; catalogue no. ZM0022], 20 mM dithiothreitol [DTT], 0.002% bromophenol blue) and clarified by centrifugation at 16,100 × g for 10 min. Protein (200 μg) was loaded onto a polyacrylamide gel strip (Novex) by soaking the sample with the strip for 2 h at room temperature. Isoelectric focusing was performed at 175 V for 15 min followed by 175–200 V ramp for 45 min and 2000 V for 105 min. The strips were then soaked in 1× lithium dodecyl sulfate (LDS) sample buffer with 50 mM DTT for 15 min and resolved by SDS–PAGE. Western blots were performed as described above.
Constructs and transfection
FL cDNAs of Drosophila Asl, Plk4, and Ana2 were subcloned into a pMT vector containing in-frame coding sequences for EGFP, V5, or myc under control of the inducible metallothionein promoter. Mutants of Plk4 and Asl were generated by PCR-based site directed mutagenesis with Phusion polymerase (ThermoFisher; catalogue no. F530S). For transient transfections, (2–5) × 106 S2 cells were pelleted by centrifugation and resuspended in 100 μl of transfection solution (5 mM KCl, 15 mM MgCl2, 120 mM sodium phosphate, 50 mM d-mannitol, pH 7.2) containing 0.2–2 μg of purified plasmid. The resuspension was then transferred to a 2-mm gap cuvette and electroporated using a Nucleofector 2b (Lonza), program G-030. Transfected cells were immediately diluted in 1 ml of SF-900 II medium and placed in a six-well tissue culture plate. Cells were typically allowed to recover for ∼24 h before inducing by the addition of 0.5–2 mM CuSO4 to the culture medium.
Immunoprecipitation assays
GFP-binding protein (GBP; Rothbauer et al., 2008) was fused to the Fc domain of human IgG (pIg-Tail; R&D Systems), tagged with His6 in pET28a (EMD Biosciences), expressed in Escherichia coli, and purified on HisPur resin (ThermoFisher; catalogue no. 88221) as described previously (Buster et al., 2013). Purified GBP was bound to magnetic Dyna Beads (ThermoFisher; catalogue no. 10001D), and then cross-linked to the resin by incubating with 20 mM dimethyl pimelimidate dihydrochloride in PBS, pH 8.3, 2 h at 22°C, then quenched by incubation with 0.2 M ethanolamine, pH 8.3, 1 h at 22°C. Antibody-coated beads were washed three times with PBS-Tween20 (0.02%), then equilibrated in 1.0 ml of cell lysis buffer (CLB; 50 mM Tris, pH 7.2, 125 mM NaCl, 2 mM DTT, 0.1% Triton X-100, and 0.1 mM phenylmethylsulfonyl fluoride [PMSF]). Transfected cells expressing recombinant proteins were lysed in CLB, and the lysates clarified by centrifugation at 16,100 × g for 5 min at 4°C. Inputs (0.5–1%) were used for immunoblots. GBP-coated beads were rocked with lysate for 30 min at 4°C, washed three times with 1 ml CLB, and then boiled in Laemmli sample buffer.
Circular dichroism
Asl-A WT, 13A, and 13PM CD spectra were collected at 20 and 94°C using a Chirascan-plus CD spectrometer (Applied Photophysics). Samples were diluted to 0.1 mg/ml in CD buffer (10 mM sodium phosphate, pH 7.4, 50 mM sodium fluoride) and injected into a 1-mm–path length cuvette. CD spectra were acquired from 260 to 185 nm with a step size of 0.5 nm every 1.25 s. A buffer CD spectrum was subtracted from each Asl-A spectrum and the data were smoothed using Chirascan-plus software.
Size exclusion chromatography–multiangle light scattering
Size exclusion chromatography–multiangle light scattering (SEC-MALS) was performed as previously described (Klebba et al., 2015b). Briefly, Asl mutants (50 μM, 100 μl) were injected onto a Superdex 200 10/30 GL size exclusion column (GE Healthcare Life Sciences) at 0.5 ml/min in buffer (25 mM HEPES, pH 7.5, 300 mM NaCl, 0.1% β-mercaptoethanol, and 0.2 g/l sodium azide). Eluent was analyzed by a tandem RI detector/Dawn Heleos II multiangle static light scattering (MALS) detector (Wyatt Technology). Light scattering and refractive index data were used to calculate the weight-averaged molar mass using Wyatt Astra V software (Wyatt Technology).
In vitro binding assay
Plk4 PB1-PB2 (amino acids 382–602) was subcloned into pGEX-6p2 plasmid (GE Healthcare Life Sciences) to generate a GST-PB1-PB2 construct. Asl-A (WT, 13A, or 13PM; amino acids 1–374) was subcloned into pET28a (Life Technologies) to generate Asl-A-His6. Proteins were bacterially expressed and purified from lysates using either glutathione or HisPur resins according to manufacturer’s instructions. For each construct, optimal elution fractions (determined after SDS–PAGE analysis) were pooled, concentrated using centrifugal filters (Ultracel, Amicon), and exchanged into binding buffer (40 mM HEPES, pH 7.3, 150 mM NaCl, 5 mM MgCl2, 0.5 mM MnCl2, 1 mM DTT). Purified GST-PB1-PB2 was rebound to glutathione resin, washed, and the quantity of protein bound to the resin was determined from the difference in the quantities of soluble GST-PB1-PB2 present before and after rebinding. To assess Asl-A binding to PB1-PB2, different quantities of the GST-PB1-PB2 beads were mixed with a constant quantity of purified, soluble Asl-A-His6 (WT, 13A, or 13PM) in order to test a range of molar ratios of Asl-A to PB1-PB2 (on beads). Each assay also contained an appropriate quantity of purified GST bound to glutathione beads to keep the total molar quantity of GST and volume of beads the same in each assay. Mixtures were gently agitated for 30 min at 23°C, after which samples of the supernatant (S) and twice-washed resin (P) were resolved by SDS–PAGE.
Integrated intensities of the Coomassie-stained Asl-A bands in the gels were measured using ImageJ (National Institutes of Health [NIH]) software.
In vitro kinase assays
Bacterially expressed constructs of Drosophila Plk4 (amino acids 1–317 and FL) C-terminally tagged with FLAG-His6 and Drosophila Asl fragments A (amino acids 1–374), B (amino acids 375–630), and C (amino acids 631–994) N-terminally tagged with GST (in the pGEX-6p2 vector; GE Healthcare) were prepared as described above for in vitro binding assays. For in vitro phosphorylation assays, purified proteins (Plk4 and Asl; both were 0.25 μM in Figure 4, but were 10 μM when preparing samples for tandem mass spectrometry analysis) were incubated with 100 μM ATP for 1 h at 25°C in reaction buffer (40 mM Na HEPES, pH 7.3, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 10% [by volume] glycerol). Samples were resolved by SDS–PAGE, and proteins visualized by Coomassie staining. Phosphorylation of protein substrates was evaluated by including γ-32P-ATP in assays and, subsequently, the presence of radiolabeled substrates detected by autoradiography or phosphorimaging of dried gels. Phosphorylated residues within proteins were identified by tandem mass spectrometry (Supplemental Table S1) of purified bacterially expressed proteins phosphorylated in vitro (described above) in the presence of nonradioactive ATP.
Mass spectrometry
Samples of Asl were resolved by SDS–PAGE and Coomassie-stained. Appropriate bands were cut from gels, destained (50% acetonitrile, 25 mM ammonium bicarbonate), reduced (10 μM DTT, 55°C, 1 h), alkylated (55 mM iodoacetamide, 23°C, 45 min), dehydrated (100% acetonitrile), and then digested in-gel for 12 h with ∼1.5 ng/μl of either trypsin (37°C) or chymotrypsin (23°C). Peptides were extracted with 50% acetonitrile, 1% formic acid, dried by speed-vac (with lowest heat setting), and stored at 4°C. Mass spectrometry was performed at the NHLBI Proteomics Core Facility (NIH). Desalted peptide samples were separated on a 10-cm Picofrit Biobasic C18 analytical column (New Objective, Woburn, MA) using a 90-min linear gradient of 5–35% acetonitrile/water containing 0.1% formic acid at a flow rate of 250 nl/min, ionized by electrospray ionization (ESI) in positive mode, and analyzed on a LTQ Orbitrap Velos mass spectrometer. All LC-MS analyses were carried out in “data-dependent” mode in which the top six most intense precursor ions detected in the MS1 precursor scan (m/z 300–2000) were selected for fragmentation via collision-induced dissociation (CID). Precursor ions were measured in the Orbitrap at a resolution of 60,000 (m/z 400) and all fragment ions were measured in the ion trap. Protein sequences were matched to spectra using Mascot software (Matrix Science), and the corresponding Mascot values for peptide identification are shown (Supplemental Table S1). Ascores, phosphate localization probabilities, and spectral counts were obtained from ScaffoldPTM 3.1 (Proteome Software).
Statistics
Means of measurements were analyzed for significant differences by one-way analysis of variance followed by Tukey’s posttest (to evaluate differences between treatment pairs) using Prism 6 (GraphPad) software. Means are taken to be significantly different if P < 0.05. P values shown for pairwise comparisons of Tukey’s posttest are adjusted for multiplicity. In figures, * indicates 0.05 > P ≥ 0.01; ** indicates 0.01 > P ≥ 0.001; *** indicates 0.001 > P; and ns indicates P ≥ 0.05 for the indicated pairwise comparison. Error bars in all figures indicate SEM.
Supplementary Material
Acknowledgments
G.C.R. is grateful for support from National Cancer Institute P30CA23074, National Institute of General Medical Sciences R01 GM110166 and GM126035, National Science Foundation MCB-1158151, and the Phoenix Friends of the University of Arizona Cancer Center.
Abbreviations used:
- Ana2
anastral spindle 2
- Asl
Asterless
- CD
circular dichroism
- Cep152
centrosomal protein of 152 kDa
- DRE
downstream regulatory element
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- IP
immunoprecipitation
- KD
kinase-dead
- L1
linker 1
- MS/MS
tandem mass spectrometry
- ND
nondegradable
- PB
Polo box
- Plk1
Polo-like kinase 1
- Plk4
Polo-like kinase 4
- PLP
pericentrin-like protein
- PM
phosphomimetic
- RNAi
RNA interference
- S2
Schneider 2
- SCF
Skp1-Cullin-F-box
- SEC-MALS
size exclusion chromatography multiangle light scattering
- STAN
STil/ANa2 domain
- STIL
SCL/TAL1 interrupting locus protein.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-07-0445) on September 26, 2018.
REFERENCES
- Arquint C, Gabryjonczyk AM, Imseng S, Böhm R, Sauer E, Hiller S, Nigg EA, Maier T. (2015). STIL binding to Polo-box 3 of PLK4 regulates centriole duplication. Elife , e07888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arquint C, Nigg EA. (2016). The PLK4-STIL-SAS-6 module at the core of centriole duplication. Biochem Soc Trans , 1253–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avidor-Reiss T, Gopalakrishnan J. (2013). Building a centriole. Curr Opin Cell Biol , 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydogan MG, Wainman A, Saurya S, Steinacker TL, Caballe A, Novak ZA, Baumbach J, Muschalik N, Raff JW. (2018). A homeostatic clock sets daughter centriole size in flies. J Cell Biol , 1233–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahtz R, Seidler J, Arnold M, Haselmann-Weiss U, Antony C, Lehmann WD, Hoffmann I. (2012). GCP6 is a substrate of Plk4 and required for centriole duplication. J Cell Sci , 486–496. [DOI] [PubMed] [Google Scholar]
- Bassett AR, Tibbit C, Ponting CP, Liu JL. (2014). Mutagenesis and homologous recombination in Drosophila cell lines using CRISPR/Cas9. Biol Open , 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher R, Hollmann M, Merk K, Nitschko V, Obermaier C, Philippou-Massier J, Wieland I, Gaul U, Förstemann K. (2014). Efficient chromosomal gene modification with CRISPR/cas9 and PCR-based homologous recombination donors in cultured Drosophila cells. Nucleic Acids Res , e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buster DW, Daniel SG, Nguyen HQ, Windler SL, Skwarek LC, Peterson M, Roberts M, Meserve JH, Hartl T, Klebba JE, et al. (2013). SCFSlimb ubiquitin ligase suppresses condensin II-mediated nuclear reorganization by degrading Cap-H2. J Cell Biol , 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizmecioglu O, Arnold M, Bahtz R, Settele F, Ehret L, Haselmann-Weiss U, Antony C, Hoffmann I. (2010). Cep152 acts as a scaffold for recruitment of Plk4 and CPAP to the centrosome. J Cell Biol , 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho PA, Bury L, Shahbazi MN, Liakath-Ali K, Tate PH, Wormald S, Hindley CJ, Huch M, Archer J, Skarnes WC, et al. (2015). Over-expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol , 150209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha-Ferreira I, Bento I, Pimenta-Marques A, Jana SC, Lince-Faria M, Duarte P, Borrego-Pinto J, Gilberto S, Amado T, Brito D, et al. (2013). Regulation of autophosphorylation controls PLK4 self-destruction and centriole number. Curr Biol , 2245–2254. [DOI] [PubMed] [Google Scholar]
- Cunha-Ferreira I, Rodrigues-Martins A, Bento I, Riparbelli M, Zhang W, Laue E, Callaini G, Glover DM, Bettencourt-Dias M. (2009). The SCF/Slimb ubiquitin ligase limits centrosome amplification through degradation of SAK/PLK4. Curr Biol , 43–49. [DOI] [PubMed] [Google Scholar]
- Dzhindzhev NS, Tzolovsky G, Lipinszki Z, Schneider S, Lattao R, Fu J, Debski J, Dadlez M, Glover DM. (2014). Plk4 phosphorylates Ana2 to trigger Sas6 recruitment and procentriole formation. Curr Biol , 2526–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhindzhev NS, Yu QD, Weiskopf K, Tzolovsky G, Cunha-Ferreira I, Riparbelli M, Rodrigues-Martins A, Bettencourt-Dias M, Callaini G, Glover DM. (2010). Asterless is a scaffold for the onset of centriole assembly. Nature , 714–718. [DOI] [PubMed] [Google Scholar]
- Frieden E, Walter C. (1963). Prevalence and significance of the product inhibition of enzymes. Nature , 834–837. [DOI] [PubMed] [Google Scholar]
- Fu J, Hagan IM, Glover DM. (2015). The centrosome and its duplication cycle. Cold Spring Harb Perspect Biol , a015800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Lipinszki Z, Rangone H, Min M, Mykura C, Chao-Chu J, Schneider S, Dzhindzhev NS, Gottardo M, Riparbelli MG, et al. (2016). Conserved molecular interactions in centriole-to-centrosome conversion. Nat Cell Biol , 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galletta BJ, Fagerstrom CJ, Schoborg TA, McLamarrah TA, Ryniawec JM, Buster DW, Slep KC, Rogers GC, Rusan NM. (2016). A centrosome interactome provides insight into organelle assembly and reveals a non-duplication role for Plk4. Nat Commun , 12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D. (2009). A mechanism linking extra centrosomes to chromosomal instability. Nature , 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshima G, Wollman R, Goodwin SS, Zhang N, Scholey JM, Vale RD, Stuurman N. (2007). Genes required for mitotic spindle assembly in Drosophila S2 cells. Science , 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guderian G, Westendorf J, Uldschmid A, Nigg EA. (2010). Plk4 trans-autophosphorylation regulates centriole number by controlling β-TrCP-mediated degradation. J Cell Sci , 2163–2169. [DOI] [PubMed] [Google Scholar]
- Hatch EM, Kulukian A, Holland AJ, Cleveland DW, Stearns T. (2010). Cep152 interacts with Plk4 and is required for centriole duplication. J Cell Biol , 721–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, Lan W, Niessen S, Hoover H, Cleveland DW. (2010). Polo-like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J Cell Biol , 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YJ, Ma S, Terada Y, Erikson RL. (2002). Phosphorylation of threonine 210 and the role of serine 137 in the regulation of mammalian polo-like kinase. J Biol Chem , 44115–44120. [DOI] [PubMed] [Google Scholar]
- Kitagawa D, Busso C, Flückiger I, Gönczy P. (2009). Phosphorylation of SAS-6 by ZYG-1 is critical for centriole formation in C. elegans embryos. Dev Cell , 900–907. [DOI] [PubMed] [Google Scholar]
- Klebba JE, Buster DW, McLamarrah TA, Rusan NM, Rogers GC. (2015a). Autoinhibition and relief mechanism for Polo-like kinase 4. Proc Natl Acad Sci USA , E657–E666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebba JE, Buster DW, Nguyen AL, Swatkoski S, Gucek M, Rusan NM, Rogers GC. (2013). Polo-like kinase 4 autodestructs by generating its own Slimb-binding phosphodegron. Curr Biol , 2255–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebba JE, Galletta BJ, Nye J, Plevock KM, Buster DW, Hollingsworth NA, Slep KC, Rusan NM, Rogers GC. (2015b). Two Polo-like kinase 4 binding domains in Asterless perform distinct roles in regulating kinase stability. J Cell Biol , 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof YD, Nigg EA. (2007). Plk4-induced centriole biogenesis in human cells. Dev Cell , 190–202. [DOI] [PubMed] [Google Scholar]
- Kratz AS, Bärenz F, Richter KT, Hoffmann I. (2015). Plk4-dependent phosphorylation of STIL is required for centriole duplication. Biol Open , 370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattao R, Kovács L, Glover DM. (2017). The centrioles, centrosomes, basal bodies and cilia of Drosophila melanogaster. Genetics , 33–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Seo MY, Chang J, Hwang DS, Rhee K. (2017). PLK4 phosphorylation of CP110 is required for efficient centriole assembly. Cell Cycle , 1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MS, Bakker B, Boeckx B, Moyett J, Lu J, Vitre B, Spierings DC, Lansdorp PM, Cleveland DW, Lambrechts D, et al. (2017). Centrosome amplification is sufficient to promote spontaneous tumorigenesis in mammals. Dev Cell , 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes CA, Jana SC, Cunha-Ferreira I, Zitouni S, Bento I, Duarte P, Gilberto S, Freixo F, Guerrero A, Francia M, et al. (2015). PLK4 trans-autoactivation controls centriole biogenesis in space. Dev Cell , 222–235. [DOI] [PubMed] [Google Scholar]
- Lowery DM, Lim D, Yaffe MB. (2005). Structure and function of Polo-like kinases. Oncogene , 248–259. [DOI] [PubMed] [Google Scholar]
- Martinez-Campos M, Basto R, Baker J, Kernan M, Raff JW. (2004). The Drosophila pericentrin-like protein is essential for cilia/flagella function, but appears to be dispensable for mitosis. J Cell Biol , 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLamarrah TA, Buster DW, Galletta BJ, Boese CJ, Ryniawec JM, Hollingsworth NA, Byrnes AE, Brownlee CW, Slep KC, Rusan NM, et al. (2018). An ordered pattern of Ana2 phosphorylation by Plk4 is required for centriole assembly. J Cell Biol , 1217–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer TC, Clutario KM, Lambrus BG, Daggubati V, Holland AJ. (2015). Binding of STIL to Plk4 activates kinase activity to promote centriole assembly. J Cell Biol , 863–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta M, Ashikawa T, Nozaki Y, Kozuka-Hata H, Goto H, Inagaki M, Oyama M, Kitagawa D. (2014). Direct interaction of Plk4 with STIL ensures formation of a single procentriole per parent centriole. Nat Commun , 5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta M, Watanabe K, Ashikawa T, Nozaki Y, Yoshiba S, Kimura A, Kitagawa D. (2018). Bimodal binding of STIL to Plk4 controls proper centriole copy number. Cell Rep , 3160–3169. [DOI] [PubMed] [Google Scholar]
- Peel N, Stevens NR, Basto R, Raff JW. (2007). Overexpressing centriole-replication proteins in vivo induces centriole overduplication and de novo formation. Curr Biol , 834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puklowski A, Homsi Y, Keller D, May M, Chauhan S, Kossatz U, Grünwald V, Kubicka S, Pich A, Manns MP, et al. (2011). The SCF–FBXW5 E3-ubiquitin ligase is regulated by PLK4 and targets HsSAS-6 to control centrosome duplication. Nat Cell Biol , 1004–1009. [DOI] [PubMed] [Google Scholar]
- Rodrigues-Martins A, Riparbelli M, Callaini G, Glover DM, Bettencourt-Dias M. (2007). Revisiting the role of the mother centriole in centriole biogenesis. Science , 1046–1050. [DOI] [PubMed] [Google Scholar]
- Rogers SL, Rogers GC. (2008). Culture of Drosophila S2 cells and their use for RNAi-mediated loss-of-function studies and immunofluorescence microscopy. Nat Protoc , 606–611. [DOI] [PubMed] [Google Scholar]
- Rogers GC, Rusan NM, Roberts DM, Peifer M, Rogers SL. (2009). The SCF Slimb ubiquitin ligase regulates Plk4/Sak levels to block centriole reduplication. J Cell Biol , 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbauer U, Zolghadr K, Muyldermans S, Schepers A, Cardoso MC, Leonhardt H. (2008). A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol Cell Proteomics , 282–289. [DOI] [PubMed] [Google Scholar]
- Serc¸in Ö, Larsimont JC, Karambelas AE, Marthiens V, Moers V, Boeckx B, Le Mercier M, Lambrechts D, Basto R, Blanpain C. (2016) Transient PLK4 overexpression accelerates tumorigenesis in p53-decient epidermis. Nat Cell Biol , 100–110. [DOI] [PubMed] [Google Scholar]
- Silkworth WT, Nardi IK, Scholl LM, Cimini D. (2009). Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One , e6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillibourne JE, Tack F, Vloemans N, Boeckx A, Thambirajah S, Bonnet P, Ramaekers FC, Bornens M, Grand-Perret T. (2010). Autophosphorylation of Polo-like kinase 4 and its role in centriole duplication. Mol Biol Cell , 547–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slevin LK, Nye J, Pinkerton DC, Buster DW, Rogers GC, Slep KC. (2012). The structure of the plk4 cryptic polo box reveals two tandem polo boxes required for centriole duplication. Structure , 1905–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW, et al. (2015). Cell Biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science , 1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Shen C, Wang T, Quan J. (2013). Structural basis for the inhibition of Polo-like kinase 1. Nat Struct Mol Biol , 1047–1053. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Malone JH, Powell SK, Periwal V, Spana E, Macalpine DM, Oliver B. (2010). Expression in aneuploid Drosophila S2 cells. PLoS Biol , e1000320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt-Dias M. (2014). Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol , 433–452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.