

Abstract
The DNA topoisomerase enzymes are essential to cell function and are found ubiquitously in all domains of life. The various topoisomerase enzymes perform a wide range of functions related to the maintenance of DNA topology during DNA replication, and transcription are the targets of a wide range of antimicrobial and cancer chemotherapeutic agents. Natural product-derived agents, such as the camptothecin, anthracycline, and podophyllotoxin drugs, have seen broad use in the treatment of many types of cancer. Selective targeting of the topoisomerase enzymes for cancer treatment continues to be a highly active area of basic and clinical research. The focus of this review will be to summarize the current state of the art with respect to clinically used topoisomerase inhibitors for targeted cancer treatment and to discuss the pharmacology and chemistry of promising new topoisomerase inhibitors in clinical and pre-clinical development.
Abbreviations: ADP, adenosine diphosphate; AQD, anti-cancer quinolone derivative; ATP, adenosine triphosphate; DNA, deoxyribonucleic acid; MTD, maximum tolerated dose; NCI, National Cancer Institute; NCT, national clinical trial; PD, pharmacodynamics; PFS, progression free survival
KEY WORDS: Topoisomerase, Inhibition, Cancer, Oncology, Clinical, Pre-clinical
Graphical abstract
This review summarizes the current state of the art of topoisomerase-targeted drug discovery for cancer therapeutics. The pharmacology and chemistry of promising new topoisomerase inhibitors in clinical trials and pre-clinical development are discussed. Emphases are placed on inhibitory mechanism, isoform selectivity, and the potency of the compounds discussed.
1. Human topoisomerases—structure and function
DNA topoisomerases present ubiquitous chemotherapeutic drug targets and are under continuous investigation in the development of novel antibacterial and anticancer agents. Essential for all domains of life, the multiple types and sub-types of DNA topoisomerases have been the target of marketed drug classes for several decades. The various topoisomerases enzymes deal with the topological issues related to DNA transcription and replication and are capable of relaxing positive or negatively supercoiled DNA, introducing negative or positive supercoils into DNA, and catenating or decatenating (disentangling) circular and linear DNA1, 2. Within the last decade, developments in the structural biology and biochemistry of these enzymes have enabled the advancement of the new avenues of drug discovery targeting these agents, included structure-guided methods and novel compound screening strategies3, 4, 5, 6, 7, 8. This has resulted in the discovery of several new chemical classes of topoisomerase inhibitors with both antibacterial and anticancer properties. This review will discuss new agents that have been introduced for the treatment of cancers within the last several years and discuss their novel chemistry, pharmacology, and clinical efficacy, where relevant.
Table 1 summarizes the function and mechanism of the various types and sub-types of DNA topoisomerases found in eukaryotic organisms. The DNA topoisomerases can be categorized into two general subfamilies, type I and type II topoisomerases. Type I topoisomerases affect DNA topology by passing a single DNA strand through a break in the opposing single strand using an active site tyrosine residue to cleave the DNA strand, forming a phosphodiester bond with the protein. Type II topoisomerase create double-stranded breaks using similar active site tyrosine residues, through which another double-strand DNA segment is then passed. Further sub-divisions of the topoisomerases, types IA, IB, and IIA are categorized based upon the polarity of the DNA-protein bond (i.e., tyrosine attachment to the 5′- or 3′-phosphate), the mechanism (strand passage or rotation), and the number of overhanging DNA bases in the staggered double strand cleavage of the type II topoisomerases. Structurally, eukaryotic type I topoisomerases are monomeric protein, single-gene products, while eukaryotic type II topoisomerases are homodimers, composed of the products of a distinct gene. Functionally, type I topoisomerases are not dependent on ATP hydrolysis to power the energy required for their reaction, instead they derive the energy required from the intrinsic strain energy of the supercoiled DNA itself9. Type II topoisomerases are typically ATP-dependent and possess an ATP-binding domain separate from the DNA-binding domain. Additionally, the DNA topoisomerases can also be distinguished by their dependence, or lack of dependence, on Mg2+ as a requirement for catalytic activity. The type IA and all type II topoisomerases appear to require Mg2+, while the type IB topoisomerases are catalytically active in the absence of Mg2+10. The targets of the currently marketed cancer chemotherapeutic agents are topoisomerase I, IIα, and IIβ.
Table 1.
Summary of eukaryotic DNA topoisomerase enzyme function and mechanism.
| Type | Subtype | Protein | Gene | Function | Mechanism | Multimericity | Metal dependence | Cleavage polarity |
|---|---|---|---|---|---|---|---|---|
| I | A | Topoisomerase IIIα | top3A | (—) Supercoil relaxation | Strand passage | Monomer | Yes (Mg2+) | 5′ |
| Topoisomerase IIIβ | top3B | Unknown | ||||||
| B | Topoisomerase I | top1 | (—) and (+) supercoil relaxation | Strand rotation | Monomer | No | 3′ | |
| II | A | Topoisomerase IIα | top2A | Decatenation during replication | Strand passage (ATPase) | Homodimer | Yes (Mg2+) | 5′ |
| Topoisomerase IIβ | top2B | Various, neuronal transcription |
Several excellent reviews have summarized the current state of knowledge pertaining to the structure and function of the type I and type II topoisomerases1, 9, 11, 12, 13. Fig. 1 shows the overall structures of representative human type I and type II DNA topoisomerases14, 15. The type IA topoisomerases represented by eukaryotic topoisomerase IIIα and IIIβ consist of four domains that coordinate DNA binding, cleavage, and strand passage16, 17, 18. Domain I contains the so-called TOPRIM fold, a Rossman-fold like structure that can bind magnesium ions18, 19. Domain II consists primarily of β-strands that forms the central core and links domain III, which contains the catalytic tyrosine residue, to domain IV. The type IB topoisomerases, represented by eukaryotic topoisomerase I, is also composed of four domains including an N-terminal domain, a linker domain, a core domain, and a C-terminal domain14, 20. The core domain is responsible for DNA binding and appears to be highly conserved. The catalytic tyrosine is in the C-terminal domain and operates as part of a catalytic pentad with four residues located in the core domain. Type IIA topoisomerases, represented by eukaryotic topoisomerase IIα and IIβ, consist of a three-domain structure spanning the A and B subunits that form the homodimer (or heterotetramer in prokaryotes). The N-terminal domain contains the ATB-binding region (ATPase domain), a core domain that contains a TOPRIM fold and DNA-binding region, and a C-terminal domain with unclear function21, 22, 23.
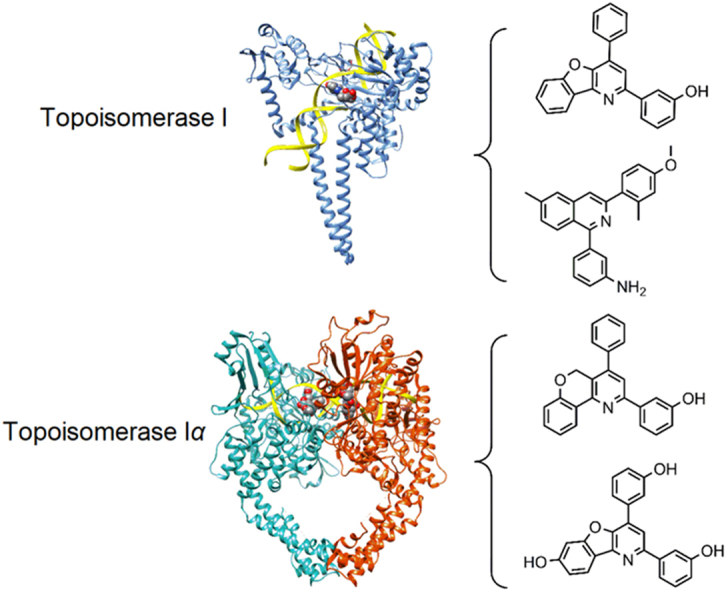
Figure 1.

Structures of human topoisomerases. Shown are the structures of the full length human topoisomerase I (left, PDB ID 1k4t) and topoisomerase IIα (right, PDB ID 5qwk) enzymes with bound DNA, representative of the overall structure and domains of the two sub-family types (DNA ribbon is colored yellow; chain A of topoisomerase I is blue; chain A of topoisomerase IIα is green; chain B of topoisomerase IIα is red).14, 15
The sub-types of topoisomerases are mechanistically distinct with respect to how they act on their DNA substrates. Type IA topoisomerases operate using a “strand-passage” mechanism, whereby a single strand of DNA is passed through a break in a second single DNA stand. In this mechanism, the first DNA single strand binds to domain I and III of the topoisomerase and is cleaved by the catalytic tyrosine residue located on domain III, creating a 5′-phosphodiester bond between the enzyme and the DNA. Domain II is then believed to act as a hinge, separating the cleaved DNA strand and permitting a second strand to pass through, after which domains I and III come back together and the cleaved DNA strand is re-joined. The type IB topoisomerases operate using a “hindered rotation mechanism”, whereby the enzyme binds to DNA and cleaves a single strand via a 3′-phosphodiester bond using an active site tyrosine residue. The 5′-end then rotates about the second DNA strand, relaxing the DNA. As discussed above, both type IA and type IB topoisomerases use the DNA torsional strain (torque) to drive the uncoiling process rather than ATP hydrolysis. Type IA topoisomerases relax negatively supercoiled DNA and perform DNA decatenation, while type IB topoisomerases can relax both negatively and positively supercoiled DNA. Like the type IA topoisomerases, the type IIA topoisomerases exert their effect through a strand passage mechanism, in this case using what is known as a “two-gate” mechanism24. Double-stranded DNA, called the “G-segment”, binds to the topoisomerase in a central DNA-binding region called the “DNA-gate”. A second double-stranded DNA, the “T-segment”, binds to the N-terminal ATPase domain facilitated by ATP binding and domain dimerization. ATP hydrolysis and the release of inorganic phosphate causes a double-stranded break with 4-base overhang in the G-segment DNA facilitated by the formation of 5′-phosphodiester bonds between two tyrosine residues and individual strands of the G-segment DNA. The DNA-binding gate separates, and the T-segment is passed through the G-segment into the C-terminal region, or “C-gate”. The C-gate opens and releases the T-segment DNA, and ADP product is released, resetting the enzyme for another catalytic cycle.
2. Targeting topoisomerases for cancer chemotherapy
Inhibition of the DNA topoisomerase enzymes can occur by one of several generally accepted molecular mechanisms5, 25, 26, 27. One potential mechanism is substrate competitive inhibition, the binding of an inhibitor compound to the topoisomerase active site that prevents the binding of the DNA substrate. There are no notable examples of topoisomerase-specific inhibitors that work by this mechanism, but recent reports of DNA-competitive inhibitors of other DNA-binding proteins suggest that this mechanism may be possible in DNA topoisomerases as well28. Another common mechanism is the formation of so-called “topoisomerase poisons”, that are composed of a ternary protein-DNA-drug complex that prevents DNA re-ligation and locks the enzyme into a “cleavage complex”. This complex results in prevention of enzyme turnover and the build-up of high levels of the cytotoxic cleavage complex within the cell. A third mechanism is by competitive inhibition of the ATP binding site, seen only in the type II topoisomerases, which prevents the ATP-hydrolysis driven enzymatic action (discussed above). Examples of ATP-site binders include novobiocin and coumermycin, which are not used clinically due to issues with potency, specificity, and poor pharmacokinetic properties29, 30. Compounds that can bind to DNA and prevent topoisomerase binding, such as the compounds aclarubicin and suramin, present another potential inhibitory mechanism, though specificity is again an issue with agents such as these31, 32. Agents such as the compound merbarone, that can bind to the DNA-protein complex and prevent cleavage, represent yet another mechanism of catalytic inhibition32, 33. Lastly, the ATP-dependent type II topoisomerases can be inhibited by agents that prevent ATP hydrolysis and DNA release after strand passage, as exemplified by the bisdioxopiperazine agent dexrazoxane34, 35, 36. These agents result in a “closed clamp complex” that is analogous to the cleavage complex generated by the topoisomerase poisons. In this section, we will review in some detail the inhibitory mechanism of the more common classes of topoisomerase inhibitors from a structural and biochemical perspective, and provide examples of clinically used agents, where applicable.
Of highest clinical relevance is the mechanism of the topoisomerase poisons27. This mechanism involves the stabilization of the cleavage complex by creation of a locked ternary complex of cleaved DNA, protein, and drug that builds up and causes a cytotoxic effect37. The mechanism is exemplified by the camptothecin-derived agents that act upon type IB topoisomerases, and the anthracycline, anthracenedione, and epipodophyllotoxin agents that act upon type IIA topoisomerases for the treatment of cancer (discussed in further detail below). These agents bind to the cleaved-DNA/protein complex and prevent the re-ligation of DNA, locking the enzyme into the cleavage complex and preventing enzyme turnover. A build-up of the cleavage complex results in DNA strand breaks and ultimately cellular death27. Most known topoisomerase poisons act via an interfacial mechanism by intercalation between the −1 and +1 DNA base pairs in the protein-DNA cleavage complex. Additional hydrophobic and electrostatic interactions with both the DNA and protein stabilize the binding of the poison and prevent DNA re-ligation by the topoisomerase37, 38, 39. A second topoisomerase poison mechanism is through the redox-dependent, covalent formation of a drug-enzyme complex. The covalent class compounds bind to a site distal to the DNA active site and work to stabilize the enzyme-DNA cleavage complex, resulting in a similar build-up and ultimate cell death39. Shown in Fig. 2, is the binding of the drug etoposide to human topoisomerase IIα. This interfacial topoisomerase poison can be seen intercalating two DNA-base pairs in the active site, facilitated by stacking interactions between the DNA bases and the connected, 4-ring system of the drug. The drug׳s adjoining ring systems are seen hydrogen bonding to the nearby residues from the protein and local DNA bases. A similar binding event (not shown) takes place 4 base pairs distal on the opposite DNA strand, reflecting the double strand cleavage of this type II topoisomerase.
Figure 2.
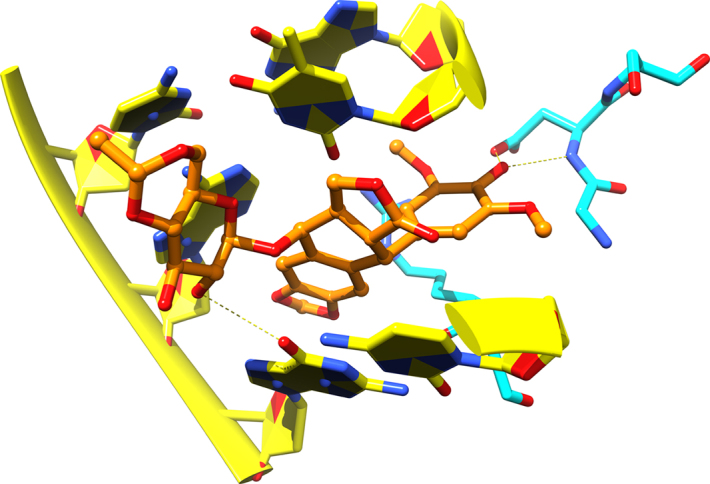
Etoposide binding to human topoisomerase IIα. Shown is the topoisomerase poison, etoposide, bound to the active site of human topoisomerase IIα. DNA is represented with yellow ribbons, yellow carbons, and filled rings. Protein is represented with cyan carbons, and the drug is shown with ball and stick representation with orange carbons. Hydrogen bonds to protein and DNA are shown as dashed yellow lines.
A second, highly relevant topoisomerase inhibition mechanism is catalytic inhibition by competitive binding of small molecules to the ATP binding site found in the N-terminal of region of type II topoisomerases5, 40. As discussed above, the energy required for the action of type I topoisomerases is derived from the torsional strain of the supercoiled DNA itself, while the energy powering the action of the type II topoisomerases is derived from ATP hydrolysis. Competitive inhibition of ATP hydrolysis by small molecules prevents the progression of the DNA T-segment through the G-gate into the C-terminal domain, resulting in a catalytic inhibition of the topoisomerase, halting DNA transcription and replication, and ultimately leads to cell death25. This mechanism does not result in the DNA damage and cellular damage responses seen with the topoisomerase poisons (discussed further below). Catalytic inhibitors of this type were first seen with the aminocoumarin antibiotics, represented by the compound novobiocin, which is no longer clinically marketed25, 41. Though there are no currently marketed anticancer agents possessing this mechanism of inhibition, there have been fairly recent reports discussing agents in development that show promise in the area42, 43. Fig. 3 shows the ATP binding site of human topoisomerase IIα with ADP bound44. The key binding interactions are highlighted. An interesting, related mechanism that bears mention is that of the bisdioxopiperazine compounds represented by the drug dexrazoxane (ICRF-187)34. This class of agents shows catalytic inhibition by binding to the enzyme in an ATP-dependent, uncompetitive manner and inhibiting the ATP to ADP conversion. This appears to lock the topoisomerase in a closed clamp conformation. Though the inhibition is catalytic in nature, there appears to be some evidence of a resulting DNA damage and cellular damage response, like that induced by the topoisomerase poisons36. Dexrazoxane is marketed under the brand name Zinecard in the USA and Cardioxane in the EU and other countries as a protectant agent to mitigate cardiotoxicity caused by anthracyclines (discussed further below).
Figure 3.
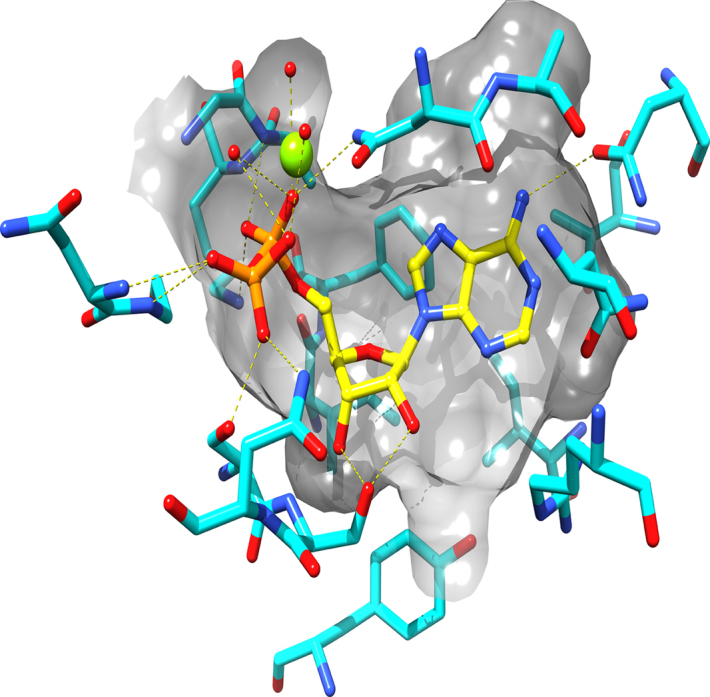
ATP binding site of Human Topoisomerase IIα. Depicted is the ATP binding site of the human topoisomerase IIα enzyme. ADP is bound to the binding site (yellow carbons) with hydrogen bonds to the protein shown in yellow, dashed lines. The active site residues are shown with cyan carbons and a translucent gray surface is used to show the shape of the binding site.
3. Clinically marketed topoisomerase inhibitors
Table 2 summarizes the currently marketed topoisomerase active agents used for cancer chemotherapy45, 46, 47. The first known class of topoisomerase inhibitors used for the treatment of cancer was the anthracycline agents. The anthracyclines were first extracted from bacterial Streptomyces species and discovered to possess antibiotic and antitumor activity48. The clinically marketed anthracycline derivatives include doxorubicin, epirubicin, valrubicin, daunorubicin, and idarubicin. Their structures are shown in Fig. 4. Doxorubicin has several current indications including treatment of breast cancer, various types of leukemia, lymphoma, sarcomas, carcinomas, and other tumors. Other anthracyclines which have indications for treatment of leukemia include daunorubicin and idarubicin. Epirubicin is indicated in breast cancer following resection, while valrubicin is indicated in urinary bladder carcinoma. The anthracycline agents affect their cytotoxic activity by acting as topoisomerase “poisons”, as discussed in the previous section. They primarily affect type IIa topoisomerases, topoisomerase IIα and topoisomerase IIβ indiscriminately, by intercalation into the bound and cleaved DNA, stabilizing the DNA and topoisomerase complex and preventing DNA re-ligation by the topoisomerase. Interestingly, this appears to be one of the mechanisms that the anthracyclines induce cell death49. The production of free radical species in an iron-dependent manner appears to be another mechanism behind the anthracyclines׳ cytotoxic effects in cancer cells. Unfortunately, this mechanism also appear to be responsible for additional toxicities associated with this class of agents50. Anthracyclines are well known for their cardiotoxic properties, causing both acute and chronic cardiac complications in a dose-dependent manner. Menna and colleagues51. discuss the mechanisms behind this toxicity, including anthracyclines׳ production of superoxide anions and hydrogen peroxide in cardiac muscle cells, which contribute to oxidative stress and eventually apoptosis. Anthracyclines also disrupt calcium and iron levels in cardiac cells, which leads to even more free-radical generation51. Some recent studies have implied another potential mechanism of the cardiotoxicity52, 53. These studies suggest that inhibition of topoisomerase IIβ, which is overexpressed in the heart, by the anthracycline agents may result cardiotoxicity from apoptosis and ROS. Dexrazoxane, discussed above, is an FDA-approved agent indicated to reduce adverse cardiac affects in women who have received a total dose of 300 mg/m2 of doxorubicin and are continuing to receive doxorubicin54. Interestingly, the mechanism of cardiotoxicity mitigation appears to be related, at least partially, to the ability of the drug to chelate iron. The drug is also known to catalytically inhibit topoisomerase IIβ, as discussed above, which may reduce cardiotoxicity related to generation of the topoisomerase IIβ poisons formed by anthracyclines. Other investigational measures to prevent cardiotoxicity include the use of the beta blocker carvedilol. Spallarossa and colleagues found a decrease in free radical production and apoptosis in heart muscle cells when treating cells with carvedilol prior to treatment with doxorubicin, due to carvedilol׳s unique antioxidant effect55.
Table 2.
Summary of marketed topoisomerase inhibitors and indications.
| Drug | Class | Approval date | Mechanism/Target | Indicationa |
|---|---|---|---|---|
| Doxorubicin | Anthracycline | 8/7/1974 (U.S.) | Type IIA poison | In combination with other chemotherapy agents to treat women after surgical resection of breast cancer with axillary lymph node involvement; acute lymphoblastic and myeloblastic leukemias; Hodgkin and non-Hodgkin lymphomas; metastatic neuroblastoma, Wilms′ tumor, cancers of the breast, soft tissue sarcoma, and bone sarcomas; metastatic ovarian, transitional cell bladder, thyroid, gastric, and bronchogenic carcinomas |
| Epirubicin | Anthracycline | 9/15/1999 (U.S.) | Type IIA poison | Combination with other chemotherapy agents to treat women after surgical resection of breast cancer with axillary lymph node involvement |
| Valrubicin | Anthracycline | 9/25/1998 (U.S.) | Type IIA poison | Intravesical administration for urinary bladder carcinoma refractory to BCG therapy in patients who are not candidates for cystectomy |
| Daunorubicin | Anthracycline | 12/19/1979 (U.S.) | Type IIA poison | In combination with other approved chemotherapy agents to induce remission in acute myelogenous, monocytic, and erythroid lymphocytic leukemias in adults and in acute lymphocytic leukemia in children |
| Idarubicin | Anthracycline | 9/27/1990 (U.S.) | Type IIA poison | In combination with other approved agents to treat adults with acute myeloid leukemia, including French-American-British M1—M7 classifications |
| Mitoxantrone | Anthracenedione | 12/23/1987 (U.S.) | Type IIA poison | For patients with secondary (chronic) multiple sclerosis with significantly abnormal neurologic status between relapses to reduce neurologic disability and or/the frequency of relapses; in combination with corticosteroids as initial chemotherapy to treat pain related to advanced hormone-refractory prostate cancer; in combination with other approved agents to as initial therapy of acute nonlymphocytic anemia in adults |
| Pixantrone | Anthracenedione | 2/16/2012 (E.U.) | Type IIA poison | Monotherapy for the treatment of adult patients with multiply relapsed or refractory aggressive Non-Hodgkin B-cell lymphomas |
| Etoposide | Epipodophyllotoxin | 11/10/1983 (U.S.) | Type IIA poison | Refractory testicular tumors in combination with other chemotherapy agents; first-line treatment in combination with cisplatin for small-cell lung cancer |
| Teniposide | Epipodophyllotoxin | 7/14/1992 (U.S.) | Type IIA poison | Induction of remission in patients with refractory childhood acute lymphoblastic leukemia |
| Amsacrine | Miscellaneous | 12/31/1983 (Canada) | Type IIA poison | Induction of remission in acute adult leukemia refractory to conventional therapy |
| Topotecan | Camptothecin | 5/28/1996 (U.S.) | Type IB poison | Small-cell lung cancer after failure of first-line chemotherapy; combination with cisplatin for persistent or recurrent stage IV-B carcinoma of the cervix not cured by surgery and/or radiation |
| Irinotecan | Camptothecin | 6/14/1996 (U.S.) | Type IB poison | First-line chemotherapy in combination with 5-fluorouracil and leucovorin in patients with metastatic carcinoma of the colon or rectum; recurrent or progressive metastatic carcinoma of the colon or rectum following initial fluorouracil-based therapy |
| Belotecan | Camptothecin | 12/10/2003 (S. Korea) | Type IB poison | Non-small-cell lung cancer; ovarian cancer |
Figure 4.

Structures of the marketed anthracycline topoisomerase inhibitors.
The anthracenedione agents include the drugs mitoxantrone and pixantrone. The anthracenedione (anthraquinone) agents are synthetic agents, designed to act similarly to the anthracycline drugs, with fewer adverse effects and toxicities. Mitoxantrone was approved in 1987 in the U.S. and pixantrone, a newer agent, was approved in 2012 in Europe. The structures of these agents are shown in Fig. 5. Mitoxantrone can cross the blood brain barrier and is indicated for the reduction of the frequency and intensity of multiple sclerosis relapses in addition to its indication as a chemotherapeutic agent in leukemia and prostate cancer56. Similar to the anthracyclines, the anthracenedione agents are topoisomerase poisons, primarily affecting type II topoisomerases. Like the previously discussed agents, mitoxantrone intercalates topoisomerase-bound DNA, preventing DNA re-ligations, and ultimately resulting in DNA strand breakage and disruption of DNA repair. Pixantrone, an aza-anthracenedione approved in treatment of non-hodgkin B-cell lymphoma, exhibits its cytotoxic effects by intercalating into DNA like the anthracyclines, but also causes long term cell damage and eventual death by causing errors in mitosis and segregation of chromosomes57. Pixantrone has shown less toxicity than doxorubicin in cardiac muscle cells because of its inability to bind iron and contribute to free radical production in the heart; as a result, animal models showed decreased heart weight in animals treated with doxorubicin as compared to pixantrone58, 59
Figure 5.

Structures of the marketed anthracenedione and acridine-derived topoisomerase inhibitors.
Clinically marketed camptothecin derivatives include topotecan, irinotecan, and belotecan. Their structures are shown in Fig. 6. The camptothecin alkaloid was first derived from the Chinese tree, Camptotheca acuminata. The camptothecin-derived agents are topoisomerase poisons which primarily affect type I topoisomerases. In vitro studies showed camptothecin, a cytotoxic alkaloid, was capable of inhibiting topoisomerase I and causing DNA strand breaks, thus preventing DNA replication60. The water-soluble forms of camptothecin include the clinically marketed irinotecan and topotecan, which reversibly bind and form a ternary complex with topoisomerase I and DNA, as discussed above61. Topotecan is approved as second-line small cell lung cancer and, in combination with cisplatin, for patients with stage IV-B cervical carcinoma not treated by surgery or radiation. Irinotecan is approved following failure or progression following treatment with fluorouracil or in combination with 5-fluorouracil and leucovorin for patients with metastatic colon or rectal carcinoma. Belotecan is a relatively new camptothecin derivative agent, approved in South Korea for treatment of non-small-cell lung cancer and ovarian cancer in 200362, 63. The mechanism of action is the same as other agents in this class. Compared with older camptothecin agents, belotecan is reported to have a similar efficacy profile, with reduced toxicities64.
Figure 6.

Structures of the camptothecin-derived topoisomerase inhibitors.
The drugs etoposide and teniposide are epipodophyllotoxin-derived agents. Epipodophyllotoxins are natural substances derived from the Mayapple plant (wild mandrake), Podophyllum peltatum65, 66. The drugs have been available in the U.S. and other countries since the early 1980׳ s. Their structures are shown in Fig. 7. Both agents act as topoisomerase poisons and cause DNA strand breaks by binding to type II topoisomerases, similar to the agents described above67. Etoposide is indicated as part of a multi-drug chemotherapy regimen for refractory testicular tumors and in combination with cisplatin to treat small-cell lung cancer. Teniposide is approved in patients with refractory childhood acute lymphoblastic leukemia in combination with other chemotherapy drugs.
Figure 7.

Structures of epipodophyllotoxin-derived topoisomerase inhibitors.
The sole marketed agent it its chemical class, amsacrine (m-AMSA, Fig. 4), is a synthetic agent composed of a planar, acridine ring system. Like the agents discussed above, amsacrine is a topoisomerase poison targeting the type II topoisomerases. Interestingly, amsacrine was the first drug proven to poison eukaryotic topoisomerase II68. The acridine ring system is the component of the drug that intercalates DNA and contributes to the activity of the drug, while the non-intercalative 4′-amino-methane-sulfon-m-anisidide (m-AMSA) headgroup imparts specificity for the DNA-topoisomerase cleavage complex69. Amsacrine is approved in Canada to induce remission in adults with acute leukemia resistant to conventional therapy70.
4. Topoisomerase inhibitors in clinical trials
This section discusses novel topoisomerase inhibitor compounds that have been investigated in human clinical trials. Table 3 summarizes the trials discussed in this section and several other trials of note that are not discussed in the section. In the table, clinical trials are categorized as phase 1, 2 and 3, and listed with the relevant National Clinical Trial (NCT) identifier, or another regulatory agency identifier where applicable. Clinical trials that are summarized here are those listed in the U.S. National Library of Medicine׳s Clinical Trials database and the WHO/ICMJE ISRCTN Registry71, 72
Table 3.
Summary of clinical trials discussed.
| Study | Study purpose | Time frame | Sample size | Outcome measures | Relevant findings | NCT# |
|---|---|---|---|---|---|---|
| Phase 1 | ||||||
| Indenoisoquinoline LMP400 for advanced solid tumors and lymphomas | Safety and efficacy of Indenoisoquinoline LMP400 for advanced solid tumors or lymphomas | February 2013–October 2017 | 21 participants | To establish the safety, tolerability, and PK profiles of weekly LMP400 in patients with refractory solid tumors and lymphomas | LMP776 is overall well tolerated | 1794104 |
| A phase I study of indenoisoquinolines LMP400 and LMP776 in adults with relapsed solid tumors and lymphomas | Study how LMP400 and LMP776 are processed by the body and how effective they are in treating difficult-to-treat types of cancer | January 2010–June 2017 | 55 participants | Define the MTD, dose-limiting toxicities, and PD endpoint (gamma-H2AX in tumor biopsy pre- and post-treatment) of LMP400 and LMP776 administered intravenously daily for 5 days | N/A | 1051635 |
| Phase-I dose finding and pharmacokinetic study of the novel hydrophilic camptothecin ST-1968 (namitecan) in patients with solid tumors | First-in-human, dose-escalation study to determine the MTD of intravenous, flat-dosed ST-1968 (namitecan), a new hydrophilic camptothecan derivative | June 2007−December 2011 | 62 participants | MTD of ST1968 given intravenously once every week for 2 consecutive weeks every 3 weeks and MTD of ST1968 given intravenously once every 3 weeks for 21 days | Neutropenia was the drug-limiting toxicity, with 15 mg being defined as the recommended dose for one group and 23 mg for the other group. Non-hematological toxicity was negligible. Namitecan exhibited fully dose-proportional PK | 1748019 |
| Phase 2 clinical trials | ||||||
| Vosaroxin and infused cytarabine in treating patients with untreated acute myeloid leukemia (VITAL) | Study how well vosaroxin and cytarabine work in treating patients with untreated acute myeloid leukemia | March 2016-Ongoing (estimated to complete in July 2019) | 61 participants | Assess the rate of complete remission after induction therapy with the vosaroxin and standard dose infused cytosine arabinoside for patients with newly diagnosed, previously untreated acute myelogenous leukemia | N/A | 2658487 |
| A phase 2 Study of CRLX101 in patients with advanced non-small cell lung cancer | Compare median overall survival of patients with advanced non-small cell lung cancer treated with CRLX101 to patients treated with best supportive care (BSC) | June 2011–October 2014 | 157 participants | Compare overall survival of patients treated with CRLX101 and BSC to patients treated with BSC only for up to 18 months | No statistical analysis provided for to compare overall survival of patients in both treatment arms | 1380769 |
| Study of AR-67 in adult patients with recurrence of glioblastoma multiforme (GBM) or gliosarcoma | Study the efficacy and determine the 6-month progression free survival (PFS) of AR-67 (7-t-butyldimethylsiltyl-10-hydroxy-camptothecin) in patients with GBM | December 2009– February 2015 | 58 participants | Determine the 6-month PFS of AR-67 administered in adults with confirmed recurrence of GBM who have not had experienced a recurrence within 90 days after receiving bevacizumab or temazolamide for treatment | N/A | 1124539 |
| Rebeccamycin analog in treating children with relapsed or refractory neuroblastoma | Study effectiveness of rebeccamycin analog in treating children diagnosed with relapsed or refractory neuroblastoma | January 1999–September 2006 | 30 participants | Determine the response rate and toxicity to rebeccamycin analogue in children with relapsed or refractory neuroblastoma | N/A | 3737 |
| Rebeccamycin analog in treating patients with metastatic or locally recurrent colorectal cancer | Study effectiveness of rebeccamycin analog in treating patients diagnosed with metastatic or locally recurrent colorectal cancer | February 2000–June 2002 | 37 participants | Determine response rate, toxicity, and overall survival of patients with metastatic or locally recurrent colorectal cancer treated with rebeccamycin analogue | N/A | 5085 |
| Rebeccamycin analogue in treating women with stage IIIB or stage IV breast cancer | Compare effectiveness of two different rebeccamycin analogue regimens in treating women diagnosed with stage IIIB or stage IV breast cancer | March 2000–May 2006 | 42 participants | Assess activity of rebeccamycin analog as therapy for advanced breast cancer administered in two different treatment regimens | N/A | 5817 |
| Intravenous edotecarin in patients with advanced gastric cancer that has progressed or recurred after chemotherapy | Study the efficacy of edotecarin in adult patients with advanced gastric cancer, reasonable performance status, good organ function, lack of serious concomitant medical conditions in repeated 3-week cycles of treatment | April 2004–June 2005 | 28 participants | Assess antitumor activity of edotecarin using repeated radiographic assessments at 6-week intervals | N/A | 87503 |
| Phase 3 | ||||||
| Comparison of pixantrone + rituximab with gemcitabine + rituximab in patients with aggressive B-cell non-Hodgkin lymphoma or follicular grade 3 lymphoma who have relapsed after therapy and are not eligible for stem cell transplant (PIX-R) | Evaluate the efficacy of Pixantrone with rituximab compared to Gemcitabine with Rituximab in patients with relapsed or refractory diffuse large B-cell lymphoma or follicular grade 3 lymphoma | April 2011–December 2017 | 260 participants | Progression free survival from randomization to the date of disease progression or death | N/A | 1321541 |
| Gemcitabine and docetaxel versus doxorubicin as first-line treatment in previously untreated advanced unresectable or metastatic soft-tissue sarcomas (GeDDiS): a randomized controlled phase 3 trial | Compare gemcitabine and docetaxel versus doxorubicin as first-line treatment for advanced or metastatic soft-tissue sarcoma | January 2010–January 2013 | 250 participants | Progression-free survival, assessed using the RECIST Criteria every six weeks, after each set of two cycles; 2-monthly following treatment assessment | The proportion of patients alive and free of cancer progression after 24 weeks did not differ between the treatment arms. The most common adverse effects were neutropenia and febrile neutropenia in approximately the same percentage of patients who received either treatment arm. Percentage of patients who died during or after this study was also similar for both treatment arms; none of the deaths were related to the treatment. The study concludes that there is significant evidence for clinicians to consider doxorubicin as a single agent in patients with locally advanced or metastatic soft-tissue sarcoma | 07742377 inISRCTNregistry |
| Phase 3 study to treat patients with soft tissue sarcomas | Determine the efficacy and safety of aldoxorubicin in subjects with metastatic, locally advanced, or unresectable soft tissue sarcomas | January 2014–May 2017 | 433 participants | Progression-free survival over 24 months | Aldoxorubicin has minimal cardiac toxicity and survival advantage in patients with leiomyosarcoma and liposarcoma | 2049905 |
4.1. Phase 1 clinical trials
The U.S. National Cancer Institute (NCI) has conducted a phase 1 clinical trial for neoplasm lymphoma using agents from a novel class of non-camptothecin type I topoisomerase inhibitors known as indenoisoquinolines (NCT-01794104, Fig. 8). Indenoisoquinolines create a stable DNA-topoisomerase cleavage complex, similar to the camptothecin derivatives, but preferring specific DNA cleavage sites which allows them to gain efficacy against camptothecin-resistant cell lines73. These compounds are chemically stable and act upon cells over-expressing ATP-binding cassette transporters ATP-binding cassette (ABC) transporters ABCG2 and P-glycoprotein (MDR1)74. Stabilization of the cleavage complex induces DNA damage, demonstrating the efficacy of these inhibitors as potent anticancer therapies. Further, indenoisoquinolines delay DNA repair, which leads to cell death. This study aimed to demonstrate that patients can respond to topoisomerase I inhibitor therapy if their tumor biopsies show topoisomerase I expression. Twenty-one adult patients with refractory solid tumors and lymphomas were enrolled in this study of the indenoisoquinoline, LMP400. LMP400 has linear pharmacokinetics (PK) with drug accumulation after five days of dosing75. It is hypothesized that weekly dosing will increase the drug׳s peak levels and lead to improvements in clinical safety and efficacy76.
Figure 8.

Representative topoisomerase inhibitors in clinical trials for cancer.
Namitecan (ST1968) is a topoisomerase I inhibitor with superior antitumor activity along with a better safety profile than irinotecan and topotecan77, 78. PK studies with repeated dosing schedules demonstrated a lack of both metabolite production and accumulation based on its short half-life. Present studies have confirmed the safety and PK profile of namitecan, including manageable neutropenia and successful antitumor activity with response in bladder and endometrium cancers79, 80.
4.2. Phase 2 clinical trials
The Vanderbilt-Ingram Cancer Center is currently recruiting patients for a phase II trial study to demonstrate how well vosaroxin and cytarabine work in treating patients with untreated acute myeloid leukemia (NCT-02658487). Vosaroxin is an anti-cancer quinolone derivative (AQD), which affects type II topoisomerases (Fig. 8)81, 82, 83. AQDs target the DNA-topoisomerase cleavage complex and intercalate DNA at specific GC rich sites to prevent DNA re-ligation by the topoisomerase. This results in site-specific DNA damage and S-phase prolongation along with G2 phase cell cycle arrest, which ultimately triggers apoptosis. Vosaroxin has a stable quinolone core, rendering it less reactive than other classes of topoisomerase inhibitors (see above)84. The class produces less toxic metabolites and reactive oxygen species decreasing the potential for off-target organ damage and cardiotoxicity. This quinolone core further allows vosaroxin to evade cellular drug efflux because it is not a substrate for the P-glycoprotein efflux pump84. Vosaroxin can also induce p53-independent apoptosis, allowing for it to combat mechanisms of drug resistance associated with the inactivation of p5385. Lastly, the stable quinolone structure of vosaroxin is not well metabolized by enzymes including the major p450 isoforms nor does it readily inhibit or induce p450 activity, reducing the potential for drug—drug interactions and even allowing for the potential to enhance anti-cancer drug activity, such as cytarabine85. The primary objective of this study is to assess the rate of complete remission after induction therapy using this drug combination in patients with newly diagnosed as well as previously untreated acute myelogenous leukemia. Secondary objectives include the following: frequency of adverse events, evaluation of the presence of minimal residual disease after induction phase(s), determination of the incomplete blood count recover rate after each treatment cycle, determination of the time to neutrophil and platelet recovery following the induction phase(s), assessing disease-free and overall survival after one year following treatment, along with determining the correlation of hematopoietic stem cell transplant comorbidity index and Wheatley index scores with response to disease.
NewLink Genetics Corporation completed a Phase 2 clinical trial to study the impact of CRLX101 on average survival of patients with advanced non-small cell lung cancer (NSCLC) compared to patients receiving best supportive care (NCT-01380769)86. CRLX101 is a camptothecin nanoparticle conjugated to a cyclodextrin-based polymer, designed as a targeted therapy regimen to increase the exposure of tumor cells to camptothecin meanwhile minimizing its side effects87, 88, 89. The cyclodextrin-based polymer improves the water-solubility of camptothecin and is designed to be hydrolyzed in vivo after localization to the tumor. Tumor-specific targeting is facilitated by the size of the drug nanoparticle, which has been designed to extravasate from the “leakier” blood vessels found in tumors90.
4.3. Phase 3 clinical trials
CTI BioPharma has conducted a phase 3 study to compare the efficacy of pixantrone with rituximab to gemcitabine, a nucleoside analog, with rituximab in 260 patients with relapsed or refractory diffuse large B-cell lymphoma or follicular grade 3 lymphoma (NCT-01321541)91. The primary outcome of this study was progression free survival (PFS), with the time frame being a randomized time to the date of disease progression or death. Secondary outcome measures were evaluated from randomization to death and included overall survival, complete, and overall response rate, as well as safety evaluation regarding the number of laboratory values falling outside of predetermined ranges and the frequency of adverse events.
Doxorubicin is currently the main treatment for soft tissue sarcomas, but its pro-drug aldoxorubicin is a promising option according to expert opinion92. Aldoxorubicin contains a carboxylic hydrazine that covalently binds to albumin in blood to reach the acidic tumor environment, which then dissolves the hydrazone linker to release doxorubicin into the tissue93. A phase 3 study sponsored by CytRx involved administering aldoxorubicin on the first day of every 21-day cycle of treatment in patients with soft tissue sarcomas until there was either tumor progression or an unacceptable toxicity occurred (NCT-02049905). The active comparator was the investigator׳s choice among darcabazine, pazopanib, gemcitabine with docetaxel, doxorubicin, or ifosfamide. Aside from overall survival over 36 months, the safety of aldoxorubicin compared to the investigator׳s choice will be assessed using the following parameters: frequency and severity of adverse events, abnormal findings during physical examinations, laboratory tests, vital signs, echocardiogram evaluations, electrocardiogram results, disease control rate, and tumor response. Preliminary results from this phase 3 study demonstrated a PFS advantage in patients with leiomyosarcoma and liposarcoma treated with aldoxorubicin92.
Other topoisomerase inhibitors of note that have entered clinical trials include silatecan, a camptothecin derivative (Fig. 8)94, 95. This agent represents a unique silicon-containing class of topoisomerase I inhibitors that have progressed into clinical trials. A phase 2 study of this compound for use in gliosarcoma has been registered by Arno Therapeutics, but no results have been posted to date (NCT-01124539). Rebeccamycin analogs have similarly progressed to phase 2 clinical trials (Fig. 8)96, 97, 98. Derived from the natural product rebeccamycin, these compounds possess an indolocarbazole ring system with an attached sugar moiety. The synthetic compounds becatecarin and edotecarin are two examples that have progressed to Phase II trials (Table 3). Rebeccamycin analogs have shown activity as dual topoisomerase I and topoisomerase II poisons. Clinical development of both becatecarin and edotecarin appears to have ceased and it remains to be seen whether any additional rebeccamycin compounds will be tested clinically.
5. Pre-clinical topoisomerase inhibitors under investigation
Several interesting new chemical classes of topoisomerase inhibitor are currently under pre-clinical investigation (summarized in Table 499, 100, 101, 102). In this section, we review some of the more notable examples and discuss their chemistry, mechanism, activity, and selectivity. Discussion is focused on agents that have been reported in the literature within the last 5 years, with an emphasis on agents that have shown favorable results in animal models, but that have not yet progressed to clinical studies.
Table 4.
Summary of pre-clinical studies reported.
| Citation | Compound(s) tested | Enzyme activity (μmol/L)a | Cell activity (μmol/L)a |
|---|---|---|---|
| Kwon et al.99 | Benzo-furo-pyridine | Topo I—65.2 (IC50) | HEK293—4.93 (IC50) |
| (Fig. 9, series A, n=0) | Topo IIα—13.4 (IC50) | DU145—1.90 (IC50) | |
| HCT15—0.15 (IC50) | |||
| T47D—0.83 (IC50) | |||
| Kwon et al.99 | Chromeno-pyridine | Topo I—not reported | HEK293—5.69 (IC50) |
| (Fig. 9, series A, n=1) | Topo IIα—60.2 (IC50) | DU145—1.43 (IC50) | |
| HCT15—0.005μM (IC50) | |||
| T47D—0.54 (IC50) | |||
| Shrestha et al.100 | Benzo-furo-pyridine | Topo I—22.4% inhibition (100) | HCT15—1.22 (IC50) |
| (Fig. 9, series B, compound I) | Topo IIα—100% inhibition (100) | T47D—0.59 (IC50) | |
| HeLA—0.86 (IC50) | |||
| Khadka et al.101 | 1,3-diarylisoquinolineb | Topo I—1.22x camptothecin (100) | MCF10A—5.74 (IC50) |
| (Fig. 9, series C, compound II) | Topo IIα—0.06x camptothecin (100) | T47D—0.74 (IC50) | |
| HeLA—5.06 (IC50) | |||
| HCT15—2.77 (IC50) | |||
| Wang et al.102 | Thio-evodiamine | Topo I—gel assay results shown onlyc | A549—0.02 (IC50) |
| (Fig. 9, series D, compound III) | Topo IIα—gel assay results shown onlyc | MDA-MB-435—<0.003 (IC50) | |
| HCT116—<0.003 (IC50) |
Activity is reported for the most active compound if a series of analogs was reported.
Topoisomerase inhibition relative to camptothecin activity was reported for 1,3-diarylisoquinolone compounds.
Gel assay results at 100 indicate inhibition of both Topo I and Topo IIα.
A recent study by Kwon and coworkers99 investigated a novel terpyridine scaffold for inhibitory activity against topoisomerase I and II. An α-terpyridine molecule served as the starting point for the development of these compounds (Fig. 9). Twenty-nine compounds were synthesized and tested for inhibition of eukaryotic topoisomerase I and II. Two compounds with unique scaffolds were identified as potent, non-intercalative catalytic inhibitors. The benzo[4,5]-furo[3,2-b]-pyridine compound (Fig. 9, Series A, n=0) was identified as a dual topoisomerase I and II catalytic inhibitor, while the chromeno[4,3-b]-pyridine compound (Fig. 9, Series A, n=1) was identified as a specific topoisomerase IIα inhibitor. Both compounds showed potent anticancer activity against MCF7, T47D, and HCT15 cells and induced apoptosis and G1 arrest in T47D human breast cancer cells. Results from xenograft mouse tumor growth model tests showed that the benzofuro-pyridine compound significantly reduced both volume and weight of the tumors, but the benzochromeno-pyridine compound, while showing a clear reduction, did not demonstrate a statistically significant difference. DNA toxicity, measured by the accumulation of DNA strand breaks, compared to etoposide in MCF7 and T47D cell lines was also significantly less for both compounds.
Figure 9.

Preclinical topoisomerase inhibitors in recent literature.
Expanding on their work discussed above, Shrestha and coworkers100 recently published the synthesis and testing of a 2nd generation of benzofuro-pyridine compounds with activity against type II topoisomerases (topoisomerase IIα). Sixteen compounds based on the benzofuro[3,2-b]pyridin-7-ol scaffold (Fig. 9, Series B) were synthesized and tested for topoisomerase I and IIα inhibition as well as antiproliferative activity using several cancer cell lines. Activity testing against topoisomerase IIβ was not reported. The cancer cell lines used for the testing were HCT15 (colorectal adenocarcinoma cell line), T47D (ductal breast cancer cell line), and HeLa (cervix tumor cell line). Camptothecin, etoposide, and doxorubicin were used as the controls. Hydroxyl ring substitutions at the R-position were investigated and compounds with ortho and para hydroxyl groups showed greater topoisomerase IIα inhibition than etoposide at both concentrations tested against all cell lines. The meta hydroxyl substituted analog (Fig. 9, compound I) showed the greatest selectivity and most potent inhibitory activity against topoisomerase IIα in the HeLa cell line100. All ring-substituted analogs, and the unsubstituted analog showed potent topoisomerase IIα inhibition and strong antiproliferative activity against the HCT15 and T47D cell lines.
The diarylisoquinolone scaffold presents another novel chemical scaffold currently under investigation for topoisomerase inhibitory potential. Expanding previous efforts in the investigation of 3-arylisoquinolones and 3,4-diarylisoquinolines (Fig. 9, Series C), Khadka and coworkers101, 103 have recently reported the synthesis and evaluation of 1,3-diarylisoquinoline compounds. The C4-aromatic ring of 3,4-diarylisoquinolone was shifted to design 1,3-diarylisoquinoline and various small substituents on both rings were investigated. Overall, 22 compounds were synthesized, tested for activity, and compared with camptothecin and etoposide as controls for topoisomerase I and topoisomerase IIα inhibition. The compounds were also compared to doxorubicin for cytotoxic activity against non-cancerous cell lines. Though the preliminary cytotoxic profile of the 1,3-diarylisoquinolines showed concerning cytotoxic activity against non-cancerous cell lines, the IC50 values for the lines tested were typically at or above those shown for doxorubicin. Compound II (Fig. 9) was shown to be the most potent inhibitor of topoisomerase I and, importantly, demonstrated non-intercalative, catalytic inhibitory activity. The authors did not report testing against topoisomerase IIβ and selectivity for IIα over IIβ is unknown. The novel mechanism of action and reported activity of this new chemical class shows significant promise.
Natural compounds have also been investigated for their potential inhibitory activity against the DNA topoisomerases104. The quinazoline-carboline alkaloid, evodiamine (Fig. 9, series D), is a naturally occurring compound that is used as a dietary supplement for many potential benefits. It is also been reported to be a catalytic inhibitor of both eukaryotic topoisomerase I and II105, 106, 107. Evodiamine has been reported to exhibit anticancer activity in cancer cells resistant to camptothecin107. A recent study by Wang and coworkers102 tested the topoisomerase inhibitor activity of compounds derived by modification of the evodiamine scaffold. These researchers reported the synthesis and testing of 11 compounds against topoisomerase I and II, and tubulin. 3-Chloro-10-hydroxyl thio-evodiamine (Fig. 9, compound III) was found to be a moderate inhibitor of topoisomerase I and II and a potent inhibitor of tubulin and was reported by the investigators to be a first-in-class triple inhibitor. The lead compounds in this study, including compound III, were tested in vivo using a mouse tumor model and the tumor growth inhibition rate was favorable (up to 48% growth inhibition). The toxicity of the compounds was compared to topotecan in the in vivo studies and were found to be lower, based upon a measure of body weight loss. The authors concluded that thio-evodiamine derivatives were a promising new class of topoisomerase/tubulin inhibitors. Notwithstanding the favorable results discussed above, another recent study by Christodoulou and coworkers108 investigating the S-enantiomer of evodiamine, and derivatives thereof, showed that the compounds they synthesized and tested were unable to affect the catalytic activity of topoisomerase I. These researchers ultimately showed that their compounds possessed selective inhibitory activity against the sirtuin, SIRT2. The studies discussed here provide justification for the further investigation of evodiamine and derivatives as potential topoisomerase inhibitors with anti-cancer potential.
Other notable reports in the recent literature of novel topoisomerase inhibitors include the chalcone-linked carbazole derivatives reported by Li and coworkers109 (Fig. 10). These compounds are reported to possess topoisomerase II inhibitory activity with a non-intercalative catalytic inhibitory mechanism. Sathish and coworkers110 have reported the synthesis of a series of podophyllotoxins linked to beta-carbolines with demonstrated anticancer activity against the A549 (lung cancer), DU-145 (prostate cancer), MDAMB-231 (breast cancer), HT-29 (colon cancer), and HeLa (cervical cancer) cell lines. Their investigations to date suggest a topoisomerase II inhibitory mechanism. Jadomycins, an interesting class of natural products, have recently been reported by Hall and coworkers111 to inhibit topoisomerase IIα. Several jadomycin analogs were tested and showed activity against a drug-resistant breast cancer cell line. Mechanistic studies showed that several of the compounds acted as selective topoisomerase IIβ poisons, a concern from a cardiotoxicity standpoint, but that jadomycin S did not act through a poisoning mechanism. Finally, the polyphenol compound, resveratrol, derived from plant sources and red wine, has recently been reported to possess topoisomerase II inhibitory activity43. The chemical similarity of resveratrol to ICRF-187 (dexrazoxane), a known catalytic inhibitor of topoisomerase II, is notable (Fig. 10). Lee and coworkers reported43, based upon a series of biochemical assays, the catalytic inhibitory activity of resveratrol against topoisomerase II via a mechanism related to ICRF-187, though resveratrol appears to prevent ATPase domain dimerization rather than stabilize it, as is seen with the latter compound.
Figure 10.

Additional topoisomerase-active agents in recent literature.
6. Concluding remarks
The DNA topoisomerases continue to represent highly relevant targets for the treatment of various types of cancer and are under continuous investigation as targets for novel classes of inhibitory agents. While nearly all clinically marketed topoisomerase inhibitors used to treat cancer fall within the topoisomerase poison mechanistic category, several very promising catalytic inhibitors have been disclosed recently and offer promise as potentially novel anti-cancer agents with significantly reduced toxicity profiles. Of note are the novel chemical classes with activity against type I topoisomerases, as there is currently only one marketed class of inhibitors in clinical use. This review has presented and discussed several notable examples of novel topoisomerase inhibitors under investigation for cancer indications, both in clinical trials and in pre-clinical studies.
Acknowledgments
This work was supported by faculty development program funding from University of Tennessee Health Science Center (UTHSC) College of Pharmacy to Kirk E. Hevener. Molecular graphics (Figure 1, Figure 2, Figure 3) were created using the University of California, San Francisco (UCSF) Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, USA (supported by U.S. National Institute of General Medical Sciences (NIGMS) P41-GM103311)112.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Champoux J.J. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 2.Bush N.G., Evans-Roberts K., Maxwell A. DNA topoisomerases. EcoSal. 2015;6:2. doi: 10.1128/ecosalplus.ESP-0010-2014. [DOI] [PubMed] [Google Scholar]
- 3.Delgado J.L., Hsieh C.M., Chan N.L., Hiasa H. Topoisomerases as anticancer targets. Biochem J. 2018;475:373–398. doi: 10.1042/BCJ20160583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cuya S.M., Bjornsti M.A., van Waardenburg R.C. DNA topoisomerase-targeting chemotherapeutics: what׳s new? Cancer Chemother Pharmacol. 2017;80:1–14. doi: 10.1007/s00280-017-3334-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem Biol. 2013;8:82–95. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drwal M.N., Agama K., Wakelin L.P., Pommier Y., Griffith R. Exploring DNA topoisomerase I ligand space in search of novel anticancer agents. PLoS One. 2011;6:e25150. doi: 10.1371/journal.pone.0025150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bugreev D.V., Nevinsky G.A. Structure and mechanism of action of type IA DNA topoisomerases. Biochemistry (Moscow) 2009;74:1467–1481. doi: 10.1134/s0006297909130045. [DOI] [PubMed] [Google Scholar]
- 8.Baker N.M., Rajan R., Mondragon A. Structural studies of type I topoisomerases. Nucleic Acids Res. 2009;37:693–701. doi: 10.1093/nar/gkn1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Capranico G., Marinello J., Chillemi G. Type I DNA topoisomerases. J Med Chem. 2017;60:2169–2192. doi: 10.1021/acs.jmedchem.6b00966. [DOI] [PubMed] [Google Scholar]
- 10.Sissi C., Palumbo M. Effects of magnesium and related divalent metal ions in topoisomerase structure and function. Nucleic Acids Res. 2009;37:702–711. doi: 10.1093/nar/gkp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bakshi R.P., Galande S., Muniyappa K. Functional and regulatory characteristics of eukaryotic type II DNA topoisomerase. Crit Rev Biochem Mol Biol. 2001;36:1–37. doi: 10.1080/20014091074165. [DOI] [PubMed] [Google Scholar]
- 12.Watt P.M., Hickson I.D. Structure and function of type II DNA topoisomerases. Biochem J. 1994;303:681–695. doi: 10.1042/bj3030681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bollimpelli V.S., Dholaniya P.S., Kondapi A.K. Topoisomerase IIβ and its role in different biological contexts. Arch Biochem Biophys. 2017;633:78–84. doi: 10.1016/j.abb.2017.06.021. [DOI] [PubMed] [Google Scholar]
- 14.Staker B.L., Hjerrild K., Feese M.D., Behnke C.A., Burgin A.B., Jr, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci U S A. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y.R., Chen S.F., Wu C.C., Liao Y.W., Lin T.S., Liu K.T. Producing irreversible topoisomerase II-mediated DNA breaks by site-specific Pt(II)-methionine coordination chemistry. Nucleic Acids Res. 2017;45:10861–10871. doi: 10.1093/nar/gkx742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lima C.D., Wang J.C., Mondragón A. Three-dimensional structure of the 67K N-terminal fragment of E. coli DNA topoisomerase I. Nature. 1994;367:138–146. doi: 10.1038/367138a0. [DOI] [PubMed] [Google Scholar]
- 17.Changela A., DiGate R.J., Mondragon A. Crystal structure of a complex of a type IA DNA topoisomerase with a single-stranded DNA molecule. Nature. 2001;411:1077–1081. doi: 10.1038/35082615. [DOI] [PubMed] [Google Scholar]
- 18.Garnier F., Debat H., Nadal M., Type I.A. DNA topoisomerases: a universal core and multiple activities. Methods Mol Biol. 2018;1703:1–20. doi: 10.1007/978-1-4939-7459-7_1. [DOI] [PubMed] [Google Scholar]
- 19.Aravind L., Leipe D.D., Koonin E.V. Toprim—a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res. 1998;26:4205–4213. doi: 10.1093/nar/26.18.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stewart L., Redinbo M.R., Qiu X., Hol W.G., Champoux J.J. A model for the mechanism of human topoisomerase I. Science. 1998;279:1534–1541. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- 21.Berger J.M., Gamblin S.J., Harrison S.C., Wang J.C. Structure and mechanism of DNA topoisomerase II. Nature. 1996;379:225–232. doi: 10.1038/379225a0. [DOI] [PubMed] [Google Scholar]
- 22.Fass D., Bogden C.E., Berger J.M. Quaternary changes in topoisomerase II may direct orthogonal movement of two DNA strands. Nat Struct Biol. 1999;6:322–326. doi: 10.1038/7556. [DOI] [PubMed] [Google Scholar]
- 23.Dong K.C., Berger J.M. Structural basis for gate-DNA recognition and bending by type IIA topoisomerases. Nature. 2007;450:1201–1205. doi: 10.1038/nature06396. [DOI] [PubMed] [Google Scholar]
- 24.Roca J., Wang J.C. DNA transport by a type II DNA topoisomerase: evidence in favor of a two-gate mechanism. Cell. 1994;77:609–616. doi: 10.1016/0092-8674(94)90222-4. [DOI] [PubMed] [Google Scholar]
- 25.Nitiss J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer. 2009;9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leelaram M.N., Bhat A.G., Godbole A.A., Bhat R.S., Manjunath R., Nagaraja V. Type IA topoisomerase inhibition by clamp closure. FASEB J. 2013;27:3030–3038. doi: 10.1096/fj.12-226118. [DOI] [PubMed] [Google Scholar]
- 27.Pommier Y., Leo E., Zhang H., Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17:421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hornyak P., Askwith T., Walker S., Komulainen E., Paradowski M., Pennicott L.E. Mode of action of DNA-competitive small molecule inhibitors of tyrosyl DNA phosphodiesterase 2. Biochem J. 2016;473:1869–1879. doi: 10.1042/BCJ20160180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ali J.A., Jackson A.P., Howells A.J., Maxwell A. The 43-kilodalton N-terminal fragment of the DNA gyrase B protein hydrolyzes ATP and binds coumarin drugs. Biochemistry. 1993;32:2717–2724. doi: 10.1021/bi00061a033. [DOI] [PubMed] [Google Scholar]
- 30.Bisacchi G.S., Manchester J.I. A new-class antibacterial-almost. Lessons in drug discovery and development: a critical analysis of more than 50 years of effort toward ATPase inhibitors of DNA gyrase and topoisomerase IV. ACS Infect Dis. 2015;1:4–41. doi: 10.1021/id500013t. [DOI] [PubMed] [Google Scholar]
- 31.Bridewell D.J., Finlay G.J., Baguley B.C. Differential actions of aclarubicin and doxorubicin: the role of topoisomerase I. Oncol Res. 1997;9:535–542. [PubMed] [Google Scholar]
- 32.Larsen A.K., Escargueil A.E., Skladanowski A. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol Ther. 2003;99:167–181. doi: 10.1016/s0163-7258(03)00058-5. [DOI] [PubMed] [Google Scholar]
- 33.Fortune J.M., Osheroff N. Merbarone inhibits the catalytic activity of human topoisomerase IIalpha by blocking DNA cleavage. J Biol Chem. 1998;273:17643–17650. doi: 10.1074/jbc.273.28.17643. [DOI] [PubMed] [Google Scholar]
- 34.Roca J., Ishida R., Berger J.M., Andoh T., Wang J.C. Antitumor bisdioxopiperazines inhibit yeast DNA topoisomerase II by trapping the enzyme in the form of a closed protein clamp. Proc Natl Acad Sci U S A. 1994;91:1781–1785. doi: 10.1073/pnas.91.5.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng S., Yan T., Jendrny C., Nemecek A., Vincetic M., Gödtel-Armbrust U. Dexrazoxane may prevent doxorubicin-induced DNA damage via depleting both topoisomerase II isoforms. BMC Cancer. 2014;14:842. doi: 10.1186/1471-2407-14-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deng S., Yan T., Nikolova T., Fuhrmann D., Nemecek A., Gödtel-Armbrust U. The catalytic topoisomerase II inhibitor dexrazoxane induces DNA breaks, ATF3 and the DNA damage response in cancer cells. Br J Pharmacol. 2015;172:2246–2257. doi: 10.1111/bph.13046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindsey R.H., Jr, Pendleton M., Ashley R.E., Mercer S.L., Deweese J.E., Osheroff N. Catalytic core of human topoisomerase IIα: insights into enzyme-DNA interactions and drug mechanism. Biochemistry. 2014;53:6595–6602. doi: 10.1021/bi5010816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deweese J.E., Osheroff N. The DNA cleavage reaction of topoisomerase II: wolf in sheep׳s clothing. Nucleic Acids Res. 2009;37:738–748. doi: 10.1093/nar/gkn937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gibson E.G., Deweese J.E. Covalent poisons of topoisomerase II. Curr Top Pharmacol. 2013;17:1–12. [Google Scholar]
- 40.Maxwell A., Lawson D.M. The ATP-binding site of type II topoisomerases as a target for antibacterial drugs. Curr Top Med Chem. 2003;3:283–303. doi: 10.2174/1568026033452500. [DOI] [PubMed] [Google Scholar]
- 41.Anderle C., Stieger M., Burrell M., Reinelt S., Maxwell A., Page M. Biological activities of novel gyrase inhibitors of the aminocoumarin class. Antimicrob Agents Chemother. 2008;52:1982–1990. doi: 10.1128/AAC.01235-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chène P., Rudloff J., Schoepfer J., Furet P., Meier P., Qian Z. Catalytic inhibition of topoisomerase II by a novel rationally designed ATP-competitive purine analogue. BMC Chem Biol. 2009;9:1. doi: 10.1186/1472-6769-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee J.H., Wendorff T.J., Berger J.M. Resveratrol: a novel type of topoisomerase II inhibitor. J Biol Chem. 2017;292:21011–21022. doi: 10.1074/jbc.M117.810580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei H., Ruthenburg A.J., Bechis S.K., Verdine G.L. Nucleotide-dependent domain movement in the ATPase domain of a human type IIA DNA topoisomerase. J Biol Chem. 2005;280:37041–37047. doi: 10.1074/jbc.M506520200. [DOI] [PubMed] [Google Scholar]
- 45.FDA. Drugs@FDA: FDA approved drug products [cited 2018 Feb 24]. Available from: 〈https://www.accessdata.fda.gov/scripts/cder/daf/〉.
- 46.Government of Canada. Drug product database online query [cited 2018 Feb 24]. Available from: 〈https://health-products.canada.ca/dpd-bdpp/index-eng.jsp〉.
- 47.European Medicines Agency. Medicines [cited 2018 Feb 24]. Available from: 〈http://www.ema.europa.eu/ema/index.jsp?Curl=pages/includes/medicines/medicines_landing_page.jsp&mid=WC0b01ac058001ce7e〉.
- 48.Oki T. Recent developments in the process improvement of production of antitumor anthracycline antibiotics. Adv Biotechnol Process. 1984;3:163–196. [PubMed] [Google Scholar]
- 49.Bodley A., Liu L.F., Israel M., Seshadri R., Koseki Y., Giuliani F.C. DNA topoisomerase II-mediated interaction of doxorubicin and daunorubicin congeners with DNA. Cancer Res. 1989;49:5969–5978. [PubMed] [Google Scholar]
- 50.Bredehorst R., Panneerselvam M., Vogel C.W. Doxorubicin enhances complement susceptibility of human melanoma cells by extracellular oxygen radical formation. J Biol Chem. 1987;262:2034–2041. [PubMed] [Google Scholar]
- 51.Menna P., Salvatorelli E., Minotti G. Cardiotoxicity of antitumor drugs. Chem Res Toxicol. 2008;21:978–989. doi: 10.1021/tx800002r. [DOI] [PubMed] [Google Scholar]
- 52.Sawyer D.B. Anthracyclines and heart failure. N Engl J Med. 2013;368:1154–1156. doi: 10.1056/NEJMcibr1214975. [DOI] [PubMed] [Google Scholar]
- 53.Zhang S., Liu X., Bawa-Khalfe T., Lu L.S., Lyu Y.L., Liu L.F. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 54.Cvetkovic R.S., Scott L.J. Dexrazoxane: a review of its use for cardioprotection during anthracycline chemotherapy. Drugs. 2005;65:1005–1024. doi: 10.2165/00003495-200565070-00008. [DOI] [PubMed] [Google Scholar]
- 55.Spallarossa P., Garibaldi S., Altieri P., Fabbi P., Manca V., Nasti S. Carvedilol prevents doxorubicin-induced free radical release and apoptosis in cardiomyocytes in vitro. J Mol Cell Cardiol. 2004;37:837–846. doi: 10.1016/j.yjmcc.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 56.Fox E.J. Mechanism of action of mitoxantrone. Neurology. 2004;63(12 Suppl 6):S15–S18. doi: 10.1212/wnl.63.12_suppl_6.s15. [DOI] [PubMed] [Google Scholar]
- 57.Beeharry N., Di Rora A.G., Smith M.R., Yen T.J. Pixantrone induces cell death through mitotic perturbations and subsequent aberrant cell divisions. Cancer Biol Ther. 2015;16:1397–1406. doi: 10.1080/15384047.2015.1070979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Salvatorelli E., Menna P., Paz O.G., Chello M., Covino E., Singer J.W. The novel anthracenedione, pixantrone, lacks redox activity and inhibits doxorubicinol formation in human myocardium: insight to explain the cardiac safety of pixantrone in doxorubicin-treated patients. J Pharmacol Exp Ther. 2013;344:467–478. doi: 10.1124/jpet.112.200568. [DOI] [PubMed] [Google Scholar]
- 59.Longo M., Della Torre P., Allievi C., Morisetti A., Al-Fayoumi S., Singer J.W. Tolerability and toxicological profile of pixantrone (Pixuvri®) in juvenile mice. Comparative study with doxorubicin. Reprod Toxicol. 2014;46:20–30. doi: 10.1016/j.reprotox.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 60.Hsiang Y.H., Hertzberg R., Hecht S., Liu L.F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem. 1985;260:14873–14878. [PubMed] [Google Scholar]
- 61.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 62.Crul M. CKD-602. Chong kun dang. Curr Opin Investig Drugs. 2003;4:1455–1459. [PubMed] [Google Scholar]
- 63.Liu Y.Q., Li W.Q., Morris-Natschke S.L., Qian K., Yang L., Zhu G.X. Perspectives on biologically active camptothecin derivatives. Med Res Rev. 2015;35:753–789. doi: 10.1002/med.21342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park Y.H., Chung C.U., Park B.M., Park M.R., Park D.I., Moon J.Y. Lesser toxicities of belotecan in patients with small cell lung cancer: a retrospective single-center study of camptothecin analogs. Can Respir J. 2016;(2016):3576201. doi: 10.1155/2016/3576201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang X., Rakesh K.P., Shantharam C.S., Manukumar H.M., Asiri A.M., Marwani H.M. Podophyllotoxin derivatives as an excellent anticancer aspirant for future chemotherapy: a key current imminent needs. Bioorg Med Chem. 2018;26:340–355. doi: 10.1016/j.bmc.2017.11.026. [DOI] [PubMed] [Google Scholar]
- 66.Yu X., Che Z., Xu H. Recent advances in the chemistry and biology of podophyllotoxins. Chemistry. 2017;23:4467–4526. doi: 10.1002/chem.201602472. [DOI] [PubMed] [Google Scholar]
- 67.Ross W., Rowe T., Glisson B., Yalowich J., Liu L. Role of topoisomerase II in mediating epipodophyllotoxin-induced DNA cleavage. Cancer Res. 1984;44:5857–5860. [PubMed] [Google Scholar]
- 68.Nelson E.M., Tewey K.M., Liu L.F. Mechanism of antitumor drug action: poisoning of mammalian DNA topoisomerase II on DNA by 4′-(9-acridinylamino)-methanesulfon-m-anisidide. Proc Natl Acad Sci U S A. 1984;81:1361–1365. doi: 10.1073/pnas.81.5.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ketron A.C., Denny W.A., Graves D.E., Osheroff N. Amsacrine as a topoisomerase II poison: importance of drug-DNA interactions. Biochemistry. 2012;51:1730–1739. doi: 10.1021/bi201159b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jehn U., Heinemann V. New drugs in the treatment of acute and chronic leukemia with some emphasis on m-AMSA. Anticancer Res. 1991;11:705–711. [PubMed] [Google Scholar]
- 71.U.S. National Library of Medicine. U.S. national library of medicine clinical trials database [cited 2018 Feb 24]. Available from: 〈https://clinicaltrials.gov/〉.
- 72.BioMed Central Ltd. ISRCTN registry [cited 2018 Feb 24]. Available from: 〈https://www.isrctn.com/〉.
- 73.Marchand C., Antony S., Kohn K.W., Cushman M., Ioanoviciu A., Staker B.L. A novel norindenoisoquinoline structure reveals a common interfacial inhibitor paradigm for ternary trapping of the topoisomerase I-DNA covalent complex. Mol Cancer Ther. 2006;5:287–295. doi: 10.1158/1535-7163.MCT-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pommier Y., Cushman M. The indenoisoquinoline noncamptothecin topoisomerase I inhibitors: update and perspectives. Mol Cancer Ther. 2009;8:1008–1014. doi: 10.1158/1535-7163.MCT-08-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eiseman JL, Holleran J, McCormick DL, Muzzio M, Covey JM, Khanna C, et al. Plasma and tumor pharmacokinetics of IV LMP400, a novel indenoisoquinoline topoisomerase I inhibitor, in a canine phase I study. In: Proceedings of the 105th Annual Meeting of the American Association for Cancer Research. San Diego: American Association for Cancer Research; 2014.
- 76.Kummar S., Chen A., Gutierrez M., Pfister T.D., Wang L., Redon C. Clinical and pharmacologic evaluation of two dosing schedules of indotecan (LMP400), a novel indenoisoquinoline, in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016;78:73–81. doi: 10.1007/s00280-016-2998-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meco D., Di Francesco A.M., Cusano G., Bucci F., Pierri F., Patriarca V. Preclinical evaluation of the novel 7-substituted camptothecin Namitecan (ST1968) in paediatric tumour models. Cancer Chemother Pharmacol. 2012;70:811–822. doi: 10.1007/s00280-012-1973-0. [DOI] [PubMed] [Google Scholar]
- 78.Beretta G.L., Zuco V., De Cesare M., Perego P., Zaffaroni N. Namitecan: a hydrophilic camptothecin with a promising preclinical profile. Curr Med Chem. 2012;19:3488–3501. doi: 10.2174/092986712801323252. [DOI] [PubMed] [Google Scholar]
- 79.Joerger M., Hess D., Delmonte A., Gallerani E., Barbieri P., Pace S. Phase-I dose finding and pharmacokinetic study of the novel hydrophilic camptothecin ST-1968 (namitecan) in patients with solid tumors. Investig New Drugs. 2015;33:472–479. doi: 10.1007/s10637-015-0219-5. [DOI] [PubMed] [Google Scholar]
- 80.Joerger M., Hess D., Delmonte A., Gallerani E., Fasolo A., Gianni L. Integrative population pharmacokinetic and pharmacodynamic dose finding approach of the new camptothecin compound namitecan (ST1968) Br J Clin Pharmacol. 2015;80:128–138. doi: 10.1111/bcp.12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Paubelle E., Zylbersztejn F., Thomas X. The preclinical discovery of vosaroxin for the treatment of acute myeloid leukemia. Expert Opin Drug Discov. 2017;12:747–753. doi: 10.1080/17460441.2017.1331215. [DOI] [PubMed] [Google Scholar]
- 82.Freeman C., Keane N., Swords R., Giles F. Vosaroxin: a new valuable tool with the potential to replace anthracyclines in the treatment of AML? Expert Opin Pharmacother. 2013;14:1417–1427. doi: 10.1517/14656566.2013.799138. [DOI] [PubMed] [Google Scholar]
- 83.Abbas J.A., Stuart R.K. Vosaroxin: a novel antineoplastic quinolone. Expert Opin Investig Drugs. 2012;21:1223–1233. doi: 10.1517/13543784.2012.699038. [DOI] [PubMed] [Google Scholar]
- 84.Hawtin R.E., Stockett D.E., Byl J.A., McDowell R.S., Tan N., Arkin M.R. Voreloxin is an anticancer quinolone derivative that intercalates DNA and poisons topoisomerase II. PLoS One. 2010;5:e10186. doi: 10.1371/journal.pone.0010186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Benton C.B., Ravandi F. Targeting acute myeloid leukemia with TP53-independent vosaroxin. Future Oncol. 2017;13:125–133. doi: 10.2217/fon-2016-0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Voss M.H., Hussain A., Vogelzang N., Lee J.L., Keam B., Rha S.Y. A randomized phase II trial of CRLX101 in combination with bevacizumab versus standard of care in patients with advanced renal cell carcinoma. Ann Oncol. 2017;28:2754–2760. doi: 10.1093/annonc/mdx493. [DOI] [PubMed] [Google Scholar]
- 87.Weiss G.J., Chao J., Neidhart J.D., Ramanathan R.K., Bassett D., Neidhart J.A. First-in-human phase 1/2a trial of CRLX101, a cyclodextrin-containing polymer-camptothecin nanopharmaceutical in patients with advanced solid tumor malignancies. Investig New Drugs. 2013;31:986–1000. doi: 10.1007/s10637-012-9921-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Young C., Schluep T., Hwang J., Eliasof S. CRLX101 (formerly IT-101)-a novel nanopharmaceutical of camptothecin in clinical development. Curr Bioact Compd. 2011;7:8–14. doi: 10.2174/157340711795163866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Svenson S., Wolfgang M., Hwang J., Ryan J., Eliasof S. Preclinical to clinical development of the novel camptothecin nanopharmaceutical CRLX101. J Control Release. 2011;153:49–55. doi: 10.1016/j.jconrel.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 90.Clark A.J., Wiley D.T., Zuckerman J.E., Webster P., Chao J., Lin J. CRLX101 nanoparticles localize in human tumors and not in adjacent, nonneoplastic tissue after intravenous dosing. Proc Natl Acad Sci U S A. 2016;113:3850–3854. doi: 10.1073/pnas.1603018113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Belada D., Georgiev P., Dakhil S., Inhorn L.F., Andorsky D., Beck J.T. Pixantrone-rituximab versus gemcitabine-rituximab in relapsed/refractory aggressive non-Hodgkin lymphoma. Future Oncol. 2016;12:1759–1768. doi: 10.2217/fon-2016-0137. [DOI] [PubMed] [Google Scholar]
- 92.Sachdev E., Sachdev D., Mita M. Aldoxorubicin for the treatment of soft tissue sarcoma. Expert Opin Investig Drugs. 2017;26:1175–1179. doi: 10.1080/13543784.2017.1371134. [DOI] [PubMed] [Google Scholar]
- 93.Kohli A.G., Kierstead P.H., Venditto V.J., Walsh C.L., Szoka F.C. Designer lipids for drug delivery: from heads to tails. J Control Release. 2014;190:274–287. doi: 10.1016/j.jconrel.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lazareva N.F., Baryshok V.P., Lazarev I.M. Silicon-containing analogs of camptothecin as anticancer agents. Arch Pharm (Weinheim) 2018;351:e1700297. doi: 10.1002/ardp.201700297. [DOI] [PubMed] [Google Scholar]
- 95.Chen A.Y., Shih S.J., Garriques L.N., Rothenberg M.L., Hsiao M., Curran D.P. Silatecan DB-67 is a novel DNA topoisomerase I-targeted radiation sensitizer. Mol Cancer Ther. 2005;4:317–324. [PubMed] [Google Scholar]
- 96.Prudhomme M. Recent developments of rebeccamycin analogues as topoisomerase I inhibitors and antitumor agents. Curr Med Chem. 2000;7:1189–1212. doi: 10.2174/0929867003374138. [DOI] [PubMed] [Google Scholar]
- 97.Tolcher A.W., Eckhardt S.G., Kuhn J., Hammond L., Weiss G., Rizzo J. Phase I and pharmacokinetic study of NSC 655649, a rebeccamycin analog with topoisomerase inhibitory properties. J Clin Oncol. 2001;19:2937–2947. doi: 10.1200/JCO.2001.19.11.2937. [DOI] [PubMed] [Google Scholar]
- 98.Schwandt A., Mekhail T., Halmos B., O׳Brien T., Ma P.C., Fu P. Phase-II trial of rebeccamycin analog, a dual topoisomerase-I and -II inhibitor, in relapsed "sensitive" small cell lung cancer. J Thorac Oncol. 2012;7:751–754. doi: 10.1097/JTO.0b013e31824abca2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kwon H.B., Park C., Jeon K.H., Lee E., Park S.E., Jun K.Y. A series of novel terpyridine-skeleton molecule derivants inhibit tumor growth and metastasis by targeting topoisomerases. J Med Chem. 2015;58:1100–1122. doi: 10.1021/jm501023q. [DOI] [PubMed] [Google Scholar]
- 100.Shrestha A., Jo H., Kwon Y., Lee E.S. Design. synthesis, and structure—activity relationships of new benzofuro[3,2-b.pyridin-7-ols as DNA topoisomerase II inhibitors. Bioorg Med Chem Lett. 2018;28:566–571. doi: 10.1016/j.bmcl.2018.01.048. [DOI] [PubMed] [Google Scholar]
- 101.Khadka D.B., Woo H., Yang S.H., Zhao C., Jin Y., Le T.N. Modification of 3-arylisoquinolines into 3,4-diarylisoquinolines and assessment of their cytotoxicity and topoisomerase inhibition. Eur J Med Chem. 2015;92:583–607. doi: 10.1016/j.ejmech.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 102.Wang S., Fang K., Dong G., Chen S., Liu N., Miao Z. Scaffold diversity inspired by the natural product evodiamine: discovery of highly potent and multitargeting antitumor agents. J Med Chem. 2015;58:6678–6696. doi: 10.1021/acs.jmedchem.5b00910. [DOI] [PubMed] [Google Scholar]
- 103.Khadka D.B., Park S., Jin Y., Han J., Kwon Y., Cho W.J. Design, synthesis, and biological evaluation of 1,3-diarylisoquinolines as novel topoisomerase I catalytic inhibitors. Eur J Med Chem. 2018;143:200–215. doi: 10.1016/j.ejmech.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 104.Scotti L., Bezerra Mendonca F.J., Ribeiro F.F., Tavares J.F., da Silva M.S., Barbosa Filho J.M. Natural product inhibitors of topoisomerases: review and docking study. Curr Protein Pept Sci. 2018;19:275–291. doi: 10.2174/1389203718666170111114442. [DOI] [PubMed] [Google Scholar]
- 105.Chan A.L., Chang W.S., Chen L.M., Lee C.M., Chen C.E., Lin C.M. Evodiamine stabilizes topoisomerase I-DNA cleavable complex to inhibit topoisomerase I activity. Molecules. 2009;14:1342–1352. doi: 10.3390/molecules14041342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dong G., Sheng C., Wang S., Miao Z., Yao J., Zhang W. Selection of evodiamine as a novel topoisomerase I inhibitor by structure-based virtual screening and hit optimization of evodiamine derivatives as antitumor agents. J Med Chem. 2010;53:7521–7531. doi: 10.1021/jm100387d. [DOI] [PubMed] [Google Scholar]
- 107.Pan X., Hartley J.M., Hartley J.A., White K.N., Wang Z., Bligh S.W. Evodiamine, a dual catalytic inhibitor of type I and II topoisomerases, exhibits enhanced inhibition against camptothecin resistant cells. Phytomedicine. 2012;19:618–624. doi: 10.1016/j.phymed.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 108.Christodoulou M.S., Sacchetti A., Ronchetti V., Caufin S., Silvani A., Lesma G. Quinazolinecarboline alkaloid evodiamine as scaffold for targeting topoisomerase I and sirtuins. Bioorg Med Chem. 2013;21:6920–6928. doi: 10.1016/j.bmc.2013.09.030. [DOI] [PubMed] [Google Scholar]
- 109.Li P.H., Jiang H., Zhang W.J., Li Y.L., Zhao M.C., Zhou W. Synthesis of carbazole derivatives containing chalcone analogs as non-intercalative topoisomerase II catalytic inhibitors and apoptosis inducers. Eur J Med Chem. 2018;145:498–510. doi: 10.1016/j.ejmech.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 110.Sathish M., Kavitha B., Nayak V.L., Tangella Y., Ajitha A., Nekkanti S. Synthesis of podophyllotoxin linked β-carboline congeners as potential anticancer agents and DNA topoisomerase II inhibitors. Eur J Med Chem. 2018;144:557–571. doi: 10.1016/j.ejmech.2017.12.055. [DOI] [PubMed] [Google Scholar]
- 111.Hall S.R., Toulany J., Bennett L.G., Martinez-Farina C.F., Robertson A.W., Jakeman D.L. Jadomycins inhibit type II topoisomerases and promote dna damage and apoptosis in multidrug-resistant triple-negative breast cancer cells. J Pharmacol Exp Ther. 2017;363:196–210. doi: 10.1124/jpet.117.241125. [DOI] [PubMed] [Google Scholar]
- 112.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]