

Abstract
Aims
This study aimed to determine the effect of food intake on uracil and dihydrouracil plasma levels. These levels are a promising marker for dihydropyrimidine dehydrogenase activity and for individualizing fluoropyrimidine anticancer therapy.
Methods
A randomized, cross‐over study in 16 healthy volunteers was performed, in which subjects were examined in fasted and fed state on two separate days. In fed condition, a high‐fat, high‐caloric breakfast was consumed between 8:00 h and 8:30 h. Whole blood for determination of uracil, dihydrouracil and uridine plasma levels was drawn on both test days at predefined time points between 8:00 h and 13:00 h.
Results
Uracil levels were statistically significantly different between fasting and fed state. At 13:00 h, the mean uracil level in fasting state was 12.6 ± 3.7 ng ml−1 and after a test meal 9.4 ± 2.6 ng ml−1 (P < 0.001). Dihydrouracil levels were influenced by food intake as well (mean dihydrouracil level at 13:00 h in fasting state 147.0 ± 36.4 ng ml−1 and in fed state 85.7 ± 22.1 ng ml−1, P < 0.001). Uridine plasma levels showed curves with similar patterns as for uracil.
Conclusions
It was shown that both uracil and dihydrouracil levels were higher in fasting state than in fed state. This is hypothesized to be an direct effect of uridine plasma levels, which were previously shown to be elevated in fasting state and reduced after intake of food. These findings show that, when assessing plasma uracil and dihydrouracil levels for adaptive fluoropyrimidine dosing in clinical practice, sampling should be done between 8:00 h and 9:00 h after overnight fasting to avoid bias caused by circadian rhythm and food effects.
Keywords: dihydropyrimidine dehydrogenase, dihydrouracil, food‐effect study, uracil, uridine
What is Already Known about this Subject
Dihydropyrimidine dehydrogenase (DPD) is the main metabolic enzyme for fluoropyrimidines.
The endogenous substrate and its product, uracil and dihydrouracil, can be measured as a surrogate of DPD activity and are considered promising markers for individualizing fluoropyrimidine therapy.
Currently, the effect of food intake on uracil/dihydrouracil levels is unclear.
What this Study Adds
We showed that uracil and dihydrouracil levels were significantly lower after food intake than in fasting state, which is thought to be a direct effect of uridine homeostasis.
This implies that sampling should be done in the morning and in the fasted state to avoid bias caused by food effects.
Introduction
The fluoropyrimidine anticancer drugs 5‐fluorouracil (5‐FU) and its oral prodrug capecitabine are commonly used in the treatment of solid tumours, including early and advanced breast, colorectal, gastric and head‐and‐neck cancer. The enzyme dihydropyrimidine dehydrogenase (DPD), encoded by the gene DPYD, plays an important role in the metabolism of fluoropyrimidines. Over 80% of the administered dose of 5‐FU is inactivated in the liver by DPD, which makes DPD the key metabolizing enzyme of fluoropyrimidines 1, 2. DPD enzyme activity is known to have a high interindividual variability and reduced DPD activity is present in up to 5% of the population. DPD deficiency is an important risk factor for developing severe, potentially fatal, fluoropyrimidine‐related toxicity when patients are treated with a standard fluoropyrimidine dose 3, 4, 5, 6.
DPD deficiency is often caused by single nucleotide polymorphisms (SNPs) in the DPYD gene. Pre‐treatment DPYD screening and dose individualization based on DPYD polymorphisms have shown to significantly improve patient safety 7. However, as not all DPD deficiency can be attributed to genetic DPYD variants, other methods to identify DPD‐deficient patients at risk of fluoropyrimidine‐related toxicity are being investigated, including DPD phenotyping approaches.
A frequently used phenotyping method is measuring DPD activity in peripheral blood mononuclear cells (PBMCs), as liver DPD activity correlates relatively well with DPD activity in PBMCs 8. However, this method seems less suitable for routine clinical use, as this method is expensive, laborious, logistically difficult, and requires specific equipment which is not available in most hospitals 9.
Another promising phenotyping approach to identify DPD‐deficient patients is determining the levels of uracil (U), the endogenous substrate for DPD, and its product dihydrouracil (DHU). Multiple studies have shown an association between high endogenous U levels or a low DHU/U ratio and severe fluoropyrimidine‐associated toxicity 10, 11, 12, 13, 14, 15. These results support the idea that U and DHU levels could be used to individualize fluoropyrimidine therapy in order to increase patient safety. However, an important uncertainty is that there is limited information on potential factors influencing the U and DHU levels and DHU/U ratio, such as circadian rhythm 16 or intake of food containing high levels of U. Therefore, the aim of this study was to determine the effect of oral food intake on plasma U and DHU levels, in order to investigate if a fasting state is necessary when U and DHU levels will be used as a diagnostic marker for DPD activity in routine clinical practice.
Information on food containing high levels of U or its precursor uridine is limited. Uridine can be converted in vivo to U by a phosphorolysis reaction. This reaction is catalysed by the enzyme uridine phosphorylase 17 (see Figure 1). U is also one of the four bases in RNA, so intake is also influenced by RNA contents in food. Daily RNA and DNA intake is typically in the range of 0.1–1 g person−1 day−1 18. In the gastrointestinal tract, RNA is broken down into nucleic bases including U. Relatively high concentrations of RNA and DNA can be found in edible offal, animal muscle tissues and mushrooms, whereas plant‐derived foods contain lower concentrations 18, 19.
Figure 1.
Metabolism of uridine, uracil and dihydrouracil
In this study, a breakfast containing food expected to have a high U content was consumed by healthy volunteers in a randomized, cross‐over study. It was hypothesized that U levels, and potentially also DHU levels, would increase after consumption of the test breakfast, compared to the fasting state. The basis of this hypothesis was the assumption that the U present in food would increase the U plasma concentrations after absorption.
Methods
Study design and sample collection
Sixteen healthy volunteers participated in the study. Enrolled subjects were aged 18 or older, not pregnant and able and willing to consume the prescribed breakfast and undergo blood sampling. The study (clinical http://trials.gov identifier: NCT02718664) was approved by the Medical Ethics Committee of The Netherlands Cancer Institute, Amsterdam, The Netherlands, and was conducted in accordance with the Declaration of Helsinki. All participants provided written informed consent prior to study assessments. The study had a randomized, cross‐over design, consisting of two test days: day A (fasting state, no food allowed from 22:00 h the previous night until 13:00 h on the test day) and day B (a test meal was consumed between 8:00 h and 8:30 h, no other food allowed from 22:00 h the previous night until 13:00 h). On both test days, consumption of tap water was allowed during the study period. The test days were planned on two consecutive days and participants were 1:1 randomized for the order of the test days (AB or BA).
Blood for determination of U, DHU and uridine plasma levels was collected on both days on 11 predefined time points between 8:00 h and 13:00 h (8:00 h, 8:45 h, 9:00 h, 9:15 h, 9:30 h, 10:00 h, 10:30 h, 11:00 h, 11:30 h, 12:00 h, 13:00 h). On one of the test days, an additional blood sample was taken at 8:00 h for determination of DPD enzyme activity in PBMCs. Also, a blood sample was collected for DPYD genotyping.
Determination of U, DHU and uridine plasma levels
Peripheral blood for assessment of U and DHU was drawn in a heparin tube (4 ml) and centrifuged directly (1500 g, 10 min, 4°C). Plasma was stored at −80°C until analysis. A validated ultra‐performance liquid chromatography–tandem mass spectrometry (UPLC‐MS/MS) assay was used for quantification of U and DHU levels as described by Jacobs et al. 20.
As an exploratory analysis, uridine levels were determined in the plasma samples that were drawn for determination of U and DHU levels. The same UPLC‐MS/MS assay developed by Jacobs et al. 20 was used for quantification of uridine levels, using the same sample pre‐treatment methods and analytical system settings. The concentration range for uridine was 50–5000 ng ml−1 and uridine‐2‐13C‐1,3‐15N2 was used as internal standard.
Determination of DPD enzyme activity in PBMCs
Ten millilitres of peripheral blood, drawn in a heparin tube, was collected for assessment of DPD activity in PBMCs. PBMCs were isolated directly, using Ficoll‐Paque density gradient centrifugation as described previously 21. Isolated PBMCs were stored at −80°C until further analysis. A validated radio‐assay was used, where DPD activity was expressed as the amount of 3H‐dihydrothymine formed per mg of protein of PBMC after 1 h of ex vivo incubation with 3H‐thymine 21.
DPYD genotyping
Genotyping for four DPYD variants was performed. DPYD variants tested were DPYD*2A (c.1905+1G>A, IVS14+1G>A, rs3918290), c.1679T>G (rs55886062), c.2846A>T (rs67376798) and c.1236G>A (rs56038477, in haplotype B3). DNA was isolated from 4 ml EDTA peripheral blood, and DPYD genotyping was performed with real‐time PCR, using the Roche LightCycler® 480II platform and commercially available primers and probes.
Test meal
On day B of the study (fed condition), a standardized breakfast had to be consumed. This test meal was in accordance with a high‐fat (approximately 50% of total caloric content of the meal) and high‐caloric (approximately 800–1000 kcal) meal as described in the guidance on food‐effect bioavailability and fed bioequivalence studies of the US Food and Drug Administration (FDA) 22. The breakfast consisted of two slices of whole‐wheat bread, two boiled eggs, two tomatoes, one portion (30 g) of liverwurst (liver sausage), one portion (30 g) of steak tartare, one portion (30 g) unsalted butter and 200 ml of whole milk. The total breakfast contained approximately 820 kcal, of which 490 kcal was provided by fat. Ingredients were included which were expected to have a potentially large effect on U levels, e.g. liverwurst containing pig liver. The test meal had to be consumed between 8:00 h and 8:30 h and whether the whole meal was finished within this time period was monitored. We estimated that the test meal would contain at least 15 mg of U, and based on the published value of 474 l for the volume of distribution divided by the bioavailability (Vd/F) 23, we calculated that intake of this amount of U could potentially result in plasma levels of 32 ng ml−1 (15/474 = 0.032 mg l−1), and therefore might significantly increase U levels.
Sample size calculation and statistical analyses
The primary objective of the study was to determine the effect of oral food intake on plasma U and DHU levels. A required sample size of 16 was calculated, which is also in compliance with the FDA guidance, where it is stated that a minimum of 12 subjects should be included in food‐effect studies 22. For sample size calculation, two null hypotheses were taken into account: first, that the difference in mean for U levels in both conditions is below −4 ng ml−1 and, second, that it is above 4 ng ml−1. A 90% power was chosen to reject both null hypotheses in favour of the alternative hypothesis that the means of the two conditions (A and B) are equivalent. This assumed that the expected difference in means is zero, the crossover ANOVA root mean squared error is 3.16 (so the standard deviation of differences is 4.47), and that each test is made at the 5.0% alpha level. The standard deviation of differences was calculated assuming that the standard deviation is two under the first condition (A), four under the second condition (B) and that the correlation between the two is zero.
Descriptive statistics were used to describe DPD activity in PBMCs, U, DHU and uridine levels and DHU/U ratio. Paired t‐tests were used for comparison of U levels, DHU levels and DHU/U ratio between condition A and B at different time points (8:00 h, 10:30 h, 13:00 h). Pearson correlation coefficients were estimated to examine the association between DPD activity in PBMCs and U levels, DHU levels, DHU/U ratio and uridine. The threshold for statistical significance was set at P < 0.05.
Results
A total of 16 participants (eight females, eight males) were included, with a median age of 27 years (range 25–46 years). All participants were Caucasian. Participants were equally randomized for the order of the test days (eight subjects randomized as AB, eight as BA). Baseline characteristics are summarized in Table 1.
Table 1.
Baseline characteristics of participants
| Characteristic | Participants (n = 16) |
|---|---|
| Age | |
| Median (range) | 27 (25–46) |
| Gender | |
| Male (%) | 8 (50%) |
| Female (%) | 8 (50%) |
| Height (m) | |
| Mean (range) | 1.77 (1.64–1.93) |
| Weight (kg) | |
| Mean (range) | 73 (58–96) |
| Body mass index (kg m −2 ) | |
| Mean (range) | 23.2 (20.4–27.5) |
| Randomization | |
| AB (%) | 8 (50%) |
| BA (%) | 8 (50%) |
| DPYD genotype | |
| Wild‐type a | 14 (87.5%) |
| DPYD *2A heterozygous | 1 (6.25%) |
| c.1236G>A heterozygous | 1 (6.25%) |
Wild‐type for four DPYD variants: DPYD*2A, c.1236G>A, c.2846A>T and c.1679T>G
DPD activity in PBMCs was shown to have a relatively high interindividual variability, with a mean value of 14.2 nmol−1 (mg*h) and standard deviation (SD) of 5.5 nmol−1 (mg*h); individual values for PBMC DPD activity are presented in Table S1. The participant with the lowest PBMC DPD activity (subject six; 3.4 nmol−1 (mg*h)) was identified as a heterozygous carrier of the DPYD*2A variant. Another participant (subject eight) carried the DPYD c.1236G>A variant heterozygously. This variant, however, did not result in decreased DPD activity in PBMCs for this subject (value of 13.4 nmol−1 (mg*h)). All other participants were tested as wild‐type for the four DPYD variants analysed in this study.
The individual and mean day curves for U levels, DHU levels, DHU/U ratio and uridine levels are shown in Figure 2. Results are shown separately for condition A (fasting state) and condition B (consumption of test meal between 8:00 h and 8:30 h). As shown in Figure 2A, mean U levels at 8:00 h were higher for fasting state compared to fed state (14.5 ± 4.3 ng ml−1 for fasting state and 13.4 ± 3.5 ng ml−1 for fed state, P = 0.03), but with a difference of 1.1 ng ml−1 between the two conditions; this was not considered clinically relevant. At this time point, participants were in fasting state on both days. In both conditions, U levels declined during the day. However, after consumption of the test meal (condition B), U levels declined significantly more than in fasting state (condition A). A drop between 8:45 h and 10:30 h was observed after consumption of the test meal, after which the mean value remained relatively constant. At 10:30 h, mean U levels in fasting state were 13.5 ± 4.7 ng ml−1 and in fed state 9.2 ± 2.4 ng ml−1 (P < 0.001). At 13:00 h, the U level had a mean value of 12.6 ± 3.7 ng ml−1 for fasting state and 9.4 ± 2.6 ng ml−1 for fed state (P < 0.001).
Figure 2.
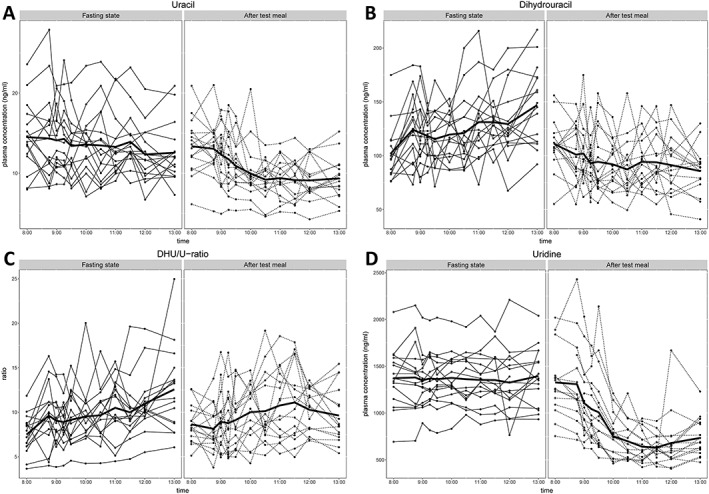
Uracil levels (A), dihydrouracil levels (B), dihydrouracil/uracil ratio (C) and uridine levels (D) in plasma. Levels were determined at predetermined time points between 8:00 h and 13:00 h in 16 participants in a fasting state (arm A) and after consuming a standardized breakfast between 8:00 h and 8:30 h (arm B). Individual levels are depicted and the mean values (bold line). DHU: dihydrouracil; U: uracil
For DHU levels, results are summarized in Figure 2B. Mean DHU levels at 8:00 h, when participants were in a fasting state on both days, were not significantly different between both test days (102.2 ± 25.2 ng ml−1 for fasting state and 111.0 ± 23.6 ng ml−1 for fed state, P = 0.23). However, in the fasting state, mean levels were found to increase over the day, with a maximum of 147.0 ± 36.4 ng ml−1 at 13:00 h, and in the fed state mean levels declined over the day, to a mean level of 85.7 ± 22.1 ng ml−1 at 13:00 h. This difference at 13:00 h was found to be statistically significant (P < 0.001). At 10:30 h, mean DHU levels in fasting state were 121.0 ± 32.1 ng ml−1 and in fed state 87.3 ± 26.9 ng ml−1 (P = 0.003).
When combining U and DHU levels into the DHU/U ratio, as depicted in Figure 2C, both at 8:00 h and 13:00 h the mean DHU/U ratio values were significantly different (8:00 h: fasting state = 7.5 ± 2.2, fed state = 8.6 ± 2.1, P = 0.012; 13:00 h: fasting state = 12.6 ± 4.7; fed state = 9.6 ± 3.1, P = 0.012), but not at 10:30 h (fasting state = 9.7 ± 3.0, fed state = 10.1 ± 4.0, P = 0.65). Individual results per subject for U levels, DHU levels and DHU/U ratio are included as supplemental information (Table S1 and Figure S1).
In an exploratory analysis, uridine levels were measured in the same plasma samples in which U and DHU levels were quantified. For nine plasma samples, insufficient plasma was available to determine uridine levels. Uridine measurements showed that curves for uridine showed similar patterns as for U, with higher (stable) levels in the fasting state, and a drop in levels after intake of the breakfast. The uridine results are depicted in Figure 2D and individual results in Figure S1.
Associations between DPD activity in PBMCs and U plasma levels, DHU plasma levels, DHU/U ratio and uridine plasma levels were investigated (Figure S2). There was a significant negative correlation coefficient between PBMC DPD activity and U plasma levels (r 2 = 0.4220; P = 0.0065) and a significant positive correlation between PBMC DPD activity and DHU/U ratio (r 2 = 0.6162; P = 0.0003). DHU levels were not significantly correlated with PBMC DPD activity (r 2 = 0.1402; P = 0.153), nor were uridine levels (r 2 = 0.2018; P = 0.093).
Discussion
As far as we know, this is the first study to investigate the effect of oral food intake on U and DHU plasma levels. U levels and DHU/U ratio are promising biomarkers for DPD enzyme activity, 5‐FU clearance and as a predictor of fluoropyrimidine‐related toxicity 10, 11, 12, 13, 14, 15, 24, 25, 26, 27. Our recent retrospective study in 550 patients showed that a high pre‐treatment U level (>16 ng ml−1) was strongly associated with severe fluoropyrimidine‐related toxicity (OR 5.3, P = 0.009) 14. Additionally, several prospective studies have been performed, in which dose reduction of fluoropyrimidine‐based chemotherapy was performed based on DHU/U ratio 25, 28 or a combination of DPYD genotype and DHU/U ratio 29, resulting in lower incidence of severe fluoropyrimidine‐related toxicity. However, a concern is that U and DHU levels might not only be influenced by systemic DPD activity, but also by other factors, e.g. circadian rhythm 16 or food containing high levels of U or uridine. In several studies investigating DHU and U levels, blood is therefore drawn in the morning and after a fasting period 12, 24, 26, 30. However, direct evidence showing the effect of oral food intake on U and DHU levels was not yet available.
In this study, we showed that U and DHU levels were not only influenced by interindividual variation and the time of the day (indicating a circadian rhythmicity), but that, indeed, intake of food was a statistically significant contributing factor as well. However, the influence had the opposite direction to what was initially hypothesized. Instead of an increase in U levels after food intake, we found that U levels both declined from 8:00 h to 13:00 h in a fasting state and after food intake, but that the decline was more pronounced after food intake compared to the fasting state. For DHU levels, the effect of oral food intake seemed even more pronounced, as the levels from 8:00 h to 13:00 h increased in the fasting state and decreased after the intake of the test breakfast. When combining U and DHU levels into the DHU/U ratio, the effect of food status is most significant after 12.00 h, influenced by the big difference between DHU levels in fasting and fed state at this time period.
These effects of food intake on U and DHU levels have not been shown previously and the exact mechanism behind these findings is uncertain. The results of our study suggest that certain metabolic processes in the body which are influenced by a prolonged fasting state or, conversely, the intake of a high‐caloric meal, influence plasma U and DHU levels. In the fasting condition, participants had to abstain from food from 22:00 h to 13:00 h the following day, meaning a period of 15 h. For uridine, the precursor of U, it has recently been shown by Deng et al. that plasma uridine levels are elevated during fasting state and show a rapid drop in a post‐prandial state 31, thus showing a similar pattern as U levels in our study. Adipose tissue dominates uridine biosynthetic activity in the fasted state, resulting in elevated plasma uridine levels, but after food intake, a rapid reduction of plasma uridine is seen, both caused by reduction of uridine synthesis in adipocytes and enhancement of its clearance through the bile 31. Assuming that endogenous U plasma levels are largely dependent on uridine homeostasis and not on the intake of U by food, this phenomenon could be an explanation for the findings in our study. It has been shown that uridine homeostasis is tightly regulated by the enzyme uridine phosphorylase, the enzyme which converts uridine to U 17, 32, 33. This supports the hypothesis that U levels are mainly dependent on uridine homeostasis. When radioactively labelled 3H‐uridine was administered intraperitoneally to mice, 3H‐uridine was metabolized rapidly with a half‐life of less than 2 min, and radioactive 3H‐uracil levels were detected in plasma already 5 min after administration 32. For uridine present in food, it was shown that gut‐derived uridine is not released in the systemic circulation, but subsequently circulates with bile within the enterohepatic circulation 31. This is also in line with our findings that food intake did not result in an increase in U levels.
The exploratory analyses in our study confirmed that uridine levels showed a similar pattern as U levels, suggesting that the differences between fed and fasted state of U and DHU are indeed likely to be the direct result of the homeostatic control of uridine. However, when comparing the uridine and U curves over the day, uridine levels show a larger drop in plasma levels after intake of food than the U levels, where the drop is more modest. This could be caused by the high uracil‐containing test meal, from which potentially some uracil is directly taken up in the systemic circulation, resulting in higher U plasma levels. Another possible explanation for this difference between uridine and U is that the formation of U from uridine is rather slow, thereby limiting the direct effect on plasma levels of U by uridine homeostasis. This is, however, in contrast to the rapid conversion from uridine to U described earlier 32.
Our study shows that mean levels of uridine, U and DHU clearly have a distinct pattern for fasting state and fed state. The mean difference for U levels between fed and fasted state was between 2 and 5 ng ml−1 (depending on the time point), showing the relevance of taking food status into account when assessing U plasma levels. The results of our study also suggest that findings of previous studies investigating U levels or the DHU/U ratio in association with fluoropyrimidine‐related toxicity might have been influenced by unknown food status. Results of our study also indicate that both inter‐ and intra‐individual variation of uridine, U and DHU levels are high, even in a homogeneous healthy volunteer group. This is in line with our previous study in healthy volunteers, where high inter‐ and intra‐individual variation for U and DHU levels were identified as well 16. When setting a threshold for dose individualization of fluoropyrimidines based on U levels or the DHU/U ratio, it is important to keep in mind this variation, as this can influence the chance of incorrectly classifying someone as DPD deficient based on a measured plasma level. In general, influences of circadian rhythm and food, among other factors that contribute to the high variation, complicate the implementation of DPD phenotyping in clinical practice, as it requires strict settings for blood drawing in patients. This is contrary to DPYD genotyping, where results inherently cannot be influenced by environmental factors. Another important aspect to take into account for feasibility in clinical practice is the method for quantification of U and DHU levels. We used a validated mass spectrometry‐based assay. Previously it was shown that there is extensive variability in reported ranges of endogenous DHU/U ratio in different studies, which is thought to be largely dependent on the method used 34. To increase the feasibility and implementation of this assay in smaller hospitals, where it is likely that no UPLC‐MS/MS equipment will be available, samples could also be shipped for quantification to another hospital if turnaround times (i.e. time between sample collection and reporting of the result for deciding on treatment individualization) can be limited.
In our study we used a relatively homogeneous group of healthy volunteers and fixed test conditions. Results can therefore not directly be extrapolated to a real‐world setting of cancer patients, where a higher interindividual variation in uridine and U metabolism can be expected. Also, it is unknown what the effect of cachexia, present in a proportion of cancer patients, would be on the uridine and U homeostasis. In addition, with our study settings, differences between the fasting state (15 h of fasting) and the fed state (intake of a high‐caloric breakfast) were large, which can also not be directly extrapolated to a clinical setting of patients. However, by using this controlled study setting, we were able to identify the significant influence of food on U and DHU levels, which is expected to be of relevance in a daily care patient setting as well. Validation of our findings in cancer patients in a clinical care setting is, however, warranted.
Of interest is the relationship between DPYD genotype and DPD phenotype (e.g. measured by DPD enzyme activity in PBMCs or U levels or DHU/U ratio). In our study we identified two DPYD variant allele carriers. The participant who carried the DPYD*2A variant heterozygously had a significantly decreased DPD enzyme activity and corresponding significantly increased U levels. This is in line with expectations, due to the known deleterious effect of the DPYD*2A variant on the DPD enzyme, leaving a heterozygous carrier with approximately 50% remaining DPD activity. The other DPYD variant allele carrier in our study was a heterozygous c.1236G>A carrier. This DPYD variant is known to result in decreased DPD enzyme activity as well, but the effect is thought to be less strong than for DPYD*2A 35. The participant in our current study had normal DPD enzyme activity in PBMCs and U levels, comparable to the other participants in our study. Therefore, this participant seems to have no functional DPD deficiency, despite the presence of a DPYD variant. A previous case series including two homozygous carriers of the c.1236G>A variant suggested that there is a relatively high variation of the effect of this genotype on DPD phenotype in patients. The exact mechanism behind this remains unclear 36.
In conclusion, with this study we showed that both U levels and DHU levels are generally lower after the intake of a high‐caloric breakfast, compared to a fasting state. This means that oral food intake of patients should be taken into account when blood is drawn for determination of U and DHU levels. We recommend choosing fixed circumstances for blood collection for measuring U and DHU levels, such as a collection time between 8:00 h and 9:00 h after overnight fasting, as this will minimize the effects of potential confounders as much as possible.
Competing Interests
There are no competing interests to declare.
Contributors
L.H., B.J., D.M., H.G., A.C., J.B. and J.S. were involved in conception and design of the study. L.H., B.J., D.P., D.B., N.V., H.R. and A.H. were involved in acquisition, analysis and interpretation of the data. L.H. drafted the manuscript and all other authors critically revised the manuscript. All authors gave final approval of the manuscript to be published. J.S. was the principal investigator.
Supporting information
Figure S1 Uracil levels (A), dihydrouracil levels (B), DHU/U ratio (C) and uridine levels (D) in plasma per individual participant
Figure S2 Correlation between DPD activity in PBMCs and uracil plasma level (A), dihydrouracil plasma level (B), DHU/U ratio (C) and uridine plasma level (D)
Table S1 Results for DPD activity, uracil and dihydrouracil levels per individual participant
Henricks, L. M. , Jacobs, B. A. W. , Meulendijks, D. , Pluim, D. , van den Broek, D. , de Vries, N. , Rosing, H. , Beijnen, J. H. , Huitema, A. D. R. , Guchelaar, H.‐J. , Cats, A. , and Schellens, J. H. M. (2018) Food‐effect study on uracil and dihydrouracil plasma levels as marker for dihydropyrimidine dehydrogenase activity in human volunteers. Br J Clin Pharmacol, 84: 2761–2769. 10.1111/bcp.13719.
References
- 1. Diasio RB, Harris BE. Clinical pharmacology of 5‐fluorouracil. Clin Pharmacokinet 1989; 16: 215–237. [DOI] [PubMed] [Google Scholar]
- 2. Longley DB, Harkin DP, Johnston PG. 5‐Fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 2003; 3: 330–338. [DOI] [PubMed] [Google Scholar]
- 3. Johnson MR, Diasio RB. Importance of dihydropyrimidine dehydrogenase (DPD) deficiency in patients exhibiting toxicity following treatment with 5‐fluorouracil. Adv Enzyme Regul 2001; 41: 151–157. [DOI] [PubMed] [Google Scholar]
- 4. Amstutz U, Froehlich TK, Largiadèr CR. Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5‐fluorouracil toxicity. Pharmacogenomics 2011; 12: 1321–1336. [DOI] [PubMed] [Google Scholar]
- 5. Morel A, Boisdron‐Celle M, Fey L, Soulie P, Craipeau MC, Traore S, et al Clinical relevance of different dihydropyrimidine dehydrogenase gene single nucleotide polymorphisms on 5‐fluorouracil tolerance. Mol Cancer Ther 2006; 5: 2895–2904. [DOI] [PubMed] [Google Scholar]
- 6. Meulendijks D, Henricks LM, Sonke GS, Deenen MJ, Froehlich TK, Amstutz U, et al Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine‐associated toxicity: a systematic review and meta‐analysis of individual patient data. Lancet Oncol 2015; 16: 1639–1650. [DOI] [PubMed] [Google Scholar]
- 7. Deenen MJ, Meulendijks D, Cats A, Sechterberger MK, Severens JL, Boot H, et al Upfront genotyping of DPYD*2A to individualize fluoropyrimidine therapy: a safety and cost analysis. J Clin Oncol 2016; 34: 227–234. [DOI] [PubMed] [Google Scholar]
- 8. Chazal M, Etienne MC, Renée N, Bourgeon A, Richelme H, Milano G. Link between dihydropyrimidine dehydrogenase activity in peripheral blood mononuclear cells and liver. Clin Cancer Res 1996; 2: 507–510. [PubMed] [Google Scholar]
- 9. Meulendijks D, Cats A, Beijnen JH, Schellens JH. Improving safety of fluoropyrimidine chemotherapy by individualizing treatment based on dihydropyrimidine dehydrogenase activity – ready for clinical practice? Cancer Treat Rev 2016; 50: 23–34. [DOI] [PubMed] [Google Scholar]
- 10. Boisdron‐Celle M, Remaud G, Traore S, Poirier AL, Gamelin L, Morel A, et al 5‐Fluorouracil‐related severe toxicity: a comparison of different methods for the pretherapeutic detection of dihydropyrimidine dehydrogenase deficiency. Cancer Lett 2007; 249: 271–282. [DOI] [PubMed] [Google Scholar]
- 11. Kristensen MH, Pedersen P, Mejer J. The value of dihydrouracil/uracil plasma ratios in predicting 5‐fluorouracil‐related toxicity in colorectal cancer patients. J Int Med Res 2010; 38: 1313–1323. [DOI] [PubMed] [Google Scholar]
- 12. Zhou ZW, Wang GQ, Wan DS, Lu ZH, Chen YB, Li S, et al The dihydrouracil/uracil ratios in plasma and toxicities of 5‐fluorouracil‐based adjuvant chemotherapy in colorectal cancer patients. Chemotherapy 2007; 53: 127–131. [DOI] [PubMed] [Google Scholar]
- 13. Ciccolini J, Mercier C, Blachon MF, Favre R, Durand A, Lacarelle B. A simple and rapid high‐performance liquid chromatographic (HPLC) method for 5‐fluorouracil (5‐FU) assay in plasma and possible detection of patients with impaired dihydropyrimidine dehydrogenase (DPD) activity. J Clin Pharm Ther 2004; 29: 307–315. [DOI] [PubMed] [Google Scholar]
- 14. Meulendijks D, Henricks LM, Jacobs BA, Aliev A, Deenen MJ, De Vries N, et al Pretreatment serum uracil concentration as a predictor of severe and fatal fluoropyrimidine‐associated toxicity. Br J Cancer 2017; 116: 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ciccolini J, Mercier C, Evrard A, Dahan L, Boyer JC, Duffaud F, et al A rapid and inexpensive method for anticipating severe toxicity to fluorouracil and fluorouracil‐based chemotherapy. Ther Drug Monit 2006; 28: 678–685. [DOI] [PubMed] [Google Scholar]
- 16. Jacobs BA, Deenen MJ, Pluim D, Van Hasselt JG, Krähenbühl MD, Van Geel RM, et al Pronounced between‐subject and circadian variability in thymidylate synthase and dihydropyrimidine dehydrogenase enzyme activity in human volunteers. Br J Clin Pharmacol 2016; 82: 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pizzorno G, Cao D, Leffert JJ, Russell RL, Zhang D, Handschumacher RE. Homeostatic control of uridine and the role of uridine phosphorylase: a biological and clinical update. Biochim Biophys Acta 2002; 1587: 133–144. [DOI] [PubMed] [Google Scholar]
- 18. Jonas DA, Elmadfa I, Engel KH, Heller KJ, Kozianowski G, König A, et al Safety considerations of DNA in food. Ann Nutr Metab 2001; 45: 235–254. [DOI] [PubMed] [Google Scholar]
- 19. Lassek E, Montag A. [Nucleic acid components in carbohydrate‐rich food]. Z Lebensm Unters Forsch 1990; 190: 17–21. [DOI] [PubMed] [Google Scholar]
- 20. Jacobs BA, Rosing H, De Vries N, Meulendijks D, Henricks LM, Schellens JH, et al Development and validation of a rapid and sensitive UPLC‐MS/MS method for determination of uracil and dihydrouracil in human plasma. J Pharm Biomed Anal 2016; 126: 75–82. [DOI] [PubMed] [Google Scholar]
- 21. Pluim D, Jacobs BA, Deenen MJ, Ruijter A, Van Geel RM, Burylo A, et al Improved pharmacodynamic assay for dihydropyrimidine dehydrogenase activity in peripheral blood mononuclear cells. Bioanalysis 2015; 7: 519–529. [DOI] [PubMed] [Google Scholar]
- 22. US Department of Health and Human Services , Food and Drug Administration , Center for Drug Evaluation and Research (CDER) . Guidance for industry: food‐effect bioavailability and fed bioequivalence studies. 2002.
- 23. European Medicines Agency . Summary of product characteristics: UFT 100 mg/224 mg hard capsules.
- 24. Galarza AF, Linden R, Antunes MV, Hahn RZ, Raymundo S, Da Silva AC, et al Endogenous plasma and salivary uracil to dihydrouracil ratios and DPYD genotyping as predictors of severe fluoropyrimidine toxicity in patients with gastrointestinal malignancies. Clin Biochem 2016; 49: 1221–1226. [DOI] [PubMed] [Google Scholar]
- 25. Yang CG, Ciccolini J, Blesius A, Dahan L, Bagarry‐Liegey D, Brunet C, et al DPD‐based adaptive dosing of 5‐FU in patients with head and neck cancer: impact on treatment efficacy and toxicity. Cancer Chemother Pharmacol 2011; 67: 49–56. [DOI] [PubMed] [Google Scholar]
- 26. Jiang H, Lu J, Jiang J, Hu P. Important role of the dihydrouracil/uracil ratio in marked interpatient variations of fluoropyrimidine pharmacokinetics and pharmacodynamics. J Clin Pharmacol 2004; 44: 1260–1272. [DOI] [PubMed] [Google Scholar]
- 27. Gamelin E, Boisdron‐Celle M, Guérin‐Meyer V, Delva R, Lortholary A, Genevieve F, et al Correlation between uracil and dihydrouracil plasma ratio, fluorouracil (5‐FU) pharmacokinetic parameters, and tolerance in patients with advanced colorectal cancer: a potential interest for predicting 5‐FU toxicity and determining optimal 5‐FU dosage. J Clin Oncol 1999; 17: 1105. [DOI] [PubMed] [Google Scholar]
- 28. Launay M, Dahan L, Duval M, Rodallec A, Milano G, Duluc M, et al Beating the odds: efficacy and toxicity of dihydropyrimidine dehydrogenase‐driven adaptive dosing of 5‐FU in patients with digestive cancer. Br J Clin Pharmacol 2016; 81: 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boisdron‐Celle M, Capitain O, Faroux R, Borg C, Metges JP, Galais MP, et al Prevention of 5‐fluorouracil‐induced early severe toxicity by pre‐therapeutic dihydropyrimidine dehydrogenase deficiency screening: assessment of a multiparametric approach. Semin Oncol 2017; 44: 13–23. [DOI] [PubMed] [Google Scholar]
- 30. Carlsson G, Odin E, Gustavsson B, Wettergren Y. Pretherapeutic uracil and dihydrouracil levels in saliva of colorectal cancer patients are associated with toxicity during adjuvant 5‐fluorouracil‐based chemotherapy. Cancer Chemother Pharmacol 2014; 74: 757–763. [DOI] [PubMed] [Google Scholar]
- 31. Deng Y, Wang ZV, Gordillo R, An Y, Zhang C, Liang Q, et al An adipo‐biliary‐uridine axis that regulates energy homeostasis. Science 2017; 355: eaaf5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cao D, Leffert JJ, McCabe J, Kim B, Pizzorno G. Abnormalities in uridine homeostatic regulation and pyrimidine nucleotide metabolism as a consequence of the deletion of the uridine phosphorylase gene. J Biol Chem 2005; 280: 21169–21175. [DOI] [PubMed] [Google Scholar]
- 33. Cao D, Pizzorno G. Uridine phosophorylase: an important enzyme in pyrimidine metabolism and fluoropyrimidine activation. Drugs Today 2004; 40: 431–443. [DOI] [PubMed] [Google Scholar]
- 34. Sistonen J, Büchel B, Froehlich TK, Kummer D, Fontana S, Joerger M, et al Predicting 5‐fluorouracil toxicity: DPD genotype and 5,6‐dihydrouracil:uracil ratio. Pharmacogenomics 2014; 15: 1653–1666. [DOI] [PubMed] [Google Scholar]
- 35. Henricks LM, Lunenburg CA, Meulendijks D, Gelderblom H, Cats A, Swen JJ, et al Translating DPYD genotype into DPD phenotype: using the DPYD gene activity score. Pharmacogenomics 2015; 16: 1277–1286. [DOI] [PubMed] [Google Scholar]
- 36. Henricks LM, Kienhuis E, De Man FM, Van der Veldt AA, Hamberg P, Van Kuilenburg AB, et al Treatment algorithm for homozygous or compound heterozygous DPYD variant allele carriers with low‐dose capecitabine. JCO Precis Oncol 2017; 10.1200/PO.17.00118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Uracil levels (A), dihydrouracil levels (B), DHU/U ratio (C) and uridine levels (D) in plasma per individual participant
Figure S2 Correlation between DPD activity in PBMCs and uracil plasma level (A), dihydrouracil plasma level (B), DHU/U ratio (C) and uridine plasma level (D)
Table S1 Results for DPD activity, uracil and dihydrouracil levels per individual participant