
Figure 6.
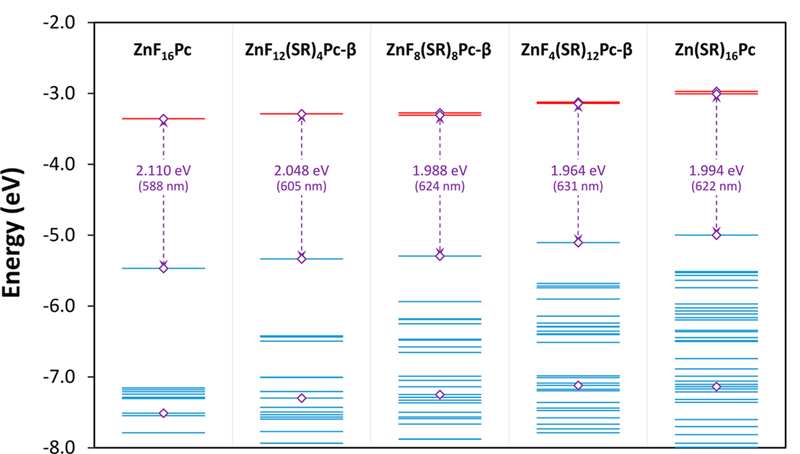
Energies for the frontier molecular orbitals of ZnF16Pc, ZnF12(SR)4Pc-β, ZnF8(SR)8Pc-β, ZnF4(SR)12Pc-β, and Zn(SR)16Pc between −2.0 and −8.0 eV, as calculated by DFT using the B3LYP hybrid functional and the 6–31G(d,p) basis set. The highest occupied orbitals are shown as blue lines, and the lowest unoccupied virtual orbitals are shown as red lines. There are two LUMOs shown for every compound, though they appear as a single line when the energies are degenerate or nearly so. Purple diamonds (◇) mark the orbitals associated with Gouterman’s original model. The HOMO—LUMO energy gaps are also shown in both electron volts and nanometers.