
Figure 2. Age-dependent gene expression dynamics.
(left) Hierarchically clustered heatmap of transcriptome at increasing replicative ages of ~ 2900 genes (the transcriptome at 10 hr is conflated with cell beading). Different transcript types are labeled in black directly to the right of the transcriptional data. (AS = antisense, SUT = stable unannotated transcript, MUT = meiotic unannotated transcript, CUT = cryptic unstable transcript, XUT = Xrn1 sensitive unstable transcript, TELO = subtelomeric, TY = TY repeat element, ESR = environmental stress response). (right) Moving window average (of 100 gene bins) of Growth Rate Slopes from Brauer et al. and log2 transcript abundances in transcripts per million during initial log-phase growth (green). Red bars indicate clusters displaying early-age induction independent of the ESR.

Figure 2—figure supplement 1. Enriched GO terms in the aging transcriptome.
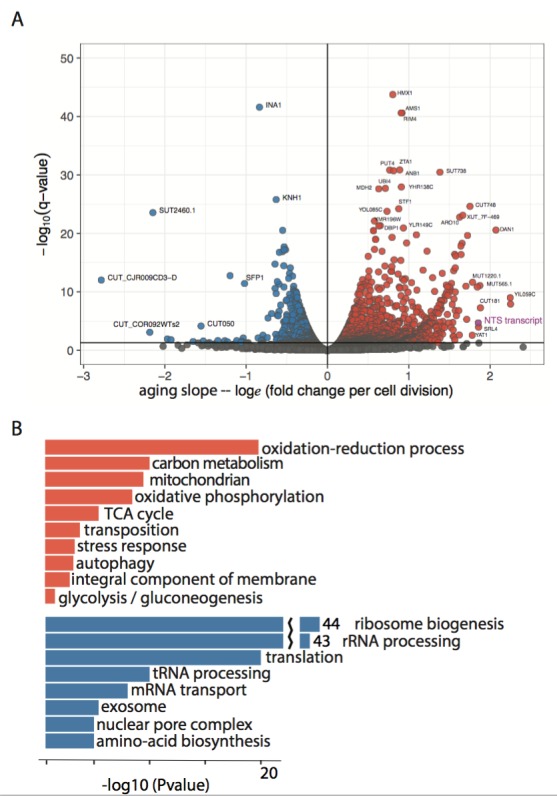
Figure 2—figure supplement 2. Further characterization of the aging transcriptome.

Figure 2—figure supplement 3. Batch effects drive the age-dependent global expression increase observed in Hu et al., 2014.
