

Abstract
Background:
Targeting specific tissues remains a major challenge to the promise of gene therapy. For example, several strategies have failed to target adeno-associated virus 2 (AAV2) vectors, to bone. We have evaluated in vitro and in vivo the affinity of an AAV2 vector to bone matrix, hydroxyapatite (HA) to treat Mucopolysacccharidosis IVA.
Methods:
To increase vector affinity to HA, an aspartic acid octapeptide (D8) was inserted immediately after the N-terminal region of VP2 capsid protein. The modified vector had physical titers and transduction efficiencies comparable to the unmodified vector.
Results:
The bone-targeting vector had significantly higher HA affinity and vector genome copies in bone than the unmodified vector. The modified vector was also released from HA, and its enzyme activity in bone, three months post-infusion, was 4.7-fold higher than the unmodified vector.
Conclusion:
Inserting a bone-targeting peptide into the vector capsid increases gene delivery and expression in bone without decreasing enzyme expression. This approach could be a novel strategy to treat systemic bone diseases.
INTRODUCTION
The three major therapies for lysosomal storage disorders all attempt to partially restore enzyme activity: i) Hematopoietic stem cell transplantation is somewhat effective, but has a relatively high mortality rate (1); ii) Enzyme replacement therapy improves somatic manifestations and quality of life, but has limited effect on neurological and skeletal symptoms (2); and iii) Gene therapy preclinical trials show the possibility to treat patients with a single infusion of viral or non-viral vectors (3), but clinical trials are limited. In addition, targeting the vector to specific tissues such as bone or brain remains a major challenge.
Mucopolysaccharidosis IVA (MPS IVA, Morquio A disease) is an autosomal recessive disorder caused by deficiency of the lysosomal enzyme N-acetylgalatosamine-6-sulfate-sulfatase (GALNS, EC 3.1.6.4). Estimated incidence is 1 in 250,000 live births (4). GALNS deficiency leads to systemic accumulation of the glycosaminoglycans (GAGs), keratan sulfate and chondroitin-6-sulfate. Features such as marked short stature and skeletal dysplasia have focused therapy on bone manifestations, though other symptoms include laxity of joints, and corneal clouding without central nervous system impairment (4). For decades, only surgery (cervical fusion, spinal cord decompression, and hip replacement) and palliative treatments (non-steroidal anti-inflammatory drugs) have been used (4). Recently, the FDA approved enzyme replacement for Morquio A (5). This treatment showed a modest improvement in the six-minute walk test, and provided limited impact on bone lesions (6, 7).
Hydroxyapatite (HA) is a major inorganic component in bone which binds tightly to proteins via the calcium sites (8). Since bone is remodeled by resorption and formation throughout life (8), a drug absorbed by HA may be released during resorption. Therefore, targeting a drug to HA could produce selective delivery to bone (9). This is feasible only if the drug exhibits a high affinity to HA and is retained there. Bisphosphonates, tetracyclines, polymers, and negatively charged peptides have been used for bone-targeted drug delivery (10). Kasugai et al. reported that conjugating fluorescein to a hexapeptide of aspartic acid increased its osteotropicity around 100 times (11). Adding this acidic oligopeptide to estradiol markedly enhanced delivery of estrogen to bone (12).
Inspired by the success of bone targeting with small molecules, we used the same strategy to a large molecule - enzymes. We showed that the tissue-non-specific alkaline phosphatase enzyme tagged with a peptide of acidic amino acid (AAA) residues had higher affinity for HA and longer retention in bone and bone marrow (13). Thereafter, Millán et al. improved bone mineralization in mice with hypophosphatasia by using bone targeting alkaline phosphatase enzyme (14). In addition, studies with MPS VII mice showed that AAA-ß-glucuronidase enzyme was delivered to bone and bone marrow 4 to 5 times as efficiently as the unmodified enzyme (15). We also showed that adding an N-terminal AAA peptide to a recombinant GALNS enzyme markedly prolonged its circulation in the blood after intravenous infusion into an MPS IVA murine model. It also remained longer in bone and substantially cleared storage materials in bone, bone marrow, and heart valves (16). A similar strategy could alter viral vectors’ tropism. Matsumoto et al. used AAV8 vectors expressing deca-aspartates linked to the C-terminus of soluble TNALP (TNALP-D10) to treat severe infantile hypophosphatasia in mice. Results showed phenotypic improvement in mice treated with the bone targeted TNALP (17).
AAV vectors are promising tool for gene delivery. AAV vectors are non-enveloped viruses, encapsidated by 60 proteins consisting of VP1, VP2, and VP3 at a 1:1:10 ratio (18). Work on targeted delivery using AAV vectors has focused on rational insertion of defined peptide sequences into the capsid (18). Mutagenesis analysis identified capsid positions that can hold peptide insertion with reduced impact on packaging and vector transduction (19). In AAV2 vectors, most studies have used residues 138 (VP2 N-terminal), and 587 and 588 [Heparan Sulfate Proteoglycan (HSPG) binding domain] to insert peptides ranging from 5 to 272 amino acids. Capsid proteins modifications improved gene delivery to pancreatic islets (20), lungs (21), muscle (22), myocardium (23), and cancer cells (24), among others. Thus, we hypothesize that a similar approach could alter the tropism of gene therapy vectors to bone in combination with the addition of AAA peptides (15-17).
Here we report adding acidic peptide to the AAV2 capsid to improve gene delivery to bone. We have engineered an AAV2 vector by inserting an AAA peptide after the initial codon of VP2 protein. The AAV2 vectors carrying the human GALNS cDNA were tested to treat MPS IVA. The modified vector was initially tested for HA affinity and in vitro transduction of different cell types. We also systemically infused the modified vector into MPS IVA knock-out mice to evaluate bone and gene product expression at the target site.
MATERIALS AND METHODS
Hydroxyapatite-binding assay
The hydroxyapatite-binding assay was carried out as described previously (13), with slight modifications. Briefly, HA beads were suspended in 25 mM Tris-HCl buffered saline, pH 7.4, at concentration of 1,000 µg/mL. Wild type (WT) AAV2 virus, CBA-GALNS or D8/CBA-GALNS vectors were added at a final concentration of 5 × 1011 and 1 × 1012 vector genome, respectively. Virus was quantified in the supernatant after 1 hour incubation at 37°C and 300 rpm. AAV2 vectors were quantified by spectrophotometric and ELISA methods.
Vector genome biodistribution
1.5 × 1011 vector genome of CBA-GALNS or D8/CBA-GALNS vectors were intravenously injected into 7- to 8-weeks-old Galns-/- mice (n=3 for each group unless otherwise indicated). Viral titers were measured by the ELISA method for all in vivo experiments. MPS IVA control mice were injected with PBS. Mice were euthanized at 24 hours, 48 hours, and 2 weeks post-injection; and brain, liver, and bone (leg) were dissected and immediately frozen in dry ice. N number is 6 for bone at 48 hours for each treatment except at 2-weeks for CBA-GALNS treatment (n=2). Bone marrow was obtained after flushing femurs with PBS. Total DNA was extracted by using DNAzol reagent (Gibco) under manufacturer’s instructions, and a PowerGen 700 homogenizer (Fisher Scientific, Pittsburgh, PA). Vector genome was quantified by qPCR using the Fast SYBR green Master Mix (Applied Biosystems, Foster City, CA), and the primers pCXNF2:5′-CCTCTAGAGCCTCTGCTAACCATGT-3′ and GALNSR:5′-GTAGCCGTCCTGTGAGCAGT-3′, which bind to the 3′- and 5′-ends of the CBA promoter and GALNS cDNA, respectively. All mice were housed in a pathogen-free environment with normal diet. All procedures were in accordance with Institutional Animal Care and Use Committee (IACUC) guidelines under approved protocols at Saint Louis University.
In vivo analysis of GALNS gene expression
Total RNA was extracted from liver, brain and bone marrow by using Trizol reagent (Invitrogen) under manufacturer’s instructions. RNA from bone was extracted as described previously (25) after completely flushing the bone marrow. All data are from CBA-GALNS (n=2) and D8/CBA-GALNS (n=3) mice with experimental replicates. First strand cDNA was synthesized by using the SuperScript® II First-Strand Synthesis System kit (Invitrogen), according to manufacturer’s instructions. Viral cDNA was quantified by qPCR using the Fast SYBR green Master Mix (Applied Biosystems) with 20 ng of first-strand product. Viral cDNA was amplified with the primers TOMF23:5′-ACAGGGCCATTGATGGCCTCAACCTCCT-3′ and pCXN2R:5′-GATCTCAGTGGTATTTGTGAGCCA-3′. Both negative and positive controls were included in each plate. Each study sample was analyzed with two technical replicates. On average three biological replicates were performed for each treatment. The quantitation of the vector genome was normalized by the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which was amplified with the primers GAPDH-S:5′-ACCACAGTCCATGCCATCAC-3′ and GAPDH-R:5′-TCCACCACCCTGTTGCTGTA-3′. The thermal cycling conditions used for the qPCR reaction were: 95°C for 10 minutes, 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Fold change was determined by the ΔΔCt method.
GALNS enzyme activity
GALNS enzyme activity was evaluated in brain, liver, heart, bone (after removal of marrow) and bone marrow. All data are from mice injected with CBA-GALNS (n=2), D8/CBA-GALNS (n=3), or PBS (n=2), and compared to wild-type mice (n=3) two weeks post injection. In addition, GALNS enzyme activity was measured in liver and bone one- and three-months post-injection with CBA-GALNS, D8/CBA-GALNS, or PBS (n=3). Tissues were resuspended in tissue lysis buffer (1mM phenylmethanesulfonyl-fluoride, 25 mM Tris-HCl, pH7.2) and homogenized by using a PowerGen 700 homogenizer (Fisher). GALNS activity was assayed by using 4-MU-ß-D-galactopyranoside-6-sulfate (Toronto Chemicals Research, North York, Canada) as a substrate. Enzyme was assayed as described previously (26). One unit was defined as the enzyme catalyzing 1 nmol of substrate per hour. GALNS activity was expressed as U/mL (media) or U/mg protein (cell lysate or tissue homogenized) as determined by micro-Lowry assay.
Immunohistochemistry
Tissues were processed into paraffin, sections cut on a Leica RM 2135 rotary microtome and dried overnight at 36°C on Fisher Superfrost Plus slides. Sections were deparaffinized and rehydrated, and antigen retrieval performed (120°C, 3 minutes) using DIVA Decloaker (Biocare Medical). After washing in distilled water twice for 3 minutes, sections were blocked in PBS containing 5% normal goat serum, 1% bovine serum albumin, and 0.25% triton X-100 in a humidified chamber for 1 hour at RT, then incubated in primary antibody GALNS (Epitomics) 1:50, normal rabbit IgG in block solution or Collagen X (Santa Cruz Biotechnology) 1:100 for 2 hours at RT. After 3 washes for 5 minutes in PBS, sections were incubated in goat anti-rabbit IgG Rhodamine Red-X (Invitrogen) 1:300 or Alexa Fluor 488 (Jackson Immunoresearch) in block solution for 1 hour at RT in the dark, washed 3 times for 5 minutes in PBS in the dark, rinsed in distilled water, and coverslipped in Prolong Gold with DAPI (Invitrogen).
Image analysis
Immunofluorescent imaging was conducted on an Olympus BX41 fluorescent microscope. Confocal imaging was performed on an Olympus FV1000 confocal microscope. Images were analyzed by using Image J software. Total fluorescent intensity of GALNS was quantified by using the mean gray value of Image J software at a scale of 3.22 pixels/micron (27).
Statistical analysis
Kruskal Wallis multiple comparison test was used to assess treatment effect, time and tissues among the three groups. A student’s t-test was used to assess pairwise comparisons. An error level of 5% (p<0.05) was considered significant. All analyses were performed by using SPSS 23 for Windows (SPSS Inc., Chicago, IL). All results are shown as mean ± standard deviation (SD).
RESULTS
Production of modified AAV2 vector
We reported that inserting an aspartic acid octapeptide (D8) into the C-terminal of tissue-nonspecific alkaline phosphatase (13), N-terminal of β-glucuronidase (15), and N-terminal of GALNS (16), significantly increased delivery of those enzymes to bone. Therefore, we applied the same principle to engineering an AAV2 vector for bone-targeting gene delivery. A sequence encoding for the D8 peptide was inserted immediately after the initial codon of VP2 protein in the packing plasmid pXX2 (Supplementary Material and Supplemental Figure S1a). Complete sequencing of the modified packing plasmid showed that the sequence was precisely located at the desired location with no fortuitous mutations (Supplemental Figure S1b).
The unmodified (CBA-GALNS) and modified (D8/CBA-GALNS) AAV2 vectors were produced in HEK293 cells, purified by iodixanol gradient and heparin-affinity column. In previous studies, vector titers varied widely between different quantification methods (28). Thus, we have determined AAV2 titers by spectrophotometry and ELISA. Spectrophotometric titers were 6.2 × 1013 and 6.5 × 1013 vector genome/mL for the unmodified and modified vectors, respectively, while ELISA titers were 1.3 × 109 and 4.0 × 109 capsids/mL for the unmodified and modified vectors, respectively. Although there was variation between the spectrophotometric and ELISA titration methods, both methods confirmed that inserting the bone-targeting sequence into the viral capsid did not affect the packing process and the physical titers. Since the peptide was not inserted into the HSPG binding site (amino acid residues 587 and 588), we did not expect an alteration in vector affinity to its natural receptor. In fact, during the heparin-affinity purification step, both the unmodified and modified vectors were only detected after elution with 1 M NaCl, showing that heparin affinity was not altered.
Hydroxyapatite-binding assay
Two different amounts (5 × 1011 vector genome and 1 × 1012 vector genome) of unmodified (AAV2) and modified (CBA-GALNS and D8/CBA-GALNS) vectors were incubated with HA, and vector titers were quantified in the supernatant after 1 hour of incubation at 37°C. Regardless of the virus amount, AAV2 and CBA-GALNS were present in the supernatant, indicating no binding to HA (Figure 1). On the other hand, supernatant showed no vector after HA incubation with D8/CBA-GALNS, implying a 100% affinity of the vector to the HA at both vector concentrations (Figure 1).
Figure 1. Hydroxyapatite-binding assay.
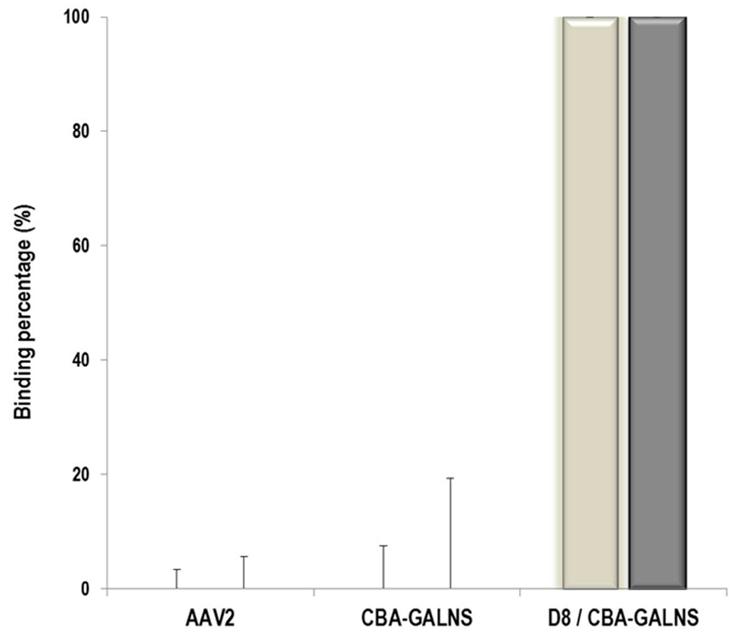
Two different virus concentrations: 5 × 1011 (white bars) and 1 × 1012 (filled bars) vector genome were incubated with 1,000 µg/mL HA, and vectors were measured in the supernatant. Results are reported as a binding percentage compared to the amount of vector found in the supernatant. Error bars represent SDs from different samples (n=3).
In vitro experiments
HA toxicity was evaluated in HEK293 cells prior to transduction experiments in the presence of HA. Cells were incubated 24 hours at 37°C with 250, 500, 750, 1,000, 2,500, or 5,000 µg/mL of HA. As shown in Supplemental Figure S2a, HA cytotoxicity increased proportionally to the HA concentration. To decrease HA cytotoxicity, HA binding was assayed with a final concentration of 100 µg/mL HA (Supplementary Material). This revised protocol eliminated the HA cytotoxicity and maintained the optimal vector concentration of 1 × 105 vector genome/cell. In fact, 48 hours after adding the HA-vector mixture, protein values in the cell lysates were comparable to those obtained in cells transduced without HA addition (data not shown), suggesting that there was no apparent effect on cell viability.
Unmodified and modified vectors were designed to encode the human GALNS cDNA to develop a gene therapy strategy for MPS IVA disease (29, 30). Vector transduction was tested in HEK293 cells, human MPS IVA fibroblasts, and murine MPS IVA chondrocytes in the presence or absence of HA. Comparable GALNS enzyme activity levels in cell lysates were observed after transduction of HEK293 cells (10.7±3.1 U/mg vs. 13.5±5.1 U/mg), human MPS IVA fibroblasts (5.9±1.1 U/mg vs. 6.0±1.2 U/mg), and murine MPS IVA chondrocytes (5.9±1.5 U/mg vs. 6.5±0.8 U/mg) with CBA-GALNS and D8/CBA-GALNS vectors, respectively (Supplemental Figures S2b-d). Enzyme activity in transduced fibroblasts and chondrocytes were 44% and 74%, respectively, of levels in WT fibroblasts and chondrocytes, regardless of presence of bone-targeting peptide. Taken together, these results show that AAA peptide in the capsid did not alter AAV2 vector transduction efficacy.
GALNS enzyme activity levels were not affected in cells transduced with CBA-GALNS vector in the presence of HA (Supplemental Figures S2b-d). GALNS tended to be more active in HEK293 cells transduced with D8/CBA-GALNS in the presence of HA, but this increment was not statistically significant (p=0.14), and activity was comparable to that observed in cells transduced with the unmodified vector (Supplemental Figure S2b). Similarly, 10% less GALNS activity in human MPS IVA fibroblasts transduced with D8/CBA-GALNS vector in the presence of HA was not statistically significant compared with those results obtained without HA (Supplemental Figure S2c). These results show that the bone-targeting vector can be released from HA under normal conditions and can transduce different cell types, suggesting that the aspartic octapeptide in the vector capsid does not alter vector transduction efficiency. PBS was used as negative control of transduction in MPS IVA human and murine deficient cells.
Vector genome biodistribution
Distribution of unmodified and modified vectors was evaluated in Galns-/- mice. Vector genome (vg) was amplified by using specific primers for the human GALNS cDNA, which do not amplify any other sequence derived from the mouse genome. Only brain, liver, bone and bone marrow are shown here, since other tissues exhibited negligible amounts of vector.
Total vector genome distribution compared to unmodified capsid (CBA-GALNS)
At 48 hours post infusion modified vector had decreased by a factor of 10 in bone marrow, which contrasted the 10 times increase in bone. At 48 hours there were more vector genome copies in the bone of mice treated with D8/CBA-GALNS than at 24 hours with an unmodified capsid (Figure 2). Vector genome levels in bone of mice treated with D8/CBA-GALNS differed significantly (p=0.034) with time from mice treated with CBA-GALNS. However, bone was the only tissue where vector genome copies differed significantly (p=0.004) between the two treatments. Vector genome distribution was also compared to liver and to 24 hours post-infusion (Supplemental Figures S3a and b).
Figure 2. High vector genome biodistribution in bone 2 weeks after infusion.
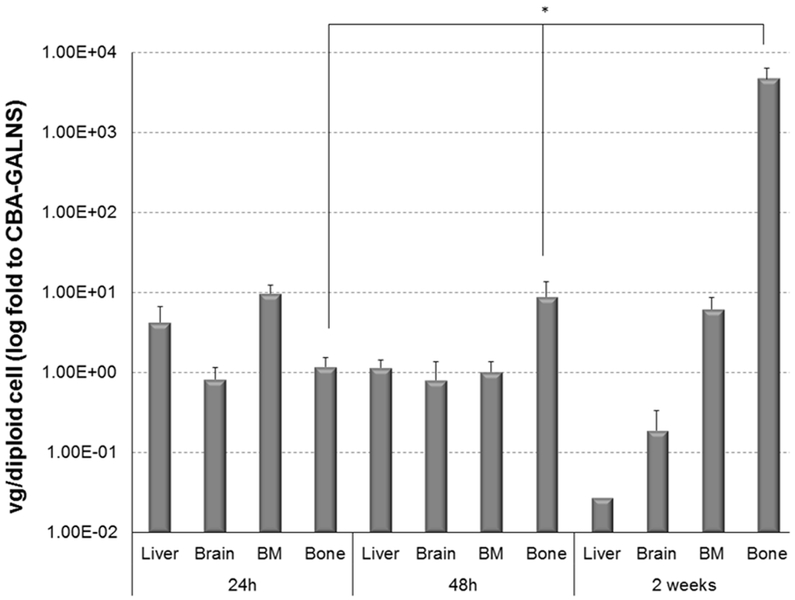
1.5 × 1011 vector genome of CBA-GALNS or D8/CBA-GALNS vectors were infused intravenously into 7- to 8-weeks-old Galns-/- mice. Vector genome was quantified in selected tissues after 24 hours, 48 hours and 2 weeks after infusion. Results are reported as the ratio of vector genome/diploid cell to the levels observed in CBA-GALNS infused mice. Vector genome was not detected in mice infused with PBS. The data was presented as geometric mean with error bars. *p<0.05. All data n=3 except, n=6 for 48 hours (CBA-GALNS and D8/CBA-GALNS) and n=2 for 2-weeks (CBA-GALNS).
These results suggest that D8/CBA-GALNS vector allows retaining vector genome copies at the target site (bone), potentially prolonging expression of its gene product.
These results were confirmed by immunofluorescence staining (DAPI, GALNS and/or Collagen X) in liver and bone. The intensity of GALNS staining derived from hepatocytes transduced with CBA-GALNS and D8/CBA-GALNS vectors showed less signal at 48 hours than at 12 hours after infusion. (CBA p=0.022; D8/CBA p=0.06) (Figure 3a).
Figure 3. GALNS expression 48 hours after vector injection.

1.5 × 1011 vector genome of CBA-GALNS or D8/CBA-GALNS vectors were infused intravenously into 7- to 8-weeks-old mice. 12 or 48 hours after injection, mice were sacrificed and tissues were evaluated. (a) Liver was stained with DAPI and GALNS (n=9). (b) Femur bone was stained with DAPI, GALNS and Collagen X (n=158). Controls were injected with PBS. Quantification was presented as geometric mean with error bars in box plots. *p<0.05.
Those liver findings contrasted with findings in the trabecular bone. In the trabecular bone, mice transduced with D8/CBA-GALNS showed higher signal at 48 hours than modified and unmodified vector at 12 hours post-infusion (p<0.001) (Figure 3b).
In summary, these results indicate that AAA peptide in the capsid vector facilitates a significant increase in gene delivery to the bone.
GALNS gene expression
GALNS gene expression was analyzed in brain, liver, bone, and bone marrow two weeks after infusion. These tissues were selected based on the vector genome distribution profile. The greatest change in GALNS gene expression of mice treated with D8/CBA-GALNS relative to CBA-GALNS was observed in bone: average fold change was 1559 (1187–1932). In brain and liver, fold change in gene expression to CBA-GALNS were 0.66 (0.53–0.79) and 0.77 (0.65–0.89), levels not considered significant for both vectors. On the other hand, GALNS gene expression in bone marrow was reduced about one-tenth: average fold change: 0.09 (0.07–0.11) (Figure 4a). GALNS gene expression was not detected in any tissue of Galns-/- mice infused with PBS.
Figure 4. In vivo transduction experiments.

1.5 × 1011 vector genome of CBA-GALNS or D8/CBA-GALNS vectors were infused intravenously into 7- to 8-weeks-old Galns-/- mice. All data are from CBA-GALNS (n=2) and D8/CBA-GALNS (n=3) mice with experimental replicates. GALNS gene expression (a) and enzyme activity were evaluated in several tissues after (b) 2 weeks p.i., (c) 1 month and 3 months p.i. . The data was presented as geometric mean with error bars. *p<0.05.
GALNS enzyme activity
Two weeks post injection:
GALNS enzyme activity in tissues two weeks after injection is shown in Figure 4b. Injections with CBA-GALNS or D8/CBA-GALNS led to activity increases in liver, brain, heart, and bone marrow (19% vs. 24%, 45% vs. 35%, 28% vs. 62%, and 12.4% vs. 13.8% of WT enzyme activity levels, respectively). Enzyme activity in bone was 4.8% (1.3±1.9 U/mg) and 42% (11.8±5.6 U/mg) in CBA-GALNS and D8/CBA-GALNS mice, respectively. Comparison of CBA-GALNS and D8/CBA-GALNS showed increased levels of enzyme activity in D8/CBA-GALNS when compared to CBA-GALNS in liver (p=0.82), heart (p=0.37), bone (p=0.07) and bone marrow (p=0.61), although not significant.
One- and three-months post injection:
Specific GALNS activity was measured in liver and bone of mice injected with CBA-GALNS or D8/CBA-GALNS, showing that activity in bone three months after injection grew to therapeutic levels (Figure 4c). Since the CBA promoter yields ubiquitous transgene expression, high activity was expected in liver (CBA-GALNS 1mo vs. D8/CBA-GALNS 1mo, p<0.05).
Thus, all parameters studied in bone (vector genome, GALNS gene expression, GALNS enzyme activity) demonstrate that AAA peptide in vector capsid not only increases gene tropism to bone but also elevates gene expression and enzyme activity in bone.
DISCUSSION
Developing systemically injectable gene delivery vectors is critical for bone disease gene therapies. This study aimed to target an AAV2 vector to bone and to evaluate the efficiency of gene delivery and expression levels in a Galns-/- mouse model. We demonstrated that including AAA peptide into the vector capsid: 1) does not affect packing and transduction, 2) enhances in vivo gene delivery, and 3) increases expression of its gene product in bone.
Here for the first time, we have demonstrated successful delivery of a gene therapy vector to bone by inserting a bone-targeting peptide into the vector capsid.
During the last decade, special attention has been paid to altering viral vectors’ natural tropism (18). AAV2 vectors have been extensively studied for peptide insertion sites, kind of peptides, and the effect on vector genome packing, transduction efficiency, and tissue targeting (18). N-terminal region of the VP2 protein represents one of the most studied capsid positions that allows peptide insertion with minimum effect on DNA packaging and virus trafficking (18). Serpin receptor ligand (KFNKPFVFLI) inserted after position 138 increased viral transduction 15-fold in IB3 cells (19). An ApoE-derived ligand inserted after position 138 led to a 90-fold increase in in vitro transduction of pancreatic islet cells, and a four-fold increase of expression of human antitrypsin (20). Such studies show progress in AAV retargeting to desired tissues, but the biodistribution and targeting of AAV vectors to bone remained unsolved.
Our present study inserting AAA peptide into the viral capsid shows successful vector targeting to bone. We also show that a highly negatively charged peptide in the N-terminal of VP2 protein was well-tolerated, without reducing physical titers and transduction efficiency. These results agree with the fact that inserting peptides after the N-terminal region of the VP2 protein does not affect the physical and infectious titers (19, 20, 32). In addition, inserting AAA peptide did not affect heparin affinity of the vector, since the HSPG binding site is distant from the VP2 N-terminal, and they do not come close together in final capsid assembly (33). This result agrees with previous findings that inserting a peptide after the N-terminal of VP2 does not affect vector binding to the natural receptor (HSPG) or to the heparin column used in purification steps (19, 20, 32). Lee et al., inserted the same acidic oligopeptide in the AAV2 capsid for muscle targeting (23). They observed no effect on vector titers, which agrees with our results. However, since the oligopeptide was inserted after amino acid 587 (the HSPG-binding site), the vector lost the ability to transduce HEK293 cells or human chondrocytes, and showed different transduction and tropism profiles to those of D8/CBA-GALNS vector (23).
Zincarelli and colleagues (34) found AAV2 vector genome in all mouse tissues investigated (heart, lung, liver, kidney, testes, brain, and muscle) at comparable levels, while gene expression was only detected in heart, liver, and muscle. However, vector distribution in bone was not assessed. Herein, we show that an AAV2 vector itself has very low affinity for HA both in vitro and in vivo, which could be associated with positively charged residues at the receptor binding site (35). More notably, although the bone-targeting vector has a marked affinity for HA both in vitro and in vivo, the vector can be released from the HA and can transduce the cells without loss of infectious efficiency. How the vector is released in vitro remains unknown, but in vivo it has been proposed that a molecule tagged to bone by using the AAA peptide could be released during bone matrix resorption (11, 12).
A single copy of AAA peptide was initially used for the bone-targeting of small molecules (estradiol) and then large molecules (recombinant enzymes: 50–100 KDa) (11-16). We have demonstrated herein that this strategy can modify tropism of even a macromolecule, a viral vector of approximately 5,000 KDa. Higher affinity to HA (100%), and better bone targeting were achieved by using multiple copies of AAA peptide, in comparison with the reports for recombinant enzymes (13-16). This efficient retargeting can be explained by a higher copy number of an AAA peptide, since AAV2 capsid is composed of 60 subunits of VP1, VP2, and VP3 at a molar ratio of approximately 1:1:10, leading to five VP1 and five VP2 proteins containing the bone-targeting sequence (18, 33).
Previously, we showed the ability to treat MPS IVA by gene therapy in vitro (29, 30, 36). We evaluate here in vivo with a new targeting strategy. Although the enzyme was less active than in WT mice, activity could be sufficient to improve lesions. Noteworthy, activity in bone from D8/CBA-GALNS treated mice were 42% of WT levels, while in mice treated with the unmodified vector these levels were only 4.8% of WT levels, indicating the marked advantage to use the AAA peptide into the vector capsid. Previous studies showed that recombinant GALNS enzyme was poorly distributed to bone (26), and that including a bone-targeting peptide to the recombinant enzyme allowed increased distribution more specific to bone, improving therapeutic effect (16). Hence, we may expect substantially improved pathology after a long-term treatment of MPS IVA mouse model with this bone-targeting approach.
Elevated enzyme activity observed in treated mice agrees with previous reports of gene therapy in adult MPS mouse models treated with AAV2 vectors (37). Although in adult mice extensive bone disease cannot be reversed, its progression can be ameliorated. Several studies show that: i) enzyme activity in plasma and tissues should be supraphysiological to correct bone pathology in adult mice (38), or that ii) treatment should begin at the neonatal stage (39). In this regard, further studies should focus on long-term evaluation of the therapy (specially the effect in growth plate), neonatal treatment of mice, evaluating other AAV serotypes (AAV8), and other bone-targeting peptides with different amino acids (such as glutamic acid) or size.
CONCLUSIONS
We have demonstrated that targeting an AAV2 vector to bone can be markedly improved by inserting eight aspartic amino acids into the N-terminal of the VP2 protein. This modification not only increases copies of viral genomes in bone but also yields substantial enzyme activity in bone. This novel capsid thus allows AAV vectors to treat a variety of other lysosomal storage disorders with bone dysplasia, as well as other bone diseases. Further long-term studies in vivo are required to validate this strategy by assessing pathological and clinical improvement in bone, the starting age for treatment, and the correlation between enzyme activity in blood and efficacy of treatment.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Dr. Guangping Gao for his critical comments to our work, to Dr. Paula Buchanan for statistical assistance; and to Eric Shelley, Dr. Qi Gan and Dr. Michael Flanagan for their technical assistance. We also thank Mike Marcinkowski for editorial assistance.
STATEMENT OF FINANCIAL SUPPORT:
This project was supported in part by International Morquio Organization (Carol Ann Foundation), and Austrian MPS Society. CJAD was supported by Departamento Administrativo de Ciencia, Tecnología e Innovación – Colciencias (Colombia, ID PRY 5174, Grant 120356933205). CJAD and LAB were supported by Pontificia Universidad Javeriana (ID PRY 3763 and 3916). S.T. and A.M.M. were supported by the National Institutes of Health grant R01HD065767. ST was supported by National Institutes of Health grant 8P20GM103464–08.
Footnotes
AUTHOR CONTRIBUTIONS
CJAD, AMM, LAB and ST designed research.
CJAD and AMM performed research.
CJAD, AMM, LAB and ST analyzed the data.
CJAD, AMM and ST contributed to report the work described.
The manuscript was written through contributions of all authors.
All authors have given approval to the final version of the manuscript.
DISCLOSURE:
CJAD, AMM, LAB and ST declare competing financial interests: they have US patent 7,972,593 issued on July 5, 2011. The patent covers the underlying concept of AAV vector with bone targeting described in the manuscript.
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint first authors.
Dr. Shunji Tomatsu, was a former employee at Saint Louis University, where the experiments were conducted and completed.
REFERENCES
- 1.Prasad VK, Kurtzberg J 2010. Transplant outcomes in mucopolysaccharidoses. Semin Hematol 47:59–69. [DOI] [PubMed] [Google Scholar]
- 2.Desnick RJ, Schuchman EH 2012. Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu Rev Genomics Hum Genet 13:307–335. [DOI] [PubMed] [Google Scholar]
- 3.Yew NS, Cheng SH 2013. Gene therapy for lysosomal storage disorders. Pediatr Endocrinol Rev 11 Suppl 1:99–109. [PubMed] [Google Scholar]
- 4.Hendriksz CJ, Harmatz P, Beck M, et al. 2013. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol Genet Metab 110:54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. 2014 FDA clears the first drug for rare Morquio A syndrome. Medscape.
- 6.Tomatsu S, Sawamoto K, Almeciga-Diaz CJ, et al. 2015. Impact of enzyme replacement therapy and hematopoietic stem cell transplantation in patients with Morquio A syndrome. Drug Des Devel Ther 9:1937–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puckett Y, Mullinder H, Montaño AM 2017. Enzyme Replacement Therapy with Elosulfase alfa for Mucopolysaccharidosis IVA (Morquio A Syndrome): Milestones and Challenges. Expert Rev Orphan Drugs 5: 741–752. [Google Scholar]
- 8.Bilezikian JP, Raisz LG, Martin J 2008. Principles of Bone Biology. Academic Press, San Diego, CA. [Google Scholar]
- 9.Fujisaki J, Tokunaga Y, Takahashi T, Shimojo F, Kimura S, Hata T 1998. Osteotropic drug delivery system (ODDS) based on bisphosphonic prodrug. I.v. effects of osteotropic estradiol on bone mineral density and uterine weight in ovariectomized rats. J Drug Target 5:129–138. [DOI] [PubMed] [Google Scholar]
- 10.Wang D, Miller SC, Kopecková P, Kopecek J 2005. Bone-targeting macromolecular therapeutics. Adv. Drug. Deliv. Rev 57:1049–1076. [DOI] [PubMed] [Google Scholar]
- 11.Kasugai S, Fujisawa R, Waki Y, Miyamoto K, Ohya K 2000. Selective drug delivery system to bone: small peptide (Asp)6 conjugation. J Bone Miner Res 15:936–943. [DOI] [PubMed] [Google Scholar]
- 12.Yokogawa K, Miya K, Sekido T, et al. 2001. Selective delivery of estradiol to bone by aspartic acid oligopeptide and its effects on ovariectomized mice. Endocrinology 142:1228–1233. [DOI] [PubMed] [Google Scholar]
- 13.Nishioka T, Tomatsu S, Gutierrez MA, et al. 2006. Enhancement of drug delivery to bone: Characterization of human tissue-nonspecific alkaline phosphatase tagged with an acidic oligopeptide. Mol Genet Metab 88:244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Millan JL, Narisawa S, Lemire I, et al. 2007. Enzyme Replacement Therapy for Murine Hypophosphatasia. J Bone Miner Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montaño AM, Oikawa H, Tomatsu S, et al. 2008. Acidic amino acid tag enhances response to enzyme replacement in mucopolysaccharidosis type VII. Mol Genet Metab 94:178–189. [DOI] [PubMed] [Google Scholar]
- 16.Tomatsu S, Montaño AM, Dung V, et al. 2010. Enhacement of drug delivery: Enzyme-replacement therapy for murine Morquio A syndrome. Mol Ther 18:1094–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsumoto T, Miyake K, Yamamoto S, et al. 2011. Rescue of severe infantile hypophosphatasia mice by AAV-mediated sustained expression of soluble alkaline phosphatase. Hum Gene Ther 22:1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michelfelder S, Trepel M 2009. Adeno-associated viral vectors and their redirection to cell-type specific receptors. Adv Genet 67:29–60. [DOI] [PubMed] [Google Scholar]
- 19.Wu P, Xiao W, Conlon T, et al. 2000. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J Virol 74:8635–8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loiler S, Conlon T, Song S, et al. 2003. Targeting recombinant adeno-associated virus vectors to enhance gene transfer to pancreatic islets and liver. Gene Ther 10:1551–1558. [DOI] [PubMed] [Google Scholar]
- 21.Kwon I, Schaffer DV 2008. Designer gene delivery vectors: molecular engineering and evolution of adeno-associated viral vectors for enhanced gene transfer. Pharm Res 25:489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu CY, Yuan Z, Cao Z, Wang B, Qiao C, Li J, Xiao X 2009. A muscle-targeting peptide displayed on AAV2 improves muscle tropism on systemic delivery. Gene Ther 16:953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee NC, Falk DJ, Byrne BJ, et al. 2012. An acidic oligopeptide displayed on AAV2 improves axial muscle tropism after systemic delivery. Genet Vaccines Ther 10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michelfelder S, Kohlschutter J, Skorupa A, Pfennings S, Muller O, Kleinschmidt JA, Trepel M 2009. Successful expansion but not complete restriction of tropism of adeno-associated virus by in vivo biopanning of random virus display peptide libraries. PLoS One 4:e5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stewart T, Mann V 2003. Extraction of nucleic acids from bone In Helfrich M, Ralston S (eds) Methodos in molecular medicine: Bone research protocols. Humana Press Inc, Totowa, NJ, pp 425–432. [DOI] [PubMed] [Google Scholar]
- 26.Tomatsu S, Montaño A, Ohashi A, et al. 2008. Enzyme replacement therapy in a murine model of Morquio A syndrome. Hum Mol Genet 17:815–824. [DOI] [PubMed] [Google Scholar]
- 27.Schneider CA, Rasband WS, Eliceiri KW 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aucoin MG, Perrier M, Kamen AA 2008. Critical assessment of current adeno-associated viral vector production and quantification methods. Biotechnol Adv 26:73–88. [DOI] [PubMed] [Google Scholar]
- 29.Almeciga-Diaz CJ, Rueda-Paramo MA, Espejo AJ, Echeverri OY, Montano A, Tomatsu S, Barrera LA 2009. Effect of elongation factor 1alpha promoter and SUMF1 over in vitro expression of N-acetylgalactosamine-6-sulfate sulfatase. Mol Biol Rep 36:1863–1870. [DOI] [PubMed] [Google Scholar]
- 30.Alméciga-Díaz C, Montaño AM, Tomatsu S, Barrera L 2010. Adeno-associated virus gene transfer on Morquio A: effect of promoters and sulfatase-modifying Factor 1. FEBS Journal 277:3608–3619. [DOI] [PubMed] [Google Scholar]
- 31.Ulrich-Vinther M 2007. Gene therapy methods in bone and joint disorders. Evaluation of the adeno-associated virus vector in experimental models of articular cartilage disorders, periprosthetic osteolysis and bone healing. Acta Orthop Suppl 78:1–64. [PubMed] [Google Scholar]
- 32.Perabo L, Goldnau D, White K, et al. 2006. Heparan Sulfate Proteoglycan Binding Properties of Adeno-Associated Virus Retargeting Mutants and Consequences for Their In Vivo Tropism. J Virol 80:7265–7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie Q, Bu W, Bhatia S, Hare J, Somasundaram T, Azzi A, Chapman MS 2002. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc Natl Acad Sci U S A 99:10405–10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE 2008. Analysis of AAV Serotypes 1-9 Mediated Gene Expression and Tropism in Mice After Systemic Injection. Mol Ther 16:1073–1080. [DOI] [PubMed] [Google Scholar]
- 35.Opie SR, Warrington KH Jr., Agbandje-McKenna M, Zolotukhin S, Muzyczka N 2003. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J Virol 77:6995–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toietta G, Severini G, Traversari C, et al. 2001. Various cells retrovirally transduced with N-acetylgalactosamine-6-sulfate sulfatase correct Morquio skin fibroblasts in vitro. Hum Gene Ther 12:2007–2016. [DOI] [PubMed] [Google Scholar]
- 37.Fu H, Kang L, Jennings J, et al. 2007. Significantly increased lifespan and improved behavioral performances by rAAV gene delivery in adult mucopolysaccharidosis IIIB mice. Gene Ther 14:1065–1077. [DOI] [PubMed] [Google Scholar]
- 38.Cardone M, Polito VA, Pepe S, et al. 2006. Correction of Hunter syndrome in the MPSII mouse model by AAV2/8-mediated gene delivery. Hum Mol Genet 15:1225–1236. [DOI] [PubMed] [Google Scholar]
- 39.Hartung SD, Frandsen JL, Pan D, et al. 2004. Correction of Metabolic, Craniofacial, and Neurologic Abnormalities in MPS I Mice Treated at Birth with Adeno-associated Virus Vector Transducing the Human A-L-Iduronidase Gene. Mol Ther 9:866–875. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.