

Abstract
A facile synthesis for novel loperamide analogs as potential μ opioid receptors is described. The synthetic procedure for compound 5, which contains two 4-phenyl piperidine scaffolds, was optimized, and this compound was synthesized in excellent yield. We also describe a mild and highly efficient protocol for the synthesis of compounds 6 and 7.
Keywords: loperamide analogs, μ opioid receptor agonist, morphine, opioid ligand receptors
1. Introduction
Opioid ligand receptors are involved in various physiological activities, including analgesia, miosis, bradycardia, general sedation, hypothermia, insensitivity and depression of the flexor reflexes and have been widely used in medicine, most prominently in the treatment of pain [1,2]. Three separate receptors—μ (mu), δ (delta), and κ (kappa)—were proven to be the basis of the pharmacologic responses using in vitro radioligand binding affinity assays and in vivo localization of labeled drug in tissue homogenates or sections [3,4]. These reports indicate that μ opioid receptor (MOR) agonists are useful analgesics in the periphery, especially for inflamed tissues [5,6]. Phenylpiperidine structures, such as loperamide (1) [7,8], N-desmethylloperamide (2) [9], diphenoxylate (3) [10], and 4 (Figure 1) [11], are typical representatives of these compounds. Although diphenoxylate and loperamide are MOR agonists and show affinity and selectivity for the cloned μ human opioid receptor, they do not easily pass through the blood–brain barrier (BBB) [12], therefore, they are currently mainly used as antidiarrheal drugs.
Figure 1.

Structures of representative MOR agonists.
More recently, MOR agonists based on the 4-phenylpiperidine scaffold have been reported and have shown promising results [13,14]. The therapeutic potential of loperamide and other compounds based on 4-phenylpiperidine scaffolds may also be of interest to the scientific community in light of the recent discovery of DiPOA, a novel, systemically available and peripherally restricted MOR agonist with antihyperalgesic activity [15].
Therefore, the compounds 1,4-bis(4-(4-chlorophenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenylbutan-1-one (5, Figure 2), 4-(4-(4-fluorophenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenylbutanamide (6, Figure 2) and (Z)-2-(2-(4-(4-fluorophenyl)-4-hydroxypiperidin-1-yl) ethyl)-3-methylene-2-phenylhex-4-enamide (7, Figure 2), which are based on loperamide and diphenoxylate, were designed and synthesized as potential MOR agonists. The novel analog 5 contains two 4-phenylpiperidine scaffolds, and analogs 6 and 7 each contain one 4-phenylpiperidine scaffold. We consider these analogs to be promising MOR agonists.
Figure 2.

Structures of novel loperamide analogs.
2. Results and Discussion
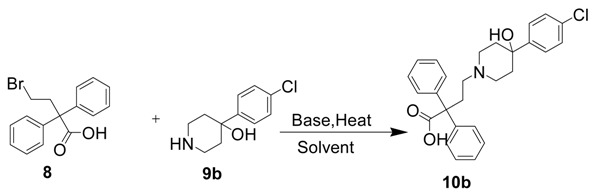
As a consequence of our interest in MORs containing 4-phenylpiperidine scaffolds, we developed a mild and highly efficient protocol for the synthesis of compounds 5, 6 and 7. We initiated our studies by preparing the key intermediate 10b by coupling commercially available compounds 8 and 9b to achieve product 10b under different reaction conditions, as shown in Table 1. The reaction using conditions based on those of a similar reaction that was previously reported in the literature was not successful for an unknown reason [16]. When the reaction time was prolonged to 30 h with 3 eq. of DIPEA at 105 °C, we successfully isolated compound 10b in a disappointing 6% yield. The possible reasons for failure may be that the hydroxyl group on compound 9b was affected. This group is also a strong nucleophile under basic conditions, as observed in our previously published study [17], and the carboxylates present under basic conditions may also compete with the secondary amine in the nucleophilic substitution reaction. Because the overall yield of 10b using this approach was poor, an alternate strategy was adopted for synthesizing a high yield of 10b.
Table 1.
Synthesis of the intermediate compound 10b.
| Entry | Ratio 8:9b:DIPEA | Reaction conditions | Yield *10b |
|---|---|---|---|
| 1 | 1:1.5:0 | THF, reflux, 2 days | - |
| 2 | 1:1:3 | THF, reflux, 2 days | - |
| 3 | 1:1:3 | CH3CN, 70 °C, 30 h | - |
| 4 | 1:1:3 | CH3CN, 85 °C, 30 h | Trace |
| 5 | 1:1:3 | CH3CN, 105 °C, 30 h | 6% |
* Isolated yield.
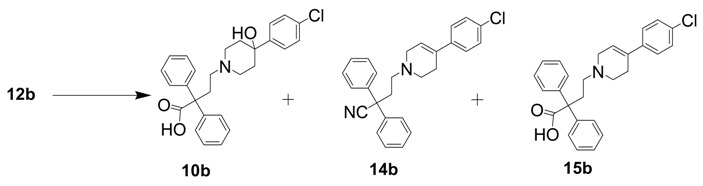
Subsequently, we designed another approach to prepare the intermediate 10b involving the alkylation of 4-(4-chlorophenyl)-4-hydroxylpiperidine with 4-bromo-2,2-diphenylbutyronitrile in the presence of DIPEA, followed by the hydrolysis of 12b with KOH in tBuOH to afford compound 13b (Scheme 1) [18]. We also prepared analogs 6 and 7 using a similar procedure for the future radiolabeling study. The slow hydrolysis of amide precursor 13b in 40% H2SO4 aq. at reflux gave a trace amount of 10b. The hydrolysis of nitrile compound 12b under different reaction conditions was also investigated (Table 2). Compound 10b was obtained in 7% yield when the reaction was catalyzed with H2O2/NaOH in H2O at 80 °C for 20 h [19,20]. A trace amount of 10b was obtained when an inorganic base (2 M sodium hydroxide) was used as a catalyst [21]. We isolated two byproducts, 14b (20%~23% yield) and 15b (66%~72% yield), when the reaction was performed under acidic conditions (37% HCl or 40% H2SO4) [22]. The principal reason was that the tertiary hydroxyl group on 13b was very easily eliminated under acidic conditions to form an alkene [17].
Scheme 1.

Synthesis of the amides 6, 13b and 7.
Table 2.
Hydrolysis of the nitrile 12b.
| Entry | Catalyst | Reaction conditions | Yield (%) * | ||
|---|---|---|---|---|---|
| 10b | 15b | 14b | |||
| 1 | NaOH a | CH3OH/H2O, Reflux, 30 h | trace | - | - |
| 2 | NaOH a | H2O, Reflux, 30 h | trace | - | - |
| 3 | HCl b | 1,4-Dioxane, Reflux, 16 h | - | 20 | 66 |
| 4 | H2SO4 c | H2O, 100 °C, 16 h | - | 23 | 72 |
| 5 | H2O2 d/NaOH a | H2O, 80 h, 20 h | 7 | - | - |
Catalyst concentration: a 2 M; b 37%; c 40%; d 30%. * Isolated yield.
The desired product 5 was obtained in low yield by coupling 10b with the amine 9b in a reaction catalyzed by TiCl4 in toluene refluxing for 20 h (Scheme 2) [23]. Unfortunately, this second approach was not feasible for use in the rest of the study.
Scheme 2.

Synthesis of compound 5.
When the first approach was analyzed in detail, it was determined that the formation of a substantial amount of salt at the start of reaction may be responsible for the low yield of 12b. Thus, a new method involving the protection of the carboxyl group of 8 with a methyl group to form an ester precursor before coupling with 9b was developed (Scheme 3). A mixture of 4-bromo-2,2-diphenylbutyric acid 8, thionyl chloride and catalytic DMF in dichloromethane was stirred and refluxed for 3 h. After concentration under reduced pressure, crude 4-bromo-2,2-diphenylbutyric acid chloride (17) was achieved as a pale yellow oil. Compound 17 was converted into the ester 18 in excellent yield (82%) by reaction with methanol at 50 °C for 4 h. The desired product, 5, was obtained as a white solid in 71% yield by condensing 18 with 9b in the presence of DIPEA in CH3CN at 80 °C for 15 h (Scheme 3). The side product 19 was also obtained as a pale orange solid in 8% yield.
Scheme 3.

The Synthesis of the ester 19 and 5.
3. Experimental
3.1. General
All reagents and organic solvents were ACS grade or higher and used without further purification. Unless otherwise noted, all chemicals were purchased from J&K Scientific (Shanghai, China). Reactions were performed under argon atmosphere with standard Schlenk techniques. Thin layer chromatography was performed on HAIYANG silica gel F254 plate, and compounds were visualized under UV light (λ = 254 nm). Column chromatography was carried out using HAIYANG silica gel (type: 200–300 mesh ZCX-2). 1H (500 MHz), 13C-NMR (126 MHz) and 19F-NMR (470 MHz) spectra were recorded on an Avance 500 spectrometer (Bruker; Billerica, MA, USA). Chemical shifts are reported in δ units (ppm) downfield relative to the chemical shift for tetramethylsilane.
4-(4-(4-Chlorophenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenylbutanoic acid (10b). Entry 2. The 4-bromo-2,2-diphenylbutanoic acid 8 (200 mg, 0.6266 mmol) was dissolved in THF (10 mL), followed by addition of 4-(4-chlorophenyl)-piperidin-4-ol (133 mg, 0.6266 mmol, 1 eq.) and DIPEA (0.242 mL, 1.8798 mmol, 3 eq.) to the solution. The mixture was refluxed for 2 days. No new product was found via TLC. Entry 5. The 4-bromo-2,2-diphenylbutanoic acid 8 (400 mg, 1.253 mmol) was suspended in acetonitrile (10 mL) and DIPEA (0.675 mL, 3.75 mmol) was added. The mixture was stirred at 105 °C for 30 h. After the solvent was removed under vacuum, the crude was dissolved in CH2Cl2 and was introduced onto a silica gel column. The product was eluted with 3% MeOH in dichloromethane to give 10b as a pale-orange solid (20 mg, 6% yield). TLC (silica gel; MeOH–CH2Cl2 (5:95 v:v); Rf = 0.40). LC-MS (M++1): found 449.88; calcd for C27H29ClNO3, 450.18.
4-(4-(4-Chlorophenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenylbutanenitrile (12b). 4-(4-Chlorophenyl)-4-hydroxypiperidine (2.1 g, 10.0 mmol) was suspended in acetonitrile (15 mL) and DIPEA (3.5 mL, 30 mmol) was added. 4-Bromo-2,2-diphenylbutanenitrile (3.00 g, 10 mmol) in acetonitrile (15 mL) was then added. The reaction mixture was stirred at 70 °C for 30 h. After the solvent was removed under vacuum, the crude material was redissolved in CH2Cl2 and introduced onto a silica gel column. The product was eluted with 5% MeOH in dichloromethane to yield 12b as a pale-orange solid (2.8 g, 66% yield). TLC (silica gel; MeOH–CH2Cl2 (5:95 v:v); Rf = 0.50). 1H-NMR (CDCl3): δ 7.37 (m, 14H), 2.80 (d, 2H, J = 11.25 Hz), 2.67 (m, 2H), 2.55 (m, 2H), 2.47 (t, 2H, J = 11.3 Hz), 2.105 (m, 2H), 1.73 (d, 2H, J = 11.9 Hz). 13C-NMR (CDCl3): δ 140.16, 132.95, 129.09, 128.57, 128.10, 126.95, 126.23, 122.27, 71.11, 54.93, 50.17, 49.71, 38.52, 36.80ppm. LC-MS (M++1) found 431.20; calcd for C27H28ClN2O, 431.19.
4-(4-(4-Fluorophenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenylbutanenitrile (12a). Compound 12a was synthesized using the same procedure that was followed the synthesis of the compound 12b, and was obtained in 35% yield as a pale-orange solid. 1H-NMR (CDCl3): δ 7.45 (m, 2H), 7.39 (d, 4H, J = 10 Hz), 7.35 (t, 4H, J = 5Hz ), 7.29 (t, 2H, J = 10 Hz ), 7.00 (t, 2H, J = 10 Hz), 2.75 (d, 2H, J = 10 Hz), 2.63 (m, 2H), 2.52 (m, 2H), 2.47 (t, 2H, J = 10 Hz), 2.08 (m, 2H), 1.71 (d, 2H, J = 15 Hz).13C-NMR: δ 162.89, 160.93, 144.27, 140.16, 129.03, 128.05, 126.91, 126.45, 126.39, 122.23, 115.18, 115.01, 70.91, 54.88, 50.19, 49.72, 38.56, 36.74. 19F-NMR: δ 116.12. LC-MS (M++1): Found: 415.06; Calcd: C27H28FN2O: 415.22.
4-(4-(4-Bromophenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenylbutanenitrile (12c). Compound 12c was synthesized using the same procedure that was followed the synthesis of the compound 12b, and was obtained in 58% yield as a pale-orange solid. 1H-NMR (CDCl3): δ 7.48 (d, 2H, J = 10 Hz), 7.41 (d, 4H, J = 10 Hz), 7.37 (m, 6H), 7.31 (t, 2H, J = 5 Hz), 2.76 (d, 2H, J = 10 Hz), 2.65 (m, 2H), 2.54 (m, 2H), 2.45 (t, 2H, J = 10 Hz), 2.08 (m, 2H), 1.68 (m, 3H). 13C-NMR: δ 147.64, 140.11, 131.47, 129.07, 128.09, 126.91, 126.66, 122.25, 121.00, 70.97, 54.90, 50.18, 49.64, 38.35, 36.67. LC-MS (M++1): Found: 476.92; Calcd: C27H28BrN2O: 477.13.
4-(4-(4-Chlorophenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenylbutanamide (13b). 4-(4-(4-Chloro-phenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenylbutanenitrile (2.5 g, 6.0 mmol) was dissolved in BuOH (40 mL) and potassium hydroxide (1.2 g, 21.3 mmol) was added. The reaction mixture was stirred at 100 °C for 3d. After concentration under vacuum, the crude material was redissolved in dichloromethane and filtered through a pad of Celite. Chromatography of the sample on a silica gel column eluted with ammonium hydroxide (2 M) solution in MeOH-CH2Cl2 (5:95 v/v) gave 13b as a pale-yellow solid (1.58 g, 55% yield). TLC (silica gel; MeOH–CH2Cl2 (5:95 v:v); Rf = 0.40). 1H-NMR (CDCl3): δ 7.36 (m, 14H), 6.58 (s, 1H), 5.46 (s, 1H), 3.91 (s, 1H), 2.83 (m, 2H), 2.69 (s, 2H), 2.39 (m, 3H), 2.10 (m, 2H), 1.74 (d, 2H, J = 12.85 Hz). 13C-NMR: 176.56, 167.25, 158.96, 146.67, 143.26, 132.82, 128.69, 128.41, 127.06, 126.10, 59.91, 54.92, 49.48, 38.28, 35.87. LC-MS (M++1): Found: 449.3; Calcd: C27H30ClN2O2: 449.20.
4-(4-(4-Fluorophenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenylbutanamide (6). Compound 6 was synthesized using the same procedure that was followed the synthesis of the compound 13b, and was obtained in 40% yield as a pale-orange solid. 1H-NMR (CDCl3): δ 7.45 (m, 2H), 7.31 (m, 10H), 6.99 (t, 2H, J = 10 Hz), 5.07 (s, 1H), 2.89 (m, 2H), 2.64 (m, 2H), 2.35 (m, 2H), 2.04 (m, 2H), 1.59 (d, 2H, J = 5 Hz), 6.22 (s, 1H), 5.82 (s, 1H), 3.02 (m, 2H), 2.82 (m, 4H), 2.63 (m, 2H), 2.36 (m, 2H), 1.76 (d, 2H, J = 10 Hz). 13C-NMR: δ 175.53, 162.43, 160.51, 145.88, 143.83, 129.27, 128.46, 127.29, 127.24, 127.07, 115.15, 114.98, 69.28, 59.54, 54.73, 49.44, 37.33, 29.55. 19F-NMR: δ 117.01.
4-(4-(4-Bromophenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenylbutanamide (7). Compound 7 was synthesized using the same procedure that was followed the synthesis of the compound 13b, and was obtained in 57% yield as a pale-orange solid.1H-NMR (CDCl3): δ 7.41 (t, 2H, J = 5 Hz), 7.31 (m, 12H), 6.45 (s, 1H), 5.87 (s, 1H), 2.82 (m, 12H), 2.31 (m, 12H), 2.50 (m, 12H), 2.42 (m, 12H), 2.11 (m, 12H), 1.69 (m, 12H). 13C-NMR: δ 176.79, 147.17, 143.14, 131.46, 128.75, 128.57, 127.26, 126.64, 121.06, 70.67, 59.88, 55.00, 49.48, 37.79, 35.46. LC-MS (M++1): Found: 494.93; Calcd: C27H30BrN2O2+: 495.14.
4-(4-(4-Chlorophenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenylbutanoic acid (10b). 4-(4-(4-Chloro-phenyl)-4-hydroxycyclohexyl)-2,2-diphenylbutanamide (1.2 g, 2.6 mmol) was added to 40%H2SO4 (50 mL) and the mixture was stirred at 100 °C for 2 days. The crude material was extracted with ethyl acetate (60 mL×3). The ethyl acetate was removed by reduced pressure distillation. After drying with MgSO4 and dissolving in CH2Cl2 (2 mL), the product was introduced onto a silica gel and eluted with 5% MeOH in CH2Cl2 to give trance amount of 10b. TLC (silica gel; MeOH–CH2Cl2 (5:95 v:v); Rf = 0.30). 1H-NMR (CDCl3): δ 7.3096(m, 14H), 5.9319 (s, 1H), 3.29 (m, 2H), 2.94 (m, 2H), 2.80 (m, 2H), 2.72 (m, 2H), 2.67 (m, 4H), 2.10 (m, 2H). LC-MS (M++1): found: 449.88; Calcd for C27H28ClNO3+, 450.18.
Procedure for Preparing 10b from 12b
Entry 3. 4-(4-(4-Chlorophenyl)-4-hydroxypiperidine-1-yl)-2,2-diphenylbutanenitrile (400 mg, 0.928 mmol) was dissolved in 1,4-dioxane (10 mL) and 37% hydrochloric acid (5 mL) was then added. The reaction mixture was refluxed for 16 h. After concentration under vacuum, the crude was dissolved in trichloromethane (10 mL) and extracted with H2O (20 mL × 3). After the organic layer was dried by MgSO4, the solvent was removed under vacuum and redissolved in CH2Cl2. The mixture was introduced onto a silica gel column and the product was eluted with 50% EtOAc in hexane first and 5% MeOH in CH2Cl2 later to give the product 14b as a yellow solid (250 mg, 66%). TLC (silica gel; EtOAc–hexane (1:1 v:v); Rf = 0.70). Compound 14b: 1H-NMR (CDCl3): δ 7.36 (m, 14H), 6.04 (m, 1H), 3.16 (m, 2H), 2.74 (m, 2H), 2.62 (m, 4H), 2.53 (m, 2H) ppm. 13C-NMR (CDCl3): δ 141.44, 140.57, 135.56, 134.26, 130.53, 129.92, 129.55, 128.27, 127.72, 123.52, 55.96, 54.81, 51.93, 51.57, 38.48, 29.44 ppm. MS (M++1): found 412.99; Calcd C27H25ClN2+, 412.95.
1,4-bis(4-(4-Chlorophenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenylbutan-1-one (5). 4-(4-Chloro-phenyl)-4-hydroxypiperidinorophenyl)-4-hydroxypiperidine (28 mg, 0.12 mmol, 2 eq.) was dissolved in toluene (5 mL) and 4-(4-(4-chlorophenyl)-4- hydroxypiperidin-1-yl)-2,2-diphenylbutanoic acid (30 mg, 0.067 mmol) and TiCl4 (1.5 mL) was then added. The mixture was refluxed for 20 h. After the solvent was removed under vacuum, the crude material was dissolved in CH2Cl2 (10 mL). After the mixture was filtered and CH2Cl2 was then removed to 2 mL, the product was introduced onto a silica gel with 5% MeOH in CH2Cl2 to give 5 as a pale-orange solid (trace amount). TLC (silica gel column; MeOH–CH2Cl2 (5:95 v:v); Rf = 0.40).
Methyl 4-bromo-2,2-diphenylbutanoate (18). 4-Bromo-2,2-diphenylbutanoic acid (2.0 g, 6.266 mmol) was dissolved in dry CH2Cl2 (20 mL) and thionyl chloride (2.27 mL, 31.33 mmol, 5 eq.) was then added slowly. A trace amount of DMF was added later. The reaction mixture was refluxed at N2 for 3 h. Methyl alcohol (2 mL, excess) was added slowly and the mixture was stirred at 50 °C for 3 h. After the solvent was removed under vacuum, the crude material was redissolved in CH2Cl2 and introduced onto a silica gel column. The product was eluted with 10% EA in hexane to give a pale-orange oil (900 mg, 43% yield).TLC (silica gel; EtAc–hexane (10:90 v:v); Rf = 0.80). 1H-NMR (CDCl3): δ 7.31 (m, 10H), 3.74 (s, 1H), 3.13 (m, 2H), 3.00 (m, 2H).13C-NMR (CDCl3): δ 174.05, 141.70, 128.70, 128.42, 127.46, 60.84, 52.78, 42.0447, 29.18 ppm. GC-MS: found: 331.99; Calcd for C17H17BrO2, 332.04.
Methyl4-(4-(4-chlorophenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenylbutanoate (19) and 1,4-bis(4-(4-chlorophenyl)-4-hydroxypiperidin-1-yl)-2,2-diphenyl -butan-1-one (5). Methyl 4-bromo-2,2-diphenylbutanoate (800 mg, 2.4 mmol) was dissolved in CH3CN (15 mL) and 4-(4-chlorophenyl)-4-hydroxypiperidine (406 mg, 1.92 mmol, 0.8 eq.) was then added. The reaction mixture was stirred at 50 °C for 30 h. After concentration under vacuum, the crude material was redissolved in CH2Cl2 and introduced onto a silica gel column. The product was eluted with 3% MeOH in CH2Cl2 first and 5% MeOH in CH2Cl2 later to get 19 as a pale-orange solid (70 mmg, 8% yield) and 20 as a pale-orange solid (437 mg, 71%). Compound 19: TLC (silica gel; MeOH-CH2Cl2 (10:90 v:v); Rf = 0.40). 1H-NMR (CDCl3): δ 7.34 (m, 14H), 3.74 (s, 3H), 3.28 (2H, m), 3.13 (2H, m), 3.12 (2H, m), 3.10 (4H, m), 2.60 (1H, s), 2.06 (2H, d, J = 14.0 Hz). 13C-NMR (CDCl3): δ 173.06, 143.62, 140.18, 132.54, 127.72, 127.52, 127.28, 126.62, 126.32, 125.04, 68.58, 58.02, 51.81, 48.22, 34.51, 31.73, 30.93, 28.36, 21.69. MS: found: 463.92; calcd for C28H31ClNO3+, 464.20. Compound 5: TLC (silica gel; MeOH–CH2Cl2 (10:90 v:v); Rf = 0.40). 1H-NMR (CDCl3): δ 7.3094 (m, 18H), 4.5945 (d, 1H, J = 11.4 Hz), 3.31 (m, 10H), 2.73 (m, 2H), 2.55 (m, 2H), 2.28 (m, 2H), 1.87 (m, 4H), 1.65 (m, 2H). 13C-NMR (CDCl3): 171.38, 146.46, 145.29, 141.45, 136.64, 133.00, 132.68, 129.45, 125.03, 128.89, 128.45, 128.31, 128.17, 127.57, 126.54, 126.08, 71.10, 69.06, 60.03, 54.44, 44.25, 42.68, 39.65, 39.21, 37.91, 35.95, 35.30, 29.77 ppm. MS: found: 644.88; Calcd for C38H42Cl2N2O3+, 644.25.
4. Conclusions
In summary, compound 5 was synthesized in excellent yield. Analogs 6 and 7 were obtained through the hydrolysis of the nitrile group in good yield. These methods have the advantages of high yields, convenient procedures, and mild reaction conditions.
Acknowledgments
This research was supported by Jiangsu Science Foundation BK2011704 and NJUST Research Funding (NO. 2011ZDJH08).We thank Wei Jiang for helpful experimental suggestions.
Supplementary Materials
Supplementary data associated with this article can be found in the online version (1H-NMR, 13C-NMR, and LC-MS for all compounds), which can be accessed at: http://www.mdpi.com/1420-3049/17/12/14288/s1.
Footnotes
Sample Availability: Samples of the compounds 5–7, 10b, 12a–c, 13b, 14b, 18 and 19 are available from the authors.
References
- 1.Davis M.P. Evidence from Basic Research for Opioid Combinations. Expert Opin. Drug Discov. 2012;7:165–178. doi: 10.1517/17460441.2012.648611. [DOI] [PubMed] [Google Scholar]
- 2.Chevlen E. Opioids: A review. Curr. Pain Headache Rep. 2003;7:15–23. doi: 10.1007/s11916-003-0005-5. [DOI] [PubMed] [Google Scholar]
- 3.Lord J.A., Waterfield A.A., Hughes J., Kosterlitz H.W. Endogenous Opioid Peptides: Multiple Agonists and Receptors. Nature. 1977;267:495–499. doi: 10.1038/267495a0. [DOI] [PubMed] [Google Scholar]
- 4.Pasternak G.W. Multiple Opiate Receptors: [3H]-Ethylketocyclazocine Receptor Binding and Ketocyclazocine Analgesia. Proc. Natl. Acad. Sci. USA. 1980;77:3691–3694. doi: 10.1073/pnas.77.6.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tokuyama S., Inoue M., Fuchigami T., Ueda H. Lack of Tolerance in Peripheral Opioid Analgesia in Mice. Life Sci. 1998;62:1677–1681. doi: 10.1016/S0024-3205(98)00127-1. [DOI] [PubMed] [Google Scholar]
- 6.Bigliardi P.L., Bigliardi-Qi M., Buechner S., Rufli T. Expression of mu-Opiate Receptor in Human Epidermis and Keratinocytes. J. Invest. Dermatol. 1998;111:297–301. doi: 10.1046/j.1523-1747.1998.00259.x. [DOI] [PubMed] [Google Scholar]
- 7.Decker M., Fulton B.F., Zhang B., Knapp B.I., Bidlack J.M., Neumeyer J.L. Univalent and Bivalent Ligands of Butorphan: Characteristics of the Linking Chain Determine the Affinity and Potency of Such Opioid Ligands. J. Med. Chem. 2009;52:7389–7396. doi: 10.1021/jm900379p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peng X., Knapp B.I., Bidlack J.M., Neumeyer J.L. Synthesis and Preliminary in Vitro Investigation of Bivalent Ligands Containing Homo- and Heterodimeric Pharmacophores at mu, delta, and kappa Opioid Receptors. J. Med. Chem. 2006;49:256–262. doi: 10.1021/jm050577x. [DOI] [PubMed] [Google Scholar]
- 9.Awouters F., Megens A., Verlinden M., Schuurkes J., Niemegeers C., Janssen P.A. Loperamide: Survey of Studies on Mechanism of its Antidiarrheal Activity. Dig. Dis. Sci. 1993;38:977–995. doi: 10.1007/BF01295711. [DOI] [PubMed] [Google Scholar]
- 10.Sato S., Komoto T., Kanamaru Y., Kawamoto N., Okada T., Kaiho T., Mogi K., Morimoto S., Umehara N., Koda T., et al. New mu-Opioid Receptor Agonists with Phenoxyacetic Acid Moiety. Chem. Pharm. Bull. 2002;50:292–297. doi: 10.1248/cpb.50.292. [DOI] [PubMed] [Google Scholar]
- 11.Chen Z., Miller W., Shan S., Valenzano K. Design and parallel synthesis of piperidine libraries targeting the nociceptin (N/OFQ) receptor. Bioorg. Med. Chem. Lett. 2003;13:3247–3252. doi: 10.1016/S0960-894X(03)00665-6. [DOI] [PubMed] [Google Scholar]
- 12.Komoto T., Okada T., Sato S., Niino Y., Oka T., Sakamoto T. New mu-Opioid Receptor Agonists with Piperazine Moiety. Chem. Pharm. Bull. 2001;49:1314–1320. doi: 10.1248/cpb.49.1314. [DOI] [PubMed] [Google Scholar]
- 13.Valenzano K.J., Miller W., Chen Z., Shan S., Crumley G., Victory S.F., Davies E., Huang J.C., Allie N., Nolan S.J., et al. DiPOA ([8-(3,3-diphenylpropyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]dec-3-yl]-acetic acid), a Novel, Systemically Available, and Peripherally Restricted Mu Opioid Agonist with Antihyperalgesic Activity: II. In Vivo Pharmacological Characterization in the Rat. J. Pharmacol. Exp. Ther. 2004;310:793–799. doi: 10.1124/jpet.103.063313. [DOI] [PubMed] [Google Scholar]
- 14.Valenzano K.J., Miller W., Chen Z., Shan S., Crumley G., Victory S.F., Davies E., Huang J.C., Allie N., Nolan S.J., et al. DiPOA ([8-(3,3-Diphenylpropyl)-4-oxo-1-Phenyl-1,3,8-Triaza-spiro[4.5]Dec-3-yl]Acetic Acid), A Novel, Systemically Available, and Peripherally Restricted mu Opioid Agonist with Antihyperalgesic Activity: I. In Vitro Pharmacological Characterization and Pharmacokinetic Properties. J. Pharmacol. Exp. Ther. 2004;310:783–792. doi: 10.1124/jpet.103.063313. [DOI] [PubMed] [Google Scholar]
- 15.Di Bosco A.M., Grieco P., Diurno M.V., Campiglia P., Novellino E., Mazzoni O. Binding Site of Loperamide: Automated Docking of Loperamide in Human mu- and delta-opioid receptors. Chem. Biol. Drug Des. 2008;71:328–335. doi: 10.1111/j.1747-0285.2008.00637.x. [DOI] [PubMed] [Google Scholar]
- 16.Khan P.M., Correa R.G., Divlianska D.B., Peddibhotla S., Sessions E.H., Magnuson G., Brown B., Suyama E., Yuan H., Mangravita-Novo A., et al. Identification of Inhibitors of NOD1-Induced Nuclear Factor-κB Activation. ACS Med. Chem. Lett. 2011;2:780–785. doi: 10.1021/ml200158b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bao X., Lu S., Simeon F.G., Victor W.P. Synthesis of a Prospective 18F-labeled Tracer for Imaging P-Glycoprotein Function. J. Labelled Comp. Radiopharm. 2009;52:S350. [Google Scholar]
- 18.Lazarova N., Zoghbi S.S., Hong J., Seneca N., Tuan E., Gladding R.L., Liow J.S., Taku A., Innis R.B., Pike V.W. Synthesis and Evaluation of [N-methyl-11C]N-Desmethyl-loperamide as a New and Improved PET Radiotracer for Imaging P-gp Function. J. Med. Chem. 2008;51:6034–6043. doi: 10.1021/jm800510m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen A., Cauchon E., Chefson A., Dolman S., Ducharme Y., Dubé D., Falgueyret J.P., Fournier P.A., Gagné S., Gallant M., et al. Renin inhibitors for the treatment of hypertension: Design and optimization of a novel series of tertiary alcohol-bearing piperidines. Bioorg. Med. Chem. Lett. 2011;21:3976–3981. doi: 10.1016/j.bmcl.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 20.Pàmies O., Bäckvall J.E. Efficient Lipase-Catalyzed Kinetic Resolution and Dynamic Kinetic Resolution of β-Hydroxy Nitriles. Correction of Absolute Configuration and Transformation to Chiral β-Hydroxy Acids and γ-Amino Alcohols. Adv. Synth. Catal. 2002;344:947–952. doi: 10.1002/1615-4169(200210)344:9<947::AID-ADSC947>3.0.CO;2-Z. [DOI] [Google Scholar]
- 21.Raparti V., Chitre T., Bothara K., Danqre S., Khachane C., Gore S., Deshmane B. Novel 4-(morpholin-4-yl)-N′-(arylidene)benzohydrazides: Synthesis, Antimycobacterial Activity and Qsar Investigations. Eur. J. Med. Chem. 2009;44:3954–3960. doi: 10.1016/j.ejmech.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 22.Einsiedel J., Schoerner C., Gmeiner P. Synthesis of Dihydrooxazole Analogues Derived from Linezolid. Tetrahedron. 2003;59:3403–3407. doi: 10.1016/S0040-4020(03)00490-3. [DOI] [Google Scholar]
- 23.Allen C.L., Chhatwal A.R., Williams J.M. Direct Amide Formation from Unactivated Carboxylic Acids and Amines. Chem. Commun. 2012;48:666–668. doi: 10.1039/C1CC15210F. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.