

Abstract
Genetic interactions occur when the combination of multiple mutations yields an unexpected phenotype, and they may confound our ability to fully understand the genetic mechanisms underlying complex diseases. Genetic interactions are challenging to study because there are millions of possible different variant combinations within a given genome. Consequently, they have primarily been systematically explored in unicellular model organisms, such as yeast, with a focus on pairwise genetic interactions between loss-of-function alleles. However, there are many different types of genetic interactions, such as those occurring between gain-of-function or heterozygous mutations. Here, we review recent advances made in the systematic analysis of such diverse genetic interactions in yeast, and briefly discuss how similar studies could be undertaken in human cells.
Keywords: genetic interactions, epistasis, dosage interactions, higher-order interactions, complex haploinsufficiency
Introduction
In this era of affordable whole-genome sequencing, causal mutations have been identified for many Mendelian or monogenic diseases. However, most common diseases cannot be traced to a single genetic cause and may result from complex combinations of genetic and environmental factors. Genome-wide association studies (GWAS) have linked thousands of variants to complex diseases [1], but these variants generally explain only a small fraction of the observed disease phenotype. Several factors may contribute to this so-called “missing heritability”, including failure to detect causal variants, either because they are rare or have very small effects. In addition, the phenotypic effects of observed variants may not combine additively but instead interact in a synergistic manner, causing the variants to be responsible for a larger fraction of the heritability than expected based on their individual effects [2,3]. Detecting genetic interactions in human genotyping datasets is a major challenge because there are so many different possible gene combinations and there are many different types of genetic interactions. However, an understanding of the diversity of the genetic interactions and their general principles as derived from model organism analysis, may provide insights that will help us identify or possibly predict interactions between human variants, which will further our understanding of the complex genetic networks underlying common diseases.
Systematic studies of genetic interactions have mainly used genetically tractable model organisms, which enable rigorous assessment of the effects of combining mutations in an otherwise isogenic background. The budding yeast Saccharomyces cerevisiae is an ideal model organism for these studies, due to its well-annotated genome and the availability of genome-wide mutant libraries and reagents [4–6]. We recently completed a survey of genetic interactions between loss-of-function alleles for nearly all possible pairs of yeast genes [6]. The resultant global genetic network provides insight into the functional organization of a yeast cell, revealing how different pathways work together to coordinate cellular functions and connecting uncharacterized genes to known pathways.
Although the global yeast genetic interaction network quantitatively mapped nearly 1 million genetic interactions and reveals the general principles underlying genetic networks, our ability to predict how genetic variants in natural populations contribute to phenotypes remains limited for several reasons. First, genetic interactions have been predominately mapped using partial or complete loss-of-function alleles, whereas naturally occurring variation can encompass a spectrum of genetic lesions, including separation-of-function and gain-of-function alleles. Second, interactions have largely been studied in haploid cells, while most organisms are naturally diploid and the majority of variants are heterozygous [7]. Finally, in general, systematic studies have focused on interactions between two alleles, but most traits, including gene essentiality [8], are complex and likely modulated by the combined effects of multiple gene variants [3,9]. These complex sets of variants can either combine additively or interact synergistically, either as digenic interactions or more complex combinations, such as trigenic interactions, to lead to profound phenotypes. Here, we review efforts to address these challenges in yeast, and briefly discuss how lessons learned from yeast genetic networks can be used to guide exploration of genetic interactions in more sophisticated biological systems, including human cells.
Mapping digenic interactions involving loss-of-function alleles
A digenic genetic interaction occurs when the combination of two mutations yields a phenotype that is unexpected given the effects of mutating each gene on its own. In yeast, fitness measured as either growth rate or colony size is often the phenotype of choice, and a multiplicative model is frequently applied to score genetic interactions (Fig. 1)[10]. According to this model, negative digenic interactions occur when the double mutant is less fit than expected based on the multiplicative combination of the single mutant fitness values. The most extreme example of a negative genetic interaction is synthetic lethality, in which the combination of two viable mutations leads to cell death (Fig. 1). By contrast, positive digenic interactions occur when the double mutant is more fit than expected (Fig. 1). Positive interactions can be further classified by their relative strength, ranging from masking, in which the double mutant fitness is higher than expected but less than or equal to that of the slowest growing single mutant, to suppression, in which the double mutant is healthier than the slowest growing single mutant and possibly has a fitness that is comparable to wild type (Fig. 1)[11].
Figure 1. Genetic interaction classes involving two genes.
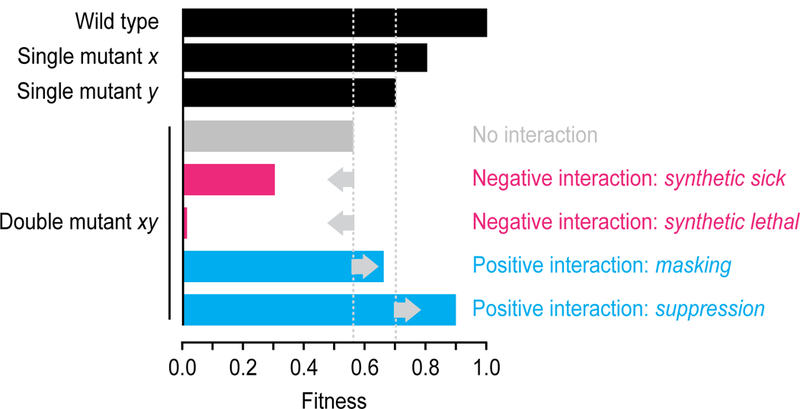
In yeast, genetic interactions are frequently scored using a multiplicative model. When two single mutants (x and y) have a fitness of 0.8 and 0.7 relative to wild-type cells, the expected double mutant (xy) fitness is 0.8 × 0.7 = 0.56. Negative and positive interactions occur when the fitness defect of a double mutant is either more or less severe, respectively, than this expected fitness. A synthetic sick negative genetic interaction occurs when the observed double mutant fitness is lower than expected, but still viable. In a synthetic lethal negative genetic interaction, the combination of two viable single mutants results in an inviable double mutant. A masking positive interaction occurs when the fitness of the double mutant is greater than expected, but lower or equal to that of the slowest growing single mutant. Suppression positive interactions occur when the double mutant fitness is greater than that of the slowest growing single mutant.
Different types of genetic interactions can reflect distinct mechanistic relationships between genes. Synthetic lethal interactions between nonessential genes often reflect the combined effect of impaired function in two parallel pathways that impinge on the same essential biological function and thus can compensate for or buffer each other (Fig. 2A)[12]. Masking positive genetic interactions are frequently observed between members of the same nonessential pathway or complex, such that in the absence of one complex or pathway member, additional loss of another member does not lead to an added fitness effect [13]. Essential genes, on the other hand, for which in general hypomorphic (partial loss-of-function) alleles are used, frequently show negative genetic interactions among genes within the same pathway or complex, as the combination of two partially functional alleles can lead to complete inactivation of the pathway or complex [6]. Finally, suppression interactions can occur between genes that have opposing biochemical roles (Fig. 2A)[14].
Figure 2. Mechanisms of genetic interactions.
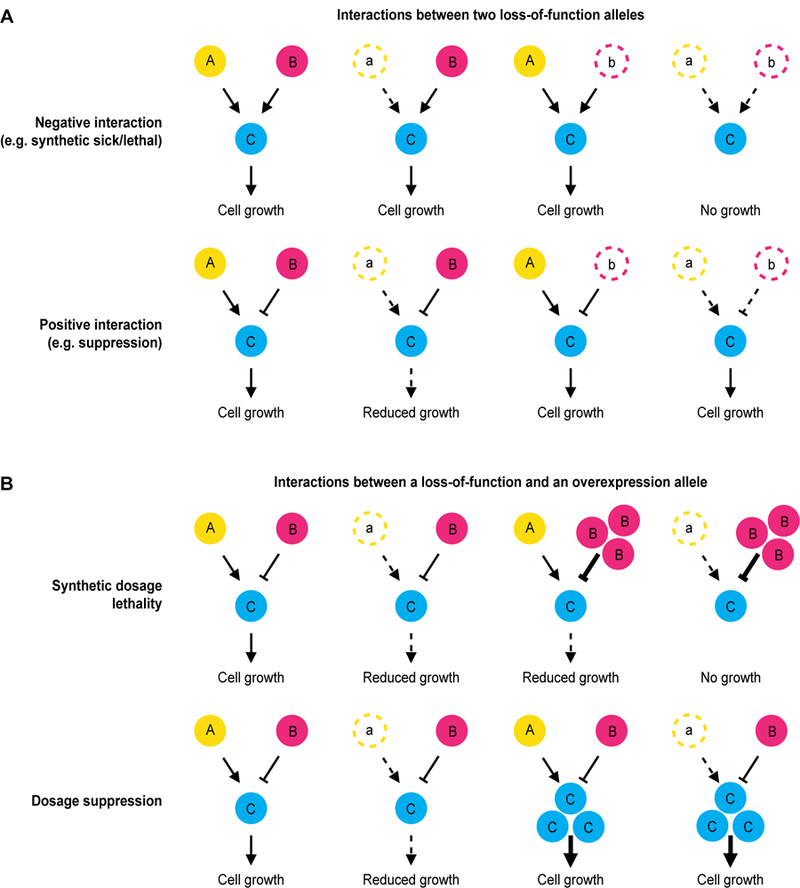
Possible mechanisms of genetic interactions between different types of alleles are illustrated. Wild-type alleles are represented as filled circles, partial or complete loss-of-function alleles as open circles with a dashed border, and overexpression alleles as multiple filled circles. A) Genetic interactions between two loss-of-function alleles. Top: A negative genetic interaction can occur between loss-of-function alleles of genes (“A” and “B”) that function in parallel pathways. Bottom: A positive genetic interaction can occur between genes (“A” and “B”) that have opposite effects on the output of a pathway. B) Genetic interactions involving one loss-of-function allele and one overexpression allele. Top: Synthetic dosage lethality can also occur between genes (“A” and “B”) that have an opposite effect on the output of a pathway. Bottom: Dosage suppression can occur when a fitness defect caused by loss-of-function mutations in a gene (“A”) is compensated for by overexpressing a downstream component of the same pathway (“C”).
The systematic analysis of digenic interactions in yeast has shown that genes encoding proteins that function together in the same pathway or complex tend to share a common set of genetic interactions [6,15]. Genetic interaction profile similarity correlates with functional relation, and can be used to form a hierarchical model of the cell, where the most similar profiles reflect members of the same complex or pathway, somewhat less similar profiles are associated with genes co-annotated to a biological process, and overlapping but more diverse profiles are associated with members of the same cellular compartment [6].
Genetic interactions between subsets of gene that are organized into pathways maps a functional wiring diagram of the cell. Coherent sets of negative or positive genetic interactions often connect all the genes within two different pathways [6]. This means that the synthetic lethality associated with two pathways can be achieved by mutation of many different pairs of genes. For example, a coherent set of genes within the 19S proteasome complex show negative, synthetically lethal, interactions with genes of the anaphase promoting complex [6]. The fact that many different pairs of genes lead to the same yeast double mutant phenotype, suggests that variation in many different pairs of genes may lead to the same disease phenotype in humans.
Mapping digenic interactions using gain-of-function alleles
Analysis of the genetic determinants of cancer and other diseases, clearly shows that gene loss-of-function and gene amplification or gain-of-function are equally important determinants of disease progression [16–18]. Several different genome-wide reagent sets are available for the systematic analysis of genetic interactions involving overexpression alleles in yeast. These include collections of strains harboring plasmids encoding a wild-type open reading frame that is overexpressed, either on a high copy plasmid or by using an inducible or constitutively active promoter [19–21]. Systematic screens using these various strain arrays have produced catalogues of genes that cause a growth defect when overexpressed in wild-type cells (toxic genes) or that make a phenotype caused by another mutation either more severe (synthetic dosage lethality or sickness) or less severe (dosage suppression) than expected (Fig. 2B)[22,23].
Experimentally, dosage genetic interaction screens involve testing the effect of overexpression of a query gene within a genome-wide loss-of-function mutant collection, or, conversely, screening a loss-of-function query mutant with genome-wide plasmid libraries [20,24,25]. Both synthetic dosage lethality and dosage suppression screens have been performed systematically [19,26,27], and typically identify a few dozen interactions for each query mutant of interest. Double dosage lethality screens, in which overexpression alleles of two genes are tested for a genetic interaction, have also been performed [28]. Mechanistically, synthetic dosage lethality can occur between genes that have opposite biological roles, for example overexpression of an inhibitor can be detrimental in the absence of an activator (Fig. 2B). Synthetic dosage lethality can also occur between enzymes and their substrates, where overexpression of the substrate leads to a fitness defect in combination with reduced activity of the enzyme [25,27,28]. Indeed, synthetic dosage lethality screens using kinase mutants are able to capture many known kinase-substrate relationships, especially when the kinase is a negative regulator of a target protein [20,27]. Dosage suppression on the other hand, can occur when overexpression of a downstream pathway member suppresses the fitness defect associated with inactivation of an upstream pathway member (Fig. 2B). Although dosage interactions show significant overlap with the global genetic interaction network of loss-of-function alleles, the majority of synthetic dosage lethality and suppression interactions identify novel interactions and thus represent distinctive genetic and functional relationships [19,24,29]. Thus, integration of genetic interaction screens between loss-of-function alleles and dosage screens enables the discovery of new biology [27]. For example, network motifs derived from an integrated network comprising both loss-of-function and dosage synthetic lethal genetic interactions identified previously unappreciated regulatory relationships between cell polarity proteins [27].
In addition to the use of plasmid libraries, spontaneous suppressor mutations can be isolated that encompass the full spectrum of loss-of-function, gain-of-function or separation-of-function events [14]. The systematic mapping of spontaneous suppressor mutations has highlighted general mechanisms of suppression [14,30]. For example, suppression of partial loss-of-function alleles was frequently achieved through deleterious mutations in the protein or mRNA degradation machineries, which led to increased expression of the partial loss-of-function allele [14]. Despite the prevalence of this type of general suppression, suppression networks are extremely rich in functional information, and thus provide a powerful tool for annotating gene function [14,19]. Finally, genome-wide genetic interaction studies using a set of query strains, each carrying a different point mutation within the same gene, allow for the functional dissection of various domains within a protein, and can link mutations at a residue level to specific cellular consequences [31–33].
Mapping complex haploinsufficiency in diploid cells
Most variation in the human genome is heterozygous [7] and, in some cases, a heterozygous mutation is sufficient to cause an obvious phenotype, a genetic situation known as haplo-insufficiency (Fig. 3)[34,35]. Complex haploinsufficiency, also known as unlinked non-complementation, refers to a genetic interaction occurring between two different heterozygous mutations (Fig. 3). A known example of complex haploinsufficiency (CHI) as a genetic mechanism of disease occurs in retinitis pigmentosa, in which heterozygous loss-of-function alleles of RDS and ROM1 cause a reduction of RDS/ROM1 tetramers, leading to a loss of rod cell production and eyesight loss over time [36]. Given the prevalence of heterozygous variation in the genome, CHI could play a major role in the genotype-to-phenotype relationship in human populations, yet the general principles of this type of genetic interaction remain poorly understood.
Figure 3. Complex haploinsufficiency.
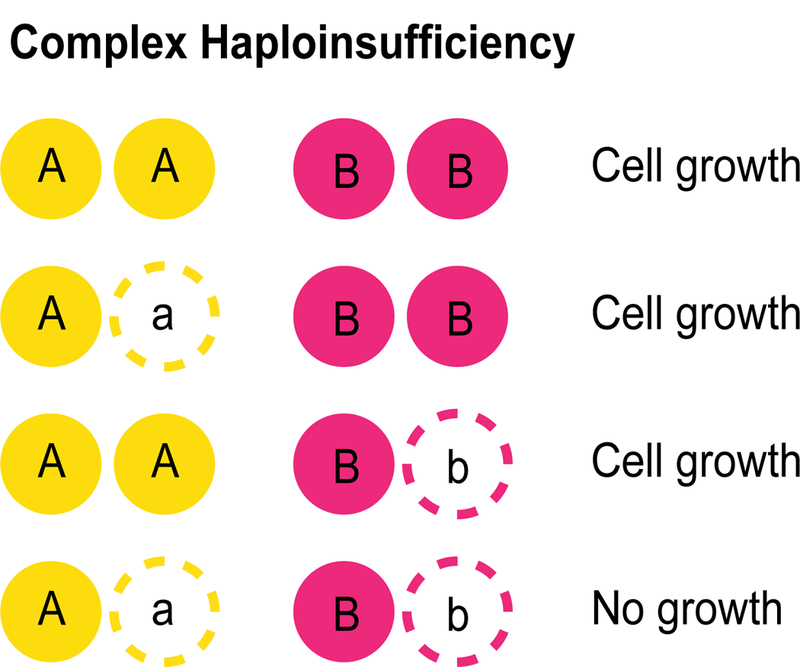
A) Haploinsufficiency occurs when a heterozygous mutation is sufficient to cause a phenotype. B) Complex haploinsufficiency occurs when the combination of two heterozygous mutations results in an unexpected phenotype.
In yeast, a few screens for interactions between two heterozygous alleles have been performed using query strains that have mutations in the essential genes encoding actin (ACT1), α- and β-tubulin (TUB1, TUB2) or kinetochore components (CTF13) [33,37–39]. Although the number of CHI screens has been limited, some universal rules regarding CHI interactions start to emerge. A systematic comparison of the number of CHI interactions between ACT1 and either essential or nonessential interaction partners suggests that essential genes are no more predisposed to CHI interactions than nonessential genes [33,37]. In addition, interaction partners are generally not haploinsufficient on their own, and CHI interactions thus appear to represent specific functional relationships, which frequently involve the reduction of a cellular component that is essential for viability [33,37]. For example, the α- and β-tubulin encoding genes TUB1, TUB2 and TUB3 show negative CHI interactions with each other, probably because mutation of two of the tubulin encoding genes leads to reduced availability of functional α/β-tubulin heterodimers [39]. In another example, ACT1 displays negative CHI interactions with mutants that cause defects in actin cytoskeleton organization or localization, which in combination with reduced actin levels likely leads to a shortage of actin [33,37].
The genome-wide CHI screens identified dozens to a few hundred hits, including both suppressive and lethal interactions [33,37,38]. These findings indicate that CHI interactions may be relatively common, but further analysis is required before we have a clear understanding of the general frequency of these interactions. Interestingly, one study compared CHI interactions to genetic interactions identified in haploid cells, and found no overlap [38]. Although this may be partially due to differences in experimental set-up, this observation suggests that CHI interactions may exhibit different fundamental properties than those mapped in haploid cells. Thus, extensive mapping of interactions between heterozygous alleles may extend our understanding of the genetic wiring of a cell.
Mapping higher-order genetic interactions
Most genetic traits are likely modulated by the combined effects of multiple gene variants [3,8,9]. To be able to fully understand the genotype-to-phenotype relationships underlying genetic traits, we thus need to map interactions involving more than two genes. This may be especially relevant for higher organisms, where gene duplication is more extensive, and pairs of partially redundant genes may buffer each other, such that inactivation of each gene on its own does need lead to genetic vulnerabilities. A recent yeast study found that 22 out of 56 tested paralog pairs can functionally compensate for each other’s absence [40]. In such cases, inactivation of both genes allows for the identification of trigenic interactions that would not have been identified by digenic interaction screens [41]. Genome-wide genetic interaction screens have been performed for a few double mutant pairs in both baker’s and fission yeast, often providing new insights into the functional connections between the genes [41–43]. For example, the genetic interaction profile of a double mutant deleted for CLB5 and CLB6, both encoding cyclins that regulate DNA replication, showed a strong resemblance to that of single mutants involved in sister chromatid cohesion, establishing a role of Clb5 and Clb6 in chromosome segregation [42]. In addition, in a screen for genetic interactions with a double mutant that lacks the histone chaperones Asf1 and Cac1, RDH54 was identified as a synthetic lethal partner that seems to functionally substitute for the two chaperones by interacting much more strongly with chromatin complexes in their absence [42].
Higher-order genetic interactions have also been identified between naturally occurring variants in genetic mapping studies [44]. For example, a cross of two S. cerevisiae strains yielded a few segregants that showed a rough colony morphology, which was traced back to a combination of variants in six different genes, most of which were involved in Ras-signaling [45]. Although these data suggest that genetic interactions involving more than two mutations may be relatively common, further systematic mapping is required to understand the frequency and general properties of these higher-order interactions. One challenge in systematically mapping higher-order interactions is the vast complexity of genetic interaction space. In yeast, there are 18 million possible gene pairs and 36 billion possible gene triads. Thus even if trigenic interactions are substantially less frequent than digenic interactions they may be numerous when explored systematically.
Mapping genetic interactions in human cells
In the past decade, genetic interaction screens have mainly been limited to model organisms that can be easily manipulated in array-based formats, such as bacteria and yeast species. Initial screens for genetic interactions in cultured mammalian cells used RNA interference technology, but these screens frequently suffered from low precision and limited sensitivity [46–49]. The development of CRISPR-Cas9 technology opened the door for efficient and precise mapping of genetic interactions in a wide range of organisms, including cultured human cells [50,51]. In the CRISPR-Cas9 system, a guide RNA forms a complex with the Cas9 nuclease protein, which specifically targets regions in the genome that are complementary to the guide RNA. Upon binding, the Cas9 protein induces a double-strand DNA break, which can either be repaired correctly, or cause a small insertion or deletion that may inactivate the targeted gene. CRISPR-Cas9 genetic interaction screening methods rely on libraries of guide RNAs, and can be performed in three ways. First, wild-type cells can be screened using double guide RNA libraries that target two genes at once [52–54]. Due to the magnitude of the combinatorial space, at most a few hundred genes can be screened for genetic interactions among each other in one experiment. Second, screens can be performed using genome-wide single guide RNA libraries in cell lines engineered to carry a query mutation of interest. By screening isogenic “wild-type” cells in parallel, differences in gene essentiality can be identified that are directly linked to the query mutation [49,55,56]. Finally, synthetic lethal relationships between mutations can also be inferred indirectly, by screening panels of cell lines using genome-wide guide RNA libraries for differential dependencies linked to the status of for example oncogenes, to identify genes that are selectively essential in cell lines carrying a particular driver mutation [56–58].
Initial screens in human cells show that genetic interactions between human genes show similar properties as those identified in yeast. For example, in both organisms, genes whose mutation causes a growth defect show more genetic interactions, and synthetic lethal interactions frequently occur between genes annotated to related biological processes, suggesting that fundamental properties of genetic interactions are conserved [49,54,55]. So far, genetic interaction screens in human cells have mainly focused on the identification of synthetic lethal interactions between loss-of-function alleles. However, as CRISPR-Cas9 screening technology further develops, increased sensitivity of the screens may lead to the detection of positive genetic interactions. In addition, continued developments in Cas9 fusion enzymes for generating point mutations or accomplishing transcriptional activation creates the possibility of genetic interactions screens using different allele types [59–62]. As Cas9 generally produces homozygous gene knockout in diploid cells, further improvements in methods to create heterozygous mutations will be required to efficiently map complex haploinsufficient interactions between heterozygous loci in human cells on a global scale [63].
Conclusion
The identification of genetic interactions between two mutant alleles provides valuable information on functional connections between genes. With a reference genetic interaction network in place, we anticipate that further genetic interaction studies in yeast will shed light on how genetic interaction networks are rewired in the presence of specific non-loss-of-function alleles and in diverse genetic backgrounds. Furthermore, the mapping of interactions between heterozygous alleles and higher-order interactions will provide insight into complex patterns of inheritance. Recent developments in CRISPR-Cas9 technology allow the study of genetic interactions in human cells. A reference network of human genetic interactions will increase the functional annotation of the human genome, and will be invaluable in understanding how variants can combine to yield observed traits, including human disease.
Highlights.
Genetic interactions may frequently affect complex genetic traits
Pairwise interactions have been mapped for most yeast loss-of-function alleles
We discuss recent efforts to map genetic interactions of increased complexity
CRISPR-Cas9 methodology allows similar studies in cultured human cells
Acknowledgements
We thank M. Costanzo for critical comments. Functional genomics work in the Boone and Andrews labs is supported primarily by the National Institutes of Health [grant number R01HG00583] and the Canadian Institutes for Health Research (CIHR) [grant numbers FDN-143264, FDN-143265]. Genetic network mapping in human cells is supported by a grant from the Ontario Research Fund (Research Excellence Award Round 7) and by the Medicine by Design program (Canada First Research Excellence Fund, University of Toronto). J.v.L. was supported by a postdoctoral fellowship from the CIHR. C.B. and B.A. are Senior Fellows and co-Director (C.B.) of the Canadian Institute for Advanced Research Genetic Networks program.
References and recommended reading
- [1].Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, et al. : The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 2014, 42:D1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zuk O, Hechter E, Sunyaev SR, Lander ES: The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A 2012, 109:1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bloom JS, Ehrenreich IM, Loo WT, Lite TL, Kruglyak L: Finding the sources of missing heritability in a yeast cross. Nature 2013, 494:234–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Botstein D, Fink GR: Yeast: an experimental organism for 21st century biology. Genetics 2011, 189:695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, et al. : Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002, 418:387–391. [DOI] [PubMed] [Google Scholar]
- [6].Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, et al. : A global genetic interaction network maps a wiring diagram of cellular function. Science 2016, 353:1381.** This study describes the first genome-wide genetic interaction network for a eukaryotic cell.
- [7].Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. : A global reference for human genetic variation. Nature 2015, 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dowell RD, Ryan O, Jansen A, Cheung D, Agarwala S, Danford T, Bernstein DA, Rolfe PA, Heisler LE, Chin B, et al. : Genotype to phenotype: a complex problem. Science 2010, 328:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Forsberg SK, Bloom JS, Sadhu MJ, Kruglyak L, Carlborg O: Accounting for genetic interactions improves modeling of individual quantitative trait phenotypes in yeast. Nat Genet 2017, 49:497–503.* This study shows that genetic interactions attribute to many quantitative traits in yeast.
- [10].Mani R, Onge RP, Hartman JLt, Giaever G, Roth FP: Defining genetic interaction. Proc Natl Acad Sci U S A 2008, 105:3461–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dixon SJ, Costanzo M, Baryshnikova A, Andrews B, Boone C: Systematic mapping of genetic interaction networks. Annu Rev Genet 2009, 43:601–625. [DOI] [PubMed] [Google Scholar]
- [12].Costanzo M, Baryshnikova A, Myers CL, Andrews B, Boone C: Charting the genetic interaction map of a cell. Curr Opin Biotechnol 2011, 22:66–74. [DOI] [PubMed] [Google Scholar]
- [13].Baryshnikova A, Costanzo M, Kim Y, Ding H, Koh J, Toufighi K, Youn JY, Ou J, San Luis BJ, Bandyopadhyay S, et al. : Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat Methods 2010, 7:1017–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Van Leeuwen J, Pons C, Mellor JC, Yamaguchi TN, Friesen H, Koschwanez J, Mattiazzi Usaj M, Pechlaner M, Takar M, Usaj M, et al. : Exploring genetic suppression interactions on a global scale. Science 2016, 354:599.* These studies systematically map global networks of suppression interactions in yeast using either gene overexpression libraries, or by isolating spontaneous suppressor mutations.
- [15].Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. : The genetic landscape of a cell. Science 2010, 327:425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Buchanan JA, Scherer SW: Contemplating effects of genomic structural variation. Genet Med 2008, 10:639–647. [DOI] [PubMed] [Google Scholar]
- [17].Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ: Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013, 155:948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yi S, Lin S, Li Y, Zhao W, Mills GB, Sahni N: Functional variomics and network perturbation: connecting genotype to phenotype in cancer. Nat Rev Genet 2017. [DOI] [PMC free article] [PubMed]
- [19].Magtanong L, Ho CH, Barker SL, Jiao W, Baryshnikova A, Bahr S, Smith AM, Heisler LE, Choy JS, Kuzmin E, et al. : Dosage suppression genetic interaction networks enhance functional wiring diagrams of the cell. Nat Biotechnol 2011, 29:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sopko R, Huang D, Preston N, Chua G, Papp B, Kafadar K, Snyder M, Oliver SG, Cyert M, Hughes TR, et al. : Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell 2006, 21:319–330. [DOI] [PubMed] [Google Scholar]
- [21].Prelich G: Gene overexpression: uses, mechanisms, and interpretation. Genetics 2012, 190:841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kroll ES, Hyland KM, Hieter P, Li JJ: Establishing genetic interactions by a synthetic dosage lethality phenotype. Genetics 1996, 143:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rine J: Gene overexpression in studies of Saccharomyces cerevisiae. Methods Enzymol 1991, 194:239–251. [DOI] [PubMed] [Google Scholar]
- [24].Measday V, Baetz K, Guzzo J, Yuen K, Kwok T, Sheikh B, Ding H, Ueta R, Hoac T, Cheng B, et al. : Systematic yeast synthetic lethal and synthetic dosage lethal screens identify genes required for chromosome segregation. Proc Natl Acad Sci U S A 2005, 102:13956–13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Duffy S, Fam HK, Wang YK, Styles EB, Kim JH, Ang JS, Singh T, Larionov V, Shah SP, Andrews B, et al. : Overexpression screens identify conserved dosage chromosome instability genes in yeast and human cancer. Proc Natl Acad Sci U S A 2016, 113:9967–9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Patra B, Kon Y, Yadav G, Sevold AW, Frumkin JP, Vallabhajosyula RR, Hintze A, Ostman B, Schossau J, Bhan A, et al. : A genome wide dosage suppressor network reveals genomic robustness. Nucleic Acids Res 2016, 45:255–270.* These studies systematically map global networks of suppression interactions in yeast using either gene overexpression libraries, or by isolating spontaneous suppressor mutations.
- [27].Sharifpoor S, van Dyk D, Costanzo M, Baryshnikova A, Friesen H, Douglas AC, Youn JY, VanderSluis B, Myers CL, Papp B, et al. : Functional wiring of the yeast kinome revealed by global analysis of genetic network motifs. Genome Res 2012, 22:791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Youn JY, Friesen H, Nguyen Ba AN, Liang W, Messier V, Cox MJ, Moses AM, Andrews B: Functional Analysis of Kinases and Transcription Factors in Saccharomyces cerevisiae Using an Integrated Overexpression Library. G3 (Bethesda) 2017, 7:911–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zimmermann C, Garcia I, Omerzu M, Chymkowitch P, Zhang B, Enserink JM: Mapping the Synthetic Dosage Lethality Network of CDK1/CDC28. G3 (Bethesda) 2017, 7:1753–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].van Leeuwen J, Pons C, Boone C, Andrews BJ: Mechanisms of suppression: The wiring of genetic resilience. Bioessays 2017, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Braberg H, Jin H, Moehle EA, Chan YA, Wang S, Shales M, Benschop JJ, Morris JH, Qiu C, Hu F, et al. : From Structure to Systems: High-Resolution, Quantitative Genetic Analysis of RNA Polymerase II. Cell 2013. [DOI] [PMC free article] [PubMed]
- [32].Beltrao P, Cagney G, Krogan NJ: Quantitative genetic interactions reveal biological modularity. Cell 2010, 141:739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Haarer B, Viggiano S, Hibbs MA, Troyanskaya OG, Amberg DC: Modeling complex genetic interactions in a simple eukaryotic genome: actin displays a rich spectrum of complex haploinsufficiencies. Genes Dev 2007, 21:148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kwabi-Addo B, Giri D, Schmidt K, Podsypanina K, Parsons R, Greenberg N, Ittmann M: Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc Natl Acad Sci U S A 2001, 98:11563–11568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Deutschbauer AM, Jaramillo DF, Proctor M, Kumm J, Hillenmeyer ME, Davis RW, Nislow C, Giaever G: Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics 2005, 169:1915–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rivolta C, Sharon D, DeAngelis MM, Dryja TP: Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns. Hum Mol Genet 2002, 11:1219–1227. [DOI] [PubMed] [Google Scholar]
- [37].Haarer B, Aggeli D, Viggiano S, Burke DJ, Amberg DC: Novel interactions between actin and the proteasome revealed by complex haploinsufficiency. PLoS Genet 2011, 7:e1002288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Baetz KK, Krogan NJ, Emili A, Greenblatt J, Hieter P: The ctf13–30/CTF13 genomic haploinsufficiency modifier screen identifies the yeast chromatin remodeling complex RSC, which is required for the establishment of sister chromatid cohesion. Mol Cell Biol 2004, 24:1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Stearns T, Botstein D: Unlinked noncomplementation: isolation of new conditional-lethal mutations in each of the tubulin genes of Saccharomyces cerevisiae. Genetics 1988, 119:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Diss G, Gagnon-Arsenault I, Dion-Cote AM, Vignaud H, Ascencio DI, Berger CM, Landry CR: Gene duplication can impart fragility, not robustness, in the yeast protein interaction network. Science 2017, 355:630–634. [DOI] [PubMed] [Google Scholar]
- [41].Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, et al. : Global mapping of the yeast genetic interaction network. Science 2004, 303:808–813. [DOI] [PubMed] [Google Scholar]
- [42].Haber JE, Braberg H, Wu Q, Alexander R, Haase J, Ryan C, Lipkin-Moore Z, Franks-Skiba KE, Johnson T, Shales M, et al. : Systematic triple-mutant analysis uncovers functional connectivity between pathways involved in chromosome regulation. Cell Rep 2013, 3:2168–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rallis C, Townsend S, Bahler J: Genetic interactions and functional analyses of the fission yeast gsk3 and amk2 single and double mutants defective in TORC1-dependent processes. Sci Rep 2017, 7:44257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Taylor MB, Ehrenreich IM: Higher-order genetic interactions and their contribution to complex traits. Trends Genet 2015, 31:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Taylor MB, Ehrenreich IM: Transcriptional Derepression Uncovers Cryptic Higher-Order Genetic Interactions. PLoS Genet 2015, 11:e1005606.* This study shows that naturally occurring variants in 6 different genes underlie a complex genetic trait in yeast.
- [46].Bassik MC, Kampmann M, Lebbink RJ, Wang S, Hein MY, Poser I, Weibezahn J, Horlbeck MA, Chen S, Mann M, et al. : A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell 2013, 152:909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kampmann M, Bassik MC, Weissman JS: Integrated platform for genome-wide screening and construction of high-density genetic interaction maps in mammalian cells. Proc Natl Acad Sci U S A 2013, 110:E2317–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Laufer C, Fischer B, Billmann M, Huber W, Boutros M: Mapping genetic interactions in human cancer cells with RNAi and multiparametric phenotyping. Nat Methods 2013, 10:427–431. [DOI] [PubMed] [Google Scholar]
- [49].Vizeacoumar FJ, Arnold R, Vizeacoumar FS, Chandrashekhar M, Buzina A, Young JT, Kwan JH, Sayad A, Mero P, Lawo S, et al. : A negative genetic interaction map in isogenic cancer cell lines reveals cancer cell vulnerabilities. Mol Syst Biol 2013, 9:696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. : Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang T, Wei JJ, Sabatini DM, Lander ES: Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343:80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wong AS, Choi GC, Cui CH, Pregernig G, Milani P, Adam M, Perli SD, Kazer SW, Gaillard A, Hermann M, et al. : Multiplexed barcoded CRISPR-Cas9 screening enabled by CombiGEM. Proc Natl Acad Sci U S A 2016, 113:2544–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Han K, Jeng EE, Hess GT, Morgens DW, Li A, Bassik MC: Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat Biotechnol 2017. [DOI] [PMC free article] [PubMed]
- [54].Shen JP, Zhao D, Sasik R, Luebeck J, Birmingham A, Bojorquez-Gomez A, Licon K, Klepper K, Pekin D, Beckett AN, et al. : Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat Methods 2017. [DOI] [PMC free article] [PubMed]
- [55].Blomen VA, Majek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A, et al. : Gene essentiality and synthetic lethality in haploid human cells. Science 2015, 350:1092–1096.** This study uses isogenic query mutant cell lines to systematically detect genetic interactions in human cells.
- [56].Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, Sabatini DM: Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 2017. [DOI] [PMC free article] [PubMed]
- [57].Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. : High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163:1515–1526. [DOI] [PubMed] [Google Scholar]
- [58].Steinhart Z, Pavlovic Z, Chandrashekhar M, Hart T, Wang X, Zhang X, Robitaille M, Brown KR, Jaksani S, Overmeer R, et al. : Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat Med 2017, 23:60–68. [DOI] [PubMed] [Google Scholar]
- [59].Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, Tabata M, Mochizuki M, Miyabe A, Araki M, Hara KY, et al. : Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016, 353. [DOI] [PubMed] [Google Scholar]
- [60].Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR: Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533:420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. : Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159:647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. : Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517:583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, Olsen KM, Gregg A, Noggle S, Tessier-Lavigne M: Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533:125–129. [DOI] [PubMed] [Google Scholar]