

Abstract
A collection of 32 structurally related N-(4-halobenzyl)amides were synthesized from cinnamic and benzoic acids through coupling reactions with 4-halobenzylamines, using (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) as a coupling agent. The compounds were identified by spectroscopic methods such as infrared, 1H- and 13C- Nuclear Magnetic Resonance (NMR) and high-resolution mass spectrometry. The compounds were then submitted to antimicrobial tests by the minimum inhibitory concentration method (MIC) and nystatin was used as a control in the antifungal assays. The purpose of the tests was to evaluate the influence of structural changes in the cinnamic and benzoic acid substructures on the inhibitory activity against strains of Candida albicans, Candida tropicalis, and Candida krusei. A quantitative structure-activity relationship (QSAR) study with KNIME v. 3.1.0 and Volsurf v. 1.0.7 softwares were realized, showing that descriptors DRDRDR, DRDRAC, L4LgS, IW4 and DD2 influence the antifungal activity of the haloamides. In general, 10 benzamides revealed fungal sensitivity, especially a vanillic amide which enjoyed the lowest MIC. The results demonstrate that a hydroxyl group in the para position, and a methoxyl at the meta position enhance antifungal activity for the amide skeletal structure. In addition, the double bond as a spacer group appears to be important for the activity of amide structures.
Keywords: halogenated amides, antimicrobial activity, Candida, vanillic acid derivatives
1. Introduction
The genus Candida includes over 200 species of human pathogens. Among the most important of these are Candida albicans, Candida tropicalis and Candida krusei. Changes in host defense mechanisms, invasive medical procedures, and anatomical barrier failures (through burns) are all factors that favor infection with these micro-organisms [1]. However, there are compounds in Nature that can inhibit such invasions. Benzoic and cinnamic acid derivatives exhibit pharmacological versatility with antitumor, anti-inflammatory, anti-microbial and immunostimulatory activities [2,3]. In the literature, cinnamic amides inhibit the growth of fungi such as Phytophthora infestans [4], Aspergillus niger, Candida albicans, and bacteria such as Escherichia coli, Bacillus subtilis and Staphylococcus aureus [5]. Cinnamic amides with cinnamic, caffeic, ferulic, sinapic, p-coumaric and 3,4,5-trimethoxycinnamic cores display proven antimicrobial activity [5]. Benzoic acid acts as an unspecific antimicrobial, inhibiting β-carbonic anhydrase in C. albicans and Cryptococcus neoformans [6]. Other benzoic derivatives such as protocatequetic, gentisic, vanillic, and p-hydroxybenzoic acids exhibit antimicrobial properties against various bacterial and fungal strains [7].
The literature reports that halogenated aromatic rings in cinnamic amides potentiate the biological activity, such as EGFR kinase inhibition [8], pesticidal effect [9,10] and microbial growth inhibition [11]. Among the halogenated amides studied, chlorinated cinnamic analogues showed higher microbial inhibition in bacterial and fungal strains than their fluorinated and brominated analogs. Furthermore, certain studies show that salicylanilides with ortho and para hydroxyls, and meta methoxyl on the aromatic ring all display increased microbial inhibitory activity [12]. Some studies also show the importance of substituents in the aromatic ring such as nitro groups, methyls and sterically bulky groups [13]. In this present study, we prepared a collection of structurally related cinnamic and benzoic acid amides with halogenated substituents in the para position, illustrated in Figure 1, in a coupling reaction using benzotriazol-1-yloxy-tris(dimethylamino) phosphonium hexafluorophosphate (BOP) as the coupling agent [14,15]. It is expected that the study will provide more information about structure-activity relationships (SAR) in this group of amides. Chemical parameters, such as lipophilicity, electronic effects, and hydrogen bonds caused by the substituents R, including the presence of the spacer (n = 1) between the carbonyl group and the aromatic ring, were analyzed in the SAR.
Figure 1.
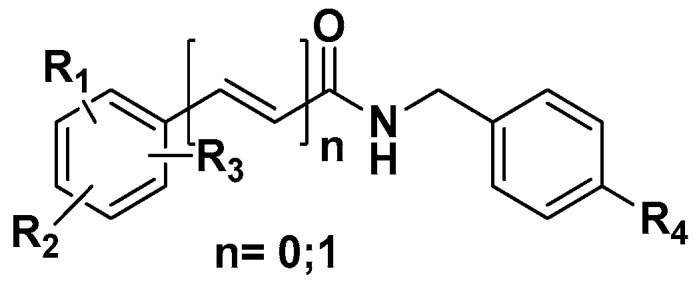
Structural skeleton of synthesized amides with substituents R1, R2 and R3 = H, Cl, OH, CH3, OMe, NO2, tert-Bu or C6H5, and R4 = F, Cl, or Br.
2. Results and Discussion
2.1. Chemistry
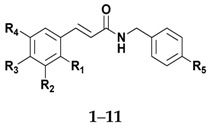
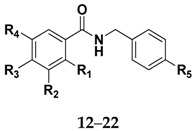
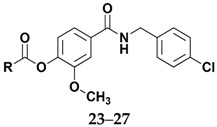
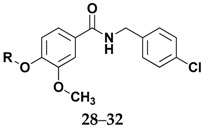
The structures of amides 1–32 (Scheme 1 and Scheme 2, Table 1 and Table 2) were consistent with the infrared spectra (IR), 1H- and 13C-NMR, and high-resolution mass spectra (MALDI) data, according to previous work [16]. The estimated purity values of the amides were at 92%–96% by 1H-NMR [17]. Compounds 1–11 were 4-chlorobenzylamides derived from cinnamic acid, and compounds 12–22 were N-(4-halobenzyl)amides derived from benzoic acid. The rings had different substituents (CH3, OH, OMe, F, Cl, Br, CH3, NO2, tert-butyl or phenyl). A vanillic acid amide had the best antifungal activity result, so five derived esters (compounds 23–27) and five derived ethers (compounds 28–32) of this vanillic acid amide (15) were prepared using reaction methods for replacing the vanillic ring OH group with an ether or ester chain. All ester and ether derivatives are reported for the first time in the literature and their spectroscopic data were in agreement with the literature data of structurally similar compounds [18,19,20,21,22,23,24,25,26].
Scheme 1.

General procedure for synthesis of halogenated amides.
Scheme 2.

General procedure for preparation of esters and ethers 23–32 derived from vanillic amide (15). R = substituents in reactions of esters and ethers.
Table 1.
Data for amides derived from cinnamic acid and benzoic acid.
 |
 |
|||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | Molecular Formula | Reaction Time (h) | Yield (%) |
| 1 | - | - | - | - | Cl | C16H14ClNO | 3 | 75 |
| 2 | - | OH | OH | - | Cl | C16H14ClNO3 | 7 | 70 |
| 3 | - | OMe | OH | - | Cl | C17H16ClNO3 | 4 | 81 |
| 4 | - | - | OMe | - | Cl | C17H16ClNO2 | 2 | 91 |
| 5 | OH | - | - | - | Cl | C16H14ClNO2 | 5 | 79 |
| 6 | - | OH | - | - | Cl | C16H14ClNO2 | 4 | 76 |
| 7 | - | - | OH | - | Cl | C16H14ClNO2 | 3 | 63 |
| 8 | - | - | Cl | - | Cl | C16H13Cl2NO | 2 | 71 |
| 9 | - | OMe | OH | OMe | Cl | C18H18ClNO4 | 6 | 60 |
| 10 | NO2 | - | - | - | Cl | C16H13ClN2O3 | 2 | 79 |
| 11 | - | OMe | OMe | OMe | Cl | C19H20ClNO4 | 3 | 86 |
| 12 | - | - | - | - | Cl | C14H12ClNO | 3 | 65 |
| 13 | - | - | C6H5 | Cl | C20H16ClNO | 6 | 56 | |
| 14 | - | OH | OH | OH | Cl | C14H12ClNO4 | 3 | 21 |
| 15 | - | OMe | OH | - | Cl | C15H14ClNO3 | 6 | 44 |
| 16 | - | OMe | OH | OMe | Cl | C16H16ClNO4 | 3 | 50 |
| 17 | - | - | OH | - | Cl | C14H12ClNO2 | 3 | 73 |
| 18 | - | C(CH3)3 | OH | C(CH3)3 | Cl | C22H28ClNO2 | 3 | 54 |
| 19 | - | OH | - | OMe | Cl | C15H14ClNO3 | 6 | 60 |
| 20 | - | Me | - | NO2 | Cl | C15H13ClN2O3 | 4 | 41 |
| 21 | - | OMe | OH | - | F | C15H14FNO3 | 2 | 27 |
| 22 | - | OMe | OH | - | Br | C15H14BrNO3 | 2 | 63 |
Table 2.
Data from amide 15 derived ethers and esters.
 |
 |
|||
|---|---|---|---|---|
| Compound | R | Molecular Formula | Reaction Time (h) | Yield (%) |
| 23 | C6H5- | C22H18ClNO4 | 1 | 76 |
| 24 | C6H5CH2- | C23H20ClNO4 | 2 | 78 |
| 25 | CH3(CH2)3- | C20H22ClNO4 | 5 | 43 |
| 26 | 3-Br-C6H4- | C22H17BrClNO4 | 1 | 86 |
| 27 | CH3 | C17H16ClNO4 | 24 | 98 |
| 28 | 4-Me-C6H4CH2- | C24H24ClNO3 | 12 | 30 |
| 29 | 4-OH-C6H4CH2- | C23H22ClNO4 | 24 | 75 |
| 30 | CH3(CH2)2- | C18H20ClNO3 | 12 | 54 |
| 31 | (CH3)2CH- | C18H20ClNO3 | 24 | 54 |
| 32 | CH3(CH2)8CH2- | C20H24ClNO3 | 12 | 81 |
2.2. Antimicrobial Activity
In the antifungal activity study, the N-(4-halobenzyl)amides 1–32 were evaluated against Candida strains. The technique used was the broth microdilution method according to published protocols [27,28] using seven strains of Candida: C. albicans ATCC 76645, C. albicans LM-106; LM-23 C. albicans, C. tropicalis ATCC 13803, C. tropicalis, LM-36, LM-13 C. krusei, and C. krusei LM-656. The control medium result showed no fungal growth, while growth of fungi in the medium without any added drug (sterile control) was detected.
The results are shown in Table 3; the minimum inhibitory concentrations (MICs) of the compounds were significantly different, ranging from 256 to more than 1024 μg·mL−1. The antimicrobial activity of the products was interpreted and considered active or not, according to the following criteria: 50–500 μg·mL−1 = strong/optimum activity; 600–1500 μg·mL−1 = moderate activity; Above 1500 μg·mL−1 = weak activity or inactive product [29,30]. The results of the control culture medium show no microbial growth occurred while control was positive in the yeast viability.
Table 3.
Evaluation of MIC (µg/mL) of the amides derived from cinnamic acid and benzoic acid in the microdilution broth assay.
| MIC b (µg·mL−1)/Yeast | |||||||
|---|---|---|---|---|---|---|---|
| Compound | C. albicans ATCC-76645 | C. albicans LM-106 | C. albicans LM-23 | C. tropicalis ATCC-13803 | C. tropicalis LM-36 | C. krusei LM-13 | C. krusei LM-656 |
| 1 | 1024 | 1024 | 1024 | 512 | 512 | + | 1024 |
| 2 | 256 | 256 | 256 | 512 | 1024 | 512 | 512 |
| 3 | 256 | 256 | 256 | 512 | 512 | 512 | 512 |
| 4 | + | + | + | + | + | + | + |
| 5 | 256 | 256 | 256 | 512 | 512 | 1024 | 1024 |
| 6 | 256 | 256 | 256 | 512 | 512 | 1024 | 1024 |
| 7 | 1024 | 1024 | 512 | + | + | + | 1024 |
| 8 | + | + | + | + | + | + | + |
| 9 | + | + | + | + | + | + | + |
| 10 | 256 | 256 | 256 | 256 | 256 | 512 | 512 |
| 11 | + | + | + | + | + | + | + |
| 12 | + | + | + | + | + | + | + |
| 13 | + | + | + | + | + | + | + |
| 14 | 256 | 256 | 512 | 512 | 512 | 256 | 256 |
| 15 | 512 | 256 | 256 | 256 | 256 | 256 | 256 |
| 16 | + | + | + | + | + | + | + |
| 17 | + | + | + | + | 1024 | + | 256 |
| 18 | + | + | + | + | + | + | + |
| 19 | + | + | + | + | + | + | + |
| 20 | + | + | + | + | + | + | + |
| 21 | 512 | 512 | 512 | + | + | 1024 | 1024 |
| 22 | + | + | + | + | + | + | + |
| 23 | + | + | + | + | + | + | + |
| 24 | + | + | + | + | + | + | + |
| 25 | + | + | + | + | + | + | + |
| 26 | + | + | + | + | + | + | + |
| 27 | + | + | + | + | + | + | + |
| 28 | + | + | + | + | + | + | + |
| 29 | + | + | + | + | + | + | + |
| 30 | + | + | + | + | + | + | + |
| 31 | + | + | + | + | + | + | + |
| 32 | + | + | + | + | + | + | + |
| Nystatin | 3.125 | 3.125 | 3.125 | 3.125 | 3.125 | 3.125 | 3.125 |
+ Growth of the microorganism b MIC defined as the lowest concentration that produced 50% reduction in fungal cell growth after 24 h of incubation.
2.3. Qualitative-Structure Activity Relantioship (QSAR)
Three-dimensional structures (3D) of amides were used as input data to generate 128 descriptors together with the dependent variable (binary classification), which describes the compound as active (A) or inactive (I). The data were used as input to the KNIME v. 3.1.0 software [31]. Importantly, the generation of descriptors is relatively fast for all 32 amides which the training data sets comprised generating all 128 descriptors for Volsurf +, it took less than 1 min, using a computer equipped with an i7, running at 3.4 GHz and equipped with 12 GB of RAM.
The KNIME software inserts the data of active and inactive compounds in mathematical algorithms that try to find the descriptors that explains the influence of the structure on microbial activity. A match is given when the software can separate the truly active and truly inactive compounds. Table 4 summarizes the statistical indices of the match model for training and cross-validation in all antifungal tests. For training set the decision tree generated high rates of correct answers for inactive compounds, up to 83.3%, and lower rates for the active compounds, 22.2%. For cross-validation, the model performed similar to the training set. The specificity (true negative) was greater than the sensitivity (true positive). Overall this means that, there was a lower false positive percentage, if it was compared with true positive prediction, which shows that the method is suitable only to screen active compounds and detect physico-chemical properties of inactive compounds.
Table 4.
Summary of training, internal cross-validation, test results and corresponding match results which were obtained using a leave-one out validation method in KNIME of the total set of 32 haloamides subjected to antifungal tests.
| C. albicans LM-23 | C. tropicalis ATCC-13803 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TRAINING | VALIDATION | TRAINING | VALIDATION | |||||||
| Samples | Match | %Match | Match | %Match | Samples | Match | %Match | Match | %Match | |
| Active | 10 | 3 | 30.0 | 3 | 30.0 | 8 | 2 | 25.0 | 2 | 25.0 |
| Inactive | 22 | 19 | 86.4 | 19 | 86.4 | 24 | 20 | 83.3 | 20 | 83.3 |
| Total | 32 | 22 | 68.8 | 22 | 68.8 | 32 | 22 | 68.8 | 22 | 68.8 |
| C. albicans ATCC-76645 | C. tropicalis LM-36 | |||||||||
| TRAINING | VALIDATION | TRAINING | VALIDATION | |||||||
| Samples | Match | %Match | Match | %Match | Samples | Match | %Match | Match | %Match | |
| Active | 10 | 3 | 30.0 | 3 | 30.0 | 9 | 2 | 22.2 | 2 | 22.2 |
| Inactive | 22 | 19 | 86.4 | 19 | 86.4 | 23 | 20 | 87.0 | 20 | 87.0 |
| Total | 32 | 22 | 68.8 | 22 | 68.8 | 32 | 22 | 68.8 | 22 | 68.8 |
| C. albicans LM-106 | C. krusei LM-656 | |||||||||
| TRAINING | VALIDATION | TRAINING | VALIDATION | |||||||
| Samples | Match | %Match | Match | %Match | Samples | Match | %Match | Match | %Match | |
| Active | 10 | 3 | 30.0 | 3 | 30.0 | 11 | 3 | 27.3% | 3 | 27.3 |
| Inactive | 22 | 19 | 86.4 | 19 | 86.4 | 21 | 19 | 90.5% | 19 | 90.5 |
| Total | 32 | 22 | 68.8 | 22 | 68.8 | 32 | 22 | 68.8% | 22 | 68.8 |
| C. krusei LM-13 | ||||||||||
| TRAINING | VALIDATION | |||||||||
| Samples | Match | %Match | Match | %Match | ||||||
| Active | 8 | 3 | 37.5 | 3 | 37.5 | |||||
| Inactive | 24 | 19 | 79.2 | 19 | 79.2 | |||||
| Total | 32 | 22 | 68.8 | 22 | 68.8 | |||||
According to the decision tree (an organization chart that compares the descriptors, to predict the biological activity), the prediction of actives and inactives was based on two Volsurf descriptors [32] of N-(4-halobenzyl)amides: DRDRDR and DRDRAC, which are pharmacophore descriptors forming the maximum triangular area DRY-DRY-DRY (three hydrophobic regions), and DRY-DRY-Acceptor (two hydrophobic regions and one H-bond acceptor region).
It was possible to establish regions for analogs that produced fungal growth sensitivity. Figure 2 shows the molecular interaction of the grid fields (Molecule Interaction Fields—MIF) around the most active compounds 14 and 15, the weakly active compound 17 and inactive compound 12. The DRY probe is shown in all active and weakly active structures. Looking at the N1 probe (dark blue), regions in the aromatic ring which show an outstanding hydrogen accepting character could be observed. This N1 probe is seen more present near the gallic (14), vanillic (15), and 4-hydroxybenzoic (17) amide hydroxyls. This study did not have a high accuracy rate for matches, decreasing the specificity of software to find descriptors related to biological activity of the evaluated amides.
Figure 2.

Gallic amide (14), vanillic amide (15), 4-hydroxybenzoic amide (17) and benzoic amide (12) interaction fields: energy level −0.6 kcal/mol DRY, and −3 kcal/mol N1 in VolSurf.
3. Materials and Methods
3.1. General Information
Purification of the compounds was performed by column chromatography on silica gel 60 (ART. 7734 Merck, Saint Louis, MO, USA) using a Hex:EtOAc solvent gradient and confirmed by analytical thin layer chromatography on silica gel 60 F254, using ultraviolet light at two wavelengths (254 and 366 nm) from a Mineralight apparatus (UVP, Upland, CA, USA) or H2SO4 in 5% ethanol for detection. FTIR spectra were recorded in a Prestige-21 FTIR spectrometer (Shimadzu, Kyoto, Japan) using KBr pellets. 1H- and 13C-NMR spectra were obtained on a MERCURY machine (200 and 50 MHz for 1H and 13C, respectively. Varian (Palo Alto, CA, USA) in deuterated solvents (CDCl3, MeOD or DMSO-d6) and tetramethylsilane (TMS) was used for the internal standard. Chemical shifts were measured in parts per million (ppm) and coupling constants (J) in Hz. Measurements of atomic mass for the compounds was carried out using an Ultraflex II TOF/TOF mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany) equipped with a high performance solid state laser (λ = 355 nm), and reflector. The system was operated by the Bruker Daltonik FlexControl 2.4 software package (Bruker, Bremen, Germany). Infrared spectrum, 1H-NMR spectrum, Expansion of the spectrum of 1H-NMR, 13C-APT NMR spectrum, expansion of the 13C-APT NMR spectrum, High resolution mass spectrum—MALDI of 2, 3, 14, 16, 23 and 28 are available at the Supplementary Materials.
3.2. Chemistry: General Procedures for the Preparation of Compounds
3.2.1. General Preparation of N-(4-Halobenzyl)amides
Procedure 1: In a 100 mL flask equipped with magnetic stirring, the organic acid (1.35 mmol, 200 mg) was dissolved in dimethylformamide (DMF, 2.7 mL) and trimethylamine (0.14 mL, 1.35 mmol). The solution was cooled in an ice bath (0 °C). Then, 4-chlorobenzylamine (1.35 mmol) was added. Soon after a 1.35 mmol solution of BOP in CH2Cl2 (10 mL) was added to the flask. The reaction was stirred at 0 °C for 30 min, and then for an additional period, at room temperature for 2 h. After the reaction, the CH2Cl2 was removed under reduced pressure and the solution was poured into a separatory funnel containing water (10 mL) and EtOAc (10 mL). The product was extracted with EtOAc (3 × 10 mL). The organic phase was washed sequentially with 1 N HCl, water, 1 M NaHCO3 and water (10 mL of each); dried with Na2SO4, filtered and concentrated in a rotavapor. The amide was purified by gel chromatography on a silica gel column using as the mobile phase an EtOAc:Hex mixture gradient of increasing polarity [11]. The following compounds were prepared by this procedure:
N-(4-Chlorobenzyl)cinnamamide (1). Crystalline solid; 75% yield (274 mg), m.p.: 152–156 °C, IR νmax (cm−1): 3253 (N-H), 3080 (CH sp2), 1654 (C=O), 1616 and 1489 (aromatic C=C), 1040 (stretching C-Cl), 1H-NMR (DMSO-d6): 7.71 (d, J = 16 Hz, 1H, H-7); 7.55–7.50 (m, 2H, H-2, H-6); 7.44–7.35 (m, 3H, H-3); 7.32–7.29 (m, 4H, H-2′, H-3′, H-5′, H-6′); 6.45 (d, J = 16 Hz, 1H, H-8); 6.03 (bs, 1H, O=C-NH); 4.57 (d, J = 4.0 Hz, H-7′). 13C-NMR (DMSO-d6) 166.0 (C=O); 141.9 (C-7); 136.9 (C-1′); 134.8 (C-1); 133.5 (C-4′); 130.0 (C-2′, C-6′); 129.4 (C-3′, C-5′); 129.0 (C-3, C-5, C-6); 128.0 (C-2, C-4); 120.2 (C-8); 43.3 (C-7′) [16].
(E)-N-(4-Chlorobenzyl)-3-(3,4-dihydroxyphenyl)acrylamide (2). Dark amorphous solid; yield: 70% (228 mg), m.p.: 103–105 °C, IR νmax (cm−1): 3460 and 3406 (OH) 3230 (NH), 3018 (CH sp2), 1651 (C=O) 1602 and 1490 (aromatic C=C), 1089 (C-Cl stretch). 1H-NMR (CD3OD): 7.55 (d, J = 15.7 Hz, 1H; H-7), 7.44–7.38 (m, 4H; H-2′, H-3′, H-5′, H-6′); 7.13 (d, J = 1.7 Hz, 1H; H-2); 7.02 (dd, J = 8.2, 1.8 Hz, 1H; H-5), 6.88 (d, J = 8.1 Hz, 1H; H-6), 6.52 (d, J = 15.7 Hz, 1H, H-8), 4.55 (bs, 2H; H-7′). 13C-NMR (CD3OD): 169.2 (C=O); 148.8 (C-4); 146.7 (C-3); 142.8 (C-7); 138.9 (C-1′); 131,4 (C-4′); 130.1 (C-2′, C-6′); 129.6 (C-3′; C-5′); 128.2 (C-1); 122.2 (C-6); 118.0 (C-8); 115.0 (C-2); 116.4 (C-5); 43.5 (C-7′) [16]. HRMS (MALDI) calculated for C16H14ClNNaO3 [M + Na]+: 326.0560; found 326.0561.
(E)-N-(4-Chlorobenzyl)-3-(4-hydroxy-3-methoxyphenyl)acrylamide (3). White amorphous solid; yield: 81% (273 mg), m.p. 129–131 °C, IR νmax (cm−1): 3414 (OH) 3275 (NH), 3010 (CH sp2), 1645 (C=O), 1612 and 1460 (aromatic C=C), 1039 (C-Cl stretch). 1H-NMR (DMSO-d6): 7.59 (d, J = 16 Hz, 1H; H-7) 7.28 (dd, J = 2.0 Hz and 6.0 Hz, 2H; H-2′, H-6′); 7.22 (dd, J = 2.0 Hz, 6.0 Hz, 2H, H-3′, H-5′) 7.13 (s, 1H; H-2); 6.95 (d, J = 8.1 Hz; 1H; H-5); 6.87 (d, J = 8.1 Hz; 1H; H-6); 6.27(d, J = 16 Hz, 1H; H-8); 4.36 (d, J = 6.0 Hz, 2H, H-7′); 3.79 (s, 3H, OCH3). 13C-NMR (DMSO-d6): 165.5 (C=O), 148.4 (C-3), 147.8 (C-4), 139.6 (C-7), 138.7 (C-1′), 131.3 (C-4′), 129.2 (C-2′, C-6′), 128.3 (C-3′, C-5′), 126.3 (C-1), 121.7 (C-6), 118.6 (C-8), 115.7 (C-5), 110.8 (C-2), 55.6 (OCH3), 41.6 (C-7′) [16]. HRMS (MALDI) calculated for C17H16ClNO3 [M + H]+: 318.0887; found 318.0870.
(E)-N-(4-Chlorobenzyl)-3-(4-methoxyphenyl)acrylamide (4). Crystalline solid; yield: 91% (311 mg), m.p. 148–150 °C, IR νmax (cm−1): 3282 (N-H), 3041 (C-H sp2), 1647 (C=O), 1602 and 1462 (aromatic C=C), 1029 (stretching C-Cl). 1H-NMR (DMSO-d6): 8.59 (t, J = 6.0 Hz, 1H, O=C-NH), 7.50 (t, J = 7.3 Hz, 2H; H-2; H-6), 7.44–7.18 (m, 5H; H-7, H-2′, H-3′, H-5′, H-6′), 6.97 (d, J = 8.7 Hz, 2H, H-3, H-5), 6.54 (d, J = 15.7 Hz, 1H; H-8), 4.38 (d, J = 6.0 Hz, 2H; H-7′), 3.77 (s, 3H, OCH3). 13C-NMR (DMSO-d6): 165.5 (C=O); 160.4 (C-4); 139.0 (C-7); 138.7 (C-1′); 131.4 (C-4′); 129.3 (C-2; C-6, C-2′; C-6′); 128.3 (C-3′, C-5′); 127.4 (C-1); 119.4 (C-8); 114.5 (C-3, C-5); 55.3 (OCH3); 41.7 (C-7′) [16]. HRMS (MALDI) calculated for C16H14ClNO2 [M + H]+: 324.0767; found 324.0771.
(E)-N-(4-Chlorobenzyl)-3-(2-hydroxyphenyl)acrylamide (5). Yellow amorphous solid; yield: 79% (277 mg), m.p.: 165–171 °C. νmax IR (cm−1): 3369 (OH), 3072 (C-H sp2), 1649 (C=O), 1589 and 1458 (aromatic C=C), 1093 (C-Cl stretch). 1H-NMR (DMSO-d6) 10.00 (bs, 1H, OH), 8.64 (t, J = 5.83 Hz, 1H; O=C-NH), 7.78 (d, J = 15.92 Hz; 1H; H-7), 7.49–7.15 (m, 6H; H-6, H-2′, H-3′, H-5′, H-6′), 6.91–6.76 (m, 2H; H-4, H-5), 6.73 (d, J = 15.92 Hz, 1H; H-8), 4.47 (d, J = 5.86 Hz, 2H; H-7′). 13C-NMR (DMSO-d6) 165.8 (C=O); 156.4 (C-2); 138.8 (C-1′); 135.2 (C-4); 135.0; 130.6 (C-4′); 129.3(C-2′, C-6′); 128.4 (C-6); 128.3 (C-3′, C-5′); 121.6 (C-1); 121.3 (C-5); 119.4 (C-8); 116.2; 41.7 (C-7′) [16]. HRMS (MALDI) calculated for C16H14ClNO2 [M + H]+: 288.0781; found 288.0785.
(E)-N-(4-Chlorobenzyl)-3-(3-hydroxyphenyl)acrylamide (6). Yellow crystalline solid; yield: 76% (268 mg), m.p.: 140–143 °C νmax IR (cm−1): 3460 (OH), 3075 (CH sp2), 1649 (C=O), 1591 and 1448 (aromatic C=C), 1089 (C-Cl stretch). 1H-NMR (DMSO-d6): 9.61 (s, 1H, OH), 8.67 (t, J = 5.9 Hz, 1H, O=C-NH), 7.47–7.14 (m, 6H; H-5, H-7, H-2′, H-3′, H-5′ e H-6′), 7.03–6.92 (m, 2H; H-4, H-6), 6.84–6.70 (m, 1H; H-2), 6.60 (d, J = 15.8 Hz, 1H; H-8), 4.38 (d, J = 5.9 Hz, 2H; H-7′). 13C-NMR (DMSO-d6): 165.1 (C=O); 157.7 (C-3); 139.4 (C-7); 138.5 (C-1′); 136.1 (C-1); 131.4 (C-4′); 130.0 (C-5); 129.3 (C-2′, C-6′); 128.3 (C-3′, C-5′); 121.7 (C-8); 118.8 (C-6); 116.8 (C-2); 113.8 (C-4); 41.7 (C-7′) [16]. HRMS (MALDI) calculated for C16H14ClNNaO2 [M + Na]+: 310.0611; found 310.0619.
(E)-N-(4-Chlorobenzyl)-3-(4-hydroxyphenyl)acrylamide (7). Crystalline solid; yield: 63% (221 mg), m.p. 157–160 °C. IV νmax (cm−1): 3479 and 3369 (OH), 3072 (C-H sp2), 1647 (C=O), 1589 and 1458 (aromatic C=C), 1093 (stretching C-Cl). 1H-NMR (DMSO-d6): 9.87 (bs, 1H, OH); 8.53 (t, J = 5.87 Hz, 1H, O=C-NH); 7.45–7.36 (m, 5H, H-2, H-6, H-7, H-2′, H-6′); 7.35–7.25 (m, 3H; H-2; H-3′e H-5′); 6.77 (d, J = 8.5 Hz, 2H; H-3 e H-5); 6.44 (d, J = 15.73 Hz, 1H; H-8); 4.36 (d, J = 6.0 Hz, 2H; H-7′). 13C-NMR (DMSO-d6) 165.6 (C=O); 159.0 (C-4); 139.4 (C-7); 138.8 (C-1′); 131.4 (C-4′); 129.4 (C-2, C-6); 129.3 (C-2′, C-6′); 128.3 (C-3′, C-5′); 125.9 (C-1); 118.3 (C-8); 115.8 (C-3, C-5); 41.6 (C-7′) [16]. HRMS (MALDI) calculated for C16H14ClNNaO2 [M + Na]+: 310.0611; found 310.0596.
(E)-N-(4-Chlorobenzyl)-3-(4-chlorophenyl) acrylamide (8). Crystalline solid; yield: 71% (240 mg), m.p. 157–161 °C. IV νmax (cm−1): 3414 (N-H), 3041 (C-H sp2), 1651 (C=O), 1614 and 1487 (aromatic C=C), 1089 (stretching C-Cl). 1H-NMR (DMSO-d6): 8.70 (t, J = 6.0 Hz, 1H, O=C-NH), 7.64–7.46 (m, 4H, H-2′, H-3′, H-5′, H-6′), 7.45–7.25 (m, 5H; H-2, H-6, H-3, H-5, H-7), 6.69 (d, J = 15.8 Hz, 1H; H-8), 4.39 (d, J = 6.0 Hz, 2H). 13C-NMR (DMSO-d6) 164.9 (C=O); 138.5 (C-1′); 137.9 (C-7); 134.0 (C-4); 133.8 (C-1); 131.5 (C-4′); 129.3 (C-2, C-6, C-2′ e C-6′); 129.0 (C-3, C-5); 128.3 (C-3′, C-5′);122.7 (C-8); 41.7 (C-7′) [16]. HRMS (MALDI) calculated for C16H13Cl2NNaO [M + Na]+: 328.0272; found 328.0273.
(E)-N-(4-Chlorobenzyl)-3-(4-hydroxy-3,5-dimethoxyphenyl)-acrylamide (9). Yellow amorphous solid; yield: 60% (193 mg), m.p.: 182–185 °C, IR νmax (cm−1): 3414 (OH) and 3358 or(NH), 3000 (CH sp2), 1658 (C=O), 1624 and 1458 (aromatic C=C), 1091 (C-Cl stretch). 1H-NMR (DMSO-d6) 8.52 (t, J = 6.0 Hz; 1H, O=C-NH), 7.28–7.48 (m, 5H; H-7, H-2′, H-3′, H-5′, H-6′), 6.86 (s, 2H; H-2, H-6), 6.55 (d, J = 15.8, 1H; H-8), 4.37 (d, J = 6 Hz, 2H; H-7′) 3.79 (s, 6H; OCH3). 13C-NMR (DMSO-d6) 165.6 (C=O); 148.1 (C-3, C-5); 140.0 (C-7) 138.7 (C-4); 137.4 (C-1′); 131.4; C-4′); 129.2 (C-2′, C-6′); 128.4 (C-3′, C-5′); 125.3 (C-1); 119.1 (C-8); 105.3 (C-2, C-6); 56.0 (OCH3); 41.7 (C-7′) [16]. HRMS (MALDI) calculated for C18H18ClNNaO4 [M + Na]+: 370.0822; found 370.0813.
(E)-N-(4-Chlorobenzyl)-3-(2-nitrophenyl)-acrylamide (10). White crystalline solid; yield: 79% (260 mg), m.p.: 164–167 °C, IR νmax (cm−1): 3290 (NH), 3030 (CH sp2), 1651 (C=O), 1624 and 1458 (aromatic C=C), 1525 and 1342 (C=O), 1091 (C-Cl stretch). 1H-NMR (DMSO-d6): 8.82 (t, J = 4.7 Hz, 1H, O=C-NH), 8.05 (d, J = 8.0 Hz, 1H; H-3), 7.78–7.75 (m, 2H; H-6, H-7), 7.72–7.57 (m, 2H; H-4, H-5), 7.43–7.27 (m, 4H, H-2′, H-3′, H-5′ e H-6′), 6.67 (d, J = 5.6 Hz, 1H; H-8), 4.39 (d, J = 5.9 Hz, 1H; H-7′). 13C-NMR (DMSO-d6): 164.3 (C=O); 148.4 (C-2); 138.3 (C-1′); 134.3 (C-7); 133.9 (C-5); 131.6 (C-4′); 130.4 (C-4); 130.0 (C-1); 129.4 (C-2′, C-6′); 128.8 (C-6); 128.4 (C-3′, C-5′); 126.6 (C-3); 124.7 (C-8); 41.8 (C-7′) [16]. HRMS (MALDI) calculated for C16H13ClN2O3 [M + H]+: 317.0683; found 317.0683.
(E)-N-(4-Chlorobenzyl)-3-(3,4,5-trimethoxyphenyl) acrylamide (11). Crystalline solid; yield: 86% (260 mg), m.p. 146–150 °C, IR νmax (KBr, cm−1): 3290 (N-H), 3070 (C-H sp2), 1651 (C=O), 1614 and 1415 (aromatic C=C), 1029 (stretching C-Cl). 1H-NMR (DMSO-d6): 8.60 (t, J = 5.9 Hz, 1H; O=C-NH), 7.46–7.26 (m, 5H; H-7, H-2′, H-3′, H-5′, H-6′), 6.90 (s, 2H; H-2, H-6), 6.64 (d, J = 15.7 Hz, 1H; H-8), 4.38 (d, J = 5.9 Hz, 1H; H-7′), 3.80 (s, 6H; m-OCH3). 3.68 (s, 3H; p-OCH3). 13C-NMR (DMSO-d6) 165.2 (C=O); 153.1 (C-3, C-5); 139.4 (C-7); 138.7 (C-4); 138.6 (C-1′); 131.4 (C-4′); 130.5 (C-1); 129.2 (C-2′, C-6′); 128.4 (C-3′, C-5′); 121.3 (C-8); 105.1 (C-2, C-6); 60.2 (C-4-OCH3); 55.9 (C-3,5-OCH3); 41.7 (C-7′) [16]. HRMS (MALDI) calculated for C19H20ClNO4 ([M + H]+: 384.0979, found 384.0913.
N-(4-Chlorobenzyl) benzamide (12). Crystalline solid; yield: 65% (260 mg), m.p. 136–139 °C, IR νmax (cm−1): 3300 (N-H), 3082 (C-H sp2), 1637 (C=O), 1618 and 1490 (aromatic C=C), 1091 (stretching C-Cl). 1H-NMR (DMSO-d6): 9.10 (t, J = 5.9 Hz, 1H, O=C-NH), 7.89 (dd, J = 8.0 e 1.6 Hz, 2H; H-2, H-6), 7.64–7.09 (m, 7H; H-3, H-4, H-5, H-2′, H-3′, H-5′, H-6′), 4.46 (d, J = 6.0 Hz, 2H; H-7′). 13C-NMR (DMSO-d6): 166.4 (C=O); 138.8 (C-1′); 134.2 (C-1); 131.4 (C-4); 131.3 (C-2′, C-6′); 129.1 (C-3′, C-5′); 128.3 (C-3, C-5); 127.3 (C-2, C-6); 42.1 (C-7′) [16].
N-(4-Chlorobenzyl)-[1,1′-biphenyl]-4-carboxamide (13). The product was prepared according to procedure 1. White amorphous solid; yield: 56% (198 mg), m.p. 222–228 °C, IR νmax (cm−1): 3271 (N-H), 3078 (C-H sp2), 1633 (C=O), 1606 and 1487 (aromatic C=C), 1089 (stretching C-Cl). 1H-NMR (DMSO-d6): 9.15 (t, J = 6.1 Hz, 1H, O=C-NH), 7.99 (d, J = 8.3 Hz, 2H; H-2, H-6), 7.84–7.65 (m, 4H; H-3, H-5, H-2″, H-6″), 7.56–7.29 (m, 7H; H-2′, H-3′, H-5′, H-6′, H-3″ H-4″, H-5″), 4.48 (d, J = 5.9 Hz, 2H). 13C-NMR (DMSO-d6) 166.1 (C=O); 143.0 (C-4); 139.2 (C-1″); 138.8 (C-1′); 133.0 (C-1); 131.4 (C-4′); 129.2 (C-2′, C-6′); 128.4 (C-2, C-6); 128.1 (C-9, C-11); 127.0 (C-3′, C-5′); 126.7 (C-10) (C-3, C-5, C-8, C-12); 42.1 (C-7′) [16]. HRMS (MALDI) calculated for C20H16ClNO3 [M + H]+: 322.0988; found 322.0969.
N-(4-Chlorobenzyl)-3,4,5-trihydroxybenzamide (14). Yellow amorphous solid; yield: 21% (73 mg), m.p. 96–100 °C, IR νmax (cm−1): 3400 (OH), 3400 (NH), 3000 (CH sp2), 1614 (C=O), 1589 and 1494 (aromatic C=C), 1043 (stretching C-Cl). 1H-NMR (MeOD): 8.12 (s, 1H; O=C-NH), 7.53–7.15 (m, 4H, H-2′, H-3′, H-5′, H-6), 6.85 (s, 2H; H-2, H-6), 4.58 (s, 2H, H-7′), 4.46 (s, 2H; m-OH), 4.35 (s, 1H, p-OH). 13C-NMR (MeOD): 170.5 (C=O), 146.7 (C-3, C-5), 139.4 (C-4), 136.1 (C-1), 134.2 (C-4), 130.5 (C-5′, C-6′), 130.1 (C-3′, C-5′), 125.9 (C-1), 107.8 (C-2, C-6), 43.6 (C-7′) [16]. HRMS (MALDI) calculated for C16H12ClNNaO3 [M + Na]+: 316.0353; found 316.0373.
N-(4-Chlorobenzyl)-4-hydroxy-3-methoxybenzamide (15). White amorphous solid; yield: 44% (151 mg), m.p.: 75–77 °C, IR νmax (cm−1): 3319 (O-H) 3251 (N-H), 3000 (C-H sp2), 1639 (C=O), 1589 and 1487 (aromatic C=C), 1091 (stretching C-Cl). 1H-NMR (DMSO-d6): 8.12 (d, J = 9.2 Hz, 1H; O=C-NH), 7.56–7.22 (m, 7H; H-2, H-4, H-6, H-3′, H-5′, H-2′, H-6′), 4.52 (s, 2H), 3.88 (s, 1H; OCH3). 13C-NMR (DMSO-d6): 169.8 (C=O); 151.3 (C-3); 148.8 (C-4); 139.3 (C-1′); 133.8 (C-4′); 130.1 (C-2′, C-6′); 129.5 (C-5′, C-3′); 126.5 (C-1); 122.1 (C-5); 115.8 (C-6); 111.9 (C-2); 56.4 (3-OMe); 43.8 (C-7′) [16]. HRMS (MALDI) calculated for C16H16ClNO4 [M + Na]+: 316.0530; found 316.0543.
N-(4-Chlorobenzyl)-4-hydroxy-3,5-dimethoxybenzamide (16). White amorphous solid; yield: 50% (164 mg), m.p. 110–115 °C, IR νmax (KBr, cm−1): 3493 (OH), 3277 (NH), 3084 (CH sp2), 1666 (C=O), 1597 and 1492 (aromatic C=C), 1016 (stretching C-Cl). 1H-NMR (MeOD): 8.15 (s, 1H, O=C-NH), 7.43 (s, 1H, OH), 7.35–7.27 (m, 4H; H-2′, H-3′, H-5′, H-6′), 7.25–7.15 (m, 2H; H-2, H-6), 4.54 (s, 2H, H-7′), 3.88 (s, 6H; OCH3). 13C-NMR (MeOD): 149.0 (C-3, C-5), 169.8 (C=O), 139.3 (C-4), 133.8 (C-1′), 130.1 (C-2′, C-6′), 129.5 (C-3′, C-5′), 125.3 (C-4′), 106.4 (C-1), 106.0 (C-2, C-6), 43.9 (C-7′), 56.8 (OCH3) [16]. HRMS (MALDI) calculated for C16H16ClNNaO4 [M + Na]+: 346.0636; found 346.0666.
N-(4-Chlorobenzyl)-4-hydroxybenzamide (17). White amorphous solid; yield: 73% (246 mg), m.p. 189–192 °C, IR νmax (cm−1): 3450 (OH), 3122 (NH), 3055 (CH sp2), 1631 (C=O), 1593 and 1440 (aromatic C=C), 1085 (stretching C-Cl). 1H-NMR (DMSO-d6): 8.83 (t, J = 5.9 Hz, 1H; O=C-NH), 7.75 (d, J = 8.6 Hz, 2H, H-2, H-6), 7.59–7.43 (m, 4H, H-2′; H-3′, H-5′, H-6′), 6.80 (d, J = 8.8 Hz, 2H, H-3, H-5), 4.42 (d, J = 6.0 Hz, 2H; H-7′). 13C-NMR (DMSO-d6): 166.0 (C=O); 160.3 (C-4); 139.2 (C-1′); 133.4 (C-4′); 130.9 (C-2, C-6); 129.3 (C-2′, C-6′); 128.3 (C-3′, C-5′); 124.9 (C-1); 114.9 (C-3, C-5); 41.7 (C-7′) [16]. HRMS (MALDI) calculated for C14H12ClNNaO2 [M + Na]+: 286.0425; found 286.0432.
3,5-Di-tert-butyl-N-(4-chlorobenzyl)-4-hydroxybenzamide (18). White amorphous solid; yield: 54% (203 mg), m.p. 184–186 °C, IR νmax (cm−1): 3450 (OH) and 3236 (NH), 3066 (CH sp2), 1680 (C=O), 1544 and 1431 (aromatic C=C), 1012 (stretching C-Cl). 1H-NMR (DMSO-d6): 8.89 (t, J = 6.0 Hz, 1H; O=C-NH), 6.09–5.94 (m, 1H), 7.70–7.54 (m, 2H, H-2, H-6), 7.54–7.20 (m, 4H, H-2′, H-3′, H-5′, H-6′), 4.42 (d, J = 5.9 Hz, 3H; H-7′), 1.39 (s, 18H; C(CH3)3). 13C-NMR (DMSO-d6): 167.0 (C=O); 156.9 (C-4); 140.0 (C-1′); 138.3 (C-3, C-5); 131.2 (C-4′); 129.2 (C-2′, C-6′); 128.3 (C-5′, C-3′); 125.3 (C-1); 124.2 (C-2, C-6); 42.0 (C-7′); 34.7 (3,5-(C(CH3)3; 30.3 (3,5-(C(CH3)3) [16] HRMS (MALDI) calculated for C22H28ClNO3 [M + H]+: 374.1877; found 374.1877.
N-(4-Chlorobenzil)-2-hydroxy-5-methoxybenzamide (19). Crystalline solid; yield: 60% (180 mg), m.p. 137–140 °C, IR νmax (cm−1): 3360 (O-H and N-H), 3076 (C-H sp2), 1651 (C=O), 1598 and 1435 (aromatic C=C), 1045 (stretching C-Cl). 1H-NMR (DMSO-d6): 9.35 (t, J = 5.8 Hz, 1H, O=C-NH), 7.48–7.26 (m, 5H, H-6, H-2′, H-3′, H-5′, H-6′), 7.04 (dd, J = 9.0, 3.0 Hz, 1H; H-3), 6.85 (d, J = 9.0 Hz, 1H; H-4), 4.49 (d, J = 5.9 Hz, 2H, H-7′), 3.72 (s, 3H; OCH3). 13C-NMR (50 MHz): 168.7 (C=O); 154.0 (C-5); 151.7 (C-2); 115.2 (C-1); 138.2 (C-1′); 131.6 (C-4′); 129.3 (C-2′, C-6′); 128.5 (C-5′, C-3′); 121.2 (C-3); 118.4 (C-4); 111.3 (C-6); 55.8 (OCH3); 41.9 (C-7′) [16]. HRMS (MALDI) calculated for C15H14ClNO3 [M + H]+: 327.0512; found 327.0516.
N-(4-Chlorobenzyl)-3-methyl-4-nitrobenzamide (20). Crystalline solid; yield: 41% (143 mg), m.p. 148–151 °C, IR νmax (cm−1): 3278 (NH), 3080 (CH sp2), 1637 (C=O), 1587 and 1423 (aromatic C=C), 1521 and 1355 (NO2 arom), 1089 (stretch C-Cl). 1H-NMR (DMSO-d6): 9.31 (t, J = 5.8 Hz, 1H, O=C-NH), 8.06 (dd, J = 8.4, 1H; H-5), 7.96 (s, 1H, H-2), 7.89 (dd, J = 8.4, 1.7 Hz, 1H; H-6), 7.42–7.31 (m, 4H, H-2′, H-3′, H-5′, H-6′), 4.47 (dd, J = 5.9, 1.7 Hz, 2H, H-7′), 2.56–2.51 (m, 3H; CH3). 13C-NMR (DMSO-d6): 164.8 (C=O); 150.5 (C-4); 138.3 (C-1); 138.1 (C-1′); 132.9 (C-4′); 131.8 (C-2′, C-6′); 131.5 (C-3); 129.3 (C-3′, C-5′); 128.4 (C-2); 126.2 (C-5); 124.6 (C-6); 42.3 (C-7′); 19.5 (CH3) [16]. HRMS (MALDI) calculated for C15H13ClN2NaO3 [M + Na]+: 327.0512; found 327.0516.
N-(4-Fluorobenzyl)-4-hydroxy-3-methoxybenzamide (21). 4-Fluorbenzylamine was used as the reagent. White amorphous solid; yield: 21% (90 mg), m.p. 161–165 °C, IR νmax (cm−1): 3304 (O-H), 3078 (N-H), 1631 (C=O), 1593 and 1423 (aromatic C=C), 1116 (C-F stretch). 1H-NMR (CDCl3): 9.60 (s, 1H; OH), 8.83 (t, J = 5.8 Hz, 1H; NH), 7.52–7.28 (m, 4H; H-2, H-6, H-2′, H-6′), 7.22–7.05 (m, 2H; H-3′, H-5′), 6.81 (d, J = 8.2 Hz, 1H; H-5), 4.43 (d, J = 5.8 Hz, 2H; H-7′), 3.80 (s, 3H; OMe). 13C-NMR (CDCl3): 166.0 (C=O), 161.1 (d, J = 242.0 Hz; C-4′), 149.6 (C-4), 147.2 (C-3), 136.2 (C-1′), 129.2 (C-2′, C-6′), 125.3 (C-1), 120.9 (C-6), 114.9 (C-3′, C-5′), 115.2 (C-5), 111.3 (C-2), 55.7 (OMe), 42.0 (C-7′) [16]. HRMS (MALDI) calculated for C15H14FNNaO3 [M + Na]+: 298.0855; found 298.0882.
N-(4-Bromobenzyl)-4-hydroxy-3-methoxybenzamide (22). 4-Bromobenzylamine was used as reactant. Red amorphous solid; yield: 63% (252 mg), m.p. 119–122 °C. IV νmax (cm−1): 3304 (OH), 3078 (NH), 3003 (CH sp2), 1631 (C=O), 1593 and 1423 (aromatic C=C), 1072 (stretching C-Br). 1H-NMR (DMSO-d6): 7.43 (d, J = 8.4 Hz, 3H; H-2, H-3′, H-5′), 7.29–7.12 (m, 3H; H-6, H-2′, H-6), 6.88 (d, J = 8.2 Hz, 1H; H-5), 6.65 (t, J = 5.2 Hz, 1H; NH), 6.21 (s, 1H; OH), 4.54 (d, J = 5.8 Hz, 2H, H-7′), 3.88 (s, 3H; OMe). 13C-NMR (DMSO-d6): 167.1 (C=O), 148.9 (C-4), 146.7 (C-3), 137.4 (C-1′), 131.7 (C-3′, C-5′), 129.4 (C-2′, C-6′), 126.1 (C-1), 119.8 (C-6, C-4′), 113.9 (C-5), 110.4 (C-2), 56.0 (OMe), 43.4 (C-7′) [16]. HRMS (MALDI) calculated for C15H14BrNO3 [M]+: 335.0157; found 335.0156.
Procedure 2: Preparation of 4-((4-chlorobenzyl) carbamoyl)-2-methoxyphenyl benzoate (23): To a 100 mL flask equipped with magnetic stirring, was added a sodium hydroxide solution 10% (0.51 mL, 0.1275 mmol NaOH) 0.100 g of the amide 15 (0.3400 mmol). Then, benzoyl chloride (0.04 mL, 0.343 mmol) was added in drops. The reaction was subjected to constant agitation for a period of one hour at room temperature. The reaction mixture was then poured into a separation funnel and extraction was completed with dichloromethane (3 × 15 mL), organic phase treated with sodium carbonate (Na2CO3) 5% (2 × 5 mL), and dried with anhydrous sodium sulfate. After filtration, the solution was concentrated under reduced pressure [18]. The product was purified by silica gel column chromatography using a mixture EtOAc:Hex (65:35) as mobile phase to give a crystalline solid compound, yield: 76% (103 mg), m.p. 122–125 °C, IR νmax (cm−1): 3334 (N-H), 3000 (C-H sp2), 1734 and 1637 (C=O), 1607 and 1425 (aromatic C=C), 1089 (stretching C-Cl). 1H-NMR (CDCl3): 9.17 (t, J = 5.9 Hz, 1H, NH), 8.13 (d, J = 7.1 Hz, 2H, H-2″, H-6″), 7.84–7.20 (m, 10H; H-2, H-5, H-6, H-2, H-3′, H-5′, H-6′, H-3″, H-4″, H-5″), 4.50 (d, J = 5.8 Hz, 2H; H-7′). 3.81 (s, J = 6.0 Hz, 3H; OMe). 13C-NMR (CDCl3): 165.9 (O=C-O), 164.2 (O=C-N), 151.2 (C-3), 142.1 (C-4), 139.1 (C-1′), 134.6 (C-2′), 133.6 (C-1), 134.6 (C-6′), 131.1 (C-4′), 130.3 (C-2″, C-4″, C-6″), 129.6 (C-3″, C-5″), 128.8 (C-1″), 128.7 (C-3′, C-5′), 123.4 (C-5), 120.4 (C-6), 112.2 (C-2), 42.5 (C-7′), 56.4 (OMe) [19]. HRMS (MALDI) calculated for C22H18ClNNaO4 [M + Na]+: 418.0822; found 418.0809. The following compounds were similarly prepared:
4-((4-Chlorobenzyl)carbamoyl)-2-methoxyphenyl 2-phenylacetate (24). Phenylacetyl chloride (0.05 mL, 0.343 mmol) was used. The reaction was subjected to constant agitation for a period of two hours at room temperature and finally extracted. Crystalline solid; yield: 78% (110 mg), m.p.: 141–145 °C, IR νmax (cm−1): 3331 (N-H), 3050 (C-H sp2), 1761 and 1633 (C=O), 1602 and 1456 (aromatic C=C), 1033 (stretching C-Cl). 1H-NMR (CDCl3): 9.12 (t, J = 5.9 Hz, 1H; NH), 7.61 (d, J= 1.7 Hz, 1H; H-2), 7.52 (dd, J = 8.2, 1.8 Hz, 1H; H-6), 7.43–7.26 (m, 10H; H-2′, H-3′, H-5′, H-6′, H-2″, H-3″, H-4″, H-5″, H-6″), 7.20 (d, J = 8.2 Hz, 1H; H-5), 4.47 (d, J = 5.9 Hz, 2H; H-7′), 3.98 (s, 2H, CH2-C6H5), 3.80 (s, 3H; OMe). 13C-NMR (CDCl3): 169.6 (O=C-O), 165.7 (O=C-N), 150.9 (C-3), 142.0 (C-4), 138.9 (C-1′), 134.1 (C-1″), 133.3 (C-4′), 131.6 (C-1), 129.8 (C-2′, C-6′), 128.7 (C-2″, C-6″), 128.6 (C-3″, C-5″), 127.3 (C-4″), 127.3 (C-3′, C-5′), 122.9 (C-5), 120.2 (C-6), 112.0 (C-2), 56.3 (OCH3), 42.4 (C-7′), 40.09 (CH2-C6H5) [20]. HRMS (MALDI) calculated for C23H20ClNNaO4 [M + Na]+: 432.0979; found 432.0914.
4-((4-Chlorobenzyl)carbamoyl)-2-methoxyphenyl valerate (25). Valeryl chloride (0.05 mL, 0.343 mmol) was used. The reaction was subjected to constant agitation for a period of five hours at room temperature and finally extracted. White amorphous solid; yield: 43% (55 mg), m.p. 97–100 °C, IR νmax (cm−1): 3253 (N-H), 3088 (C-H sp2), 1761 and 1631 (C=O), 1602 and 1450 (aromatic C=C), 1031 (stretching C-Cl). 1H-NMR (CDCl3): 7.44 (s, 1H; H-2); 7.31–7.15 (m, 5H; H-6, H-2′, H-3′, H-5′, H-6′), 6.96 (d, J = 8.2 Hz, 1H; H-5), 6.79 (t, J = 5.5 Hz, 1H; NH), 4.49 (d, J = 5.8 Hz, 2H; H-7′), 3.78 (s, 3H; OCH3), 2.56 (t, J = 7.4 Hz, 2H; H-1″), 1.71 (quint, J = 7.7 Hz, 2H; H-2″), 1.42 (sex, J = 7.3 Hz, 2H; H-3″), 0.94 (t, J = 7.2 Hz, 3H; Me). 13C-NMR (CDCl3): 171.7 (O=C-O), 166.6 (O=C-N), 151.3 (C-3), 142.4 (C-4), 136.7 (C-1′), 133.2 (C-1), 132.8 (C-4′), 129.1 (C-2′, C-6′), 128.7 (C-3′, C-5′), 122.6 (C-5), 118.7 (C-6), 111.8 (C-2), 55.9 (OMe), 43.3 (C-7′), 33.7 (C-1″), 26.9 (C-2″), 22.1 (C-3″), 13.7 (Me) [20]. HRMS (MALDI) calculated for C20H22ClNNaO4 [M + Na]+: 398.1135; found 398.1118.
4-((4-Chlorobenzyl) carbamoyl)-2-methoxyphenyl 3-bromobenzoate (26). 3-Bromobenzoyl chloride (0.05 mL, 0.343 mmol) was employed. The reaction was subjected to constant agitation for a period of one hour at room temperature and finally extracted. White amorphous solid; yield: 86% (140 mg), m.p. 143–145 °C. IV νmax (cm−1): 3334 (N-H), 3050 (C-H sp2), 1734 and 1637 (C=O), 1602 and 1490 (aromatic C=C), 1089 (stretching C-Cl). 1H-NMR (CDCl3): 8.30 (t, J = 1.7 Hz, 1H; H-2″), 8.14–8.03 (m, 1H; H-4″), 7.81–7.71 (m, 1H; H-6″), 7.51 (d, J = 1.8 Hz, 1H; H-2), 7.44–7.15 (m, 6H; H-6, H-2′, H-3′, H-5′, H-60′, H-5″), 7.09 (d, J = 8.2 Hz, 1H; H-5), 6.81 (t, J = 5.7 Hz, 1H; NH), 4.53 (d, J = 5.8 Hz, 2H; H-7′), 3.80 (s, 3H; OMe). 13C-NMR (CDCl3): 166.6 (O=C-O), 163.2 (O=C-N), 151.4 (C-3), 142.2 (C-4), 136.7 (C-4″), 136.6 (C-1′), 133.3 (C-1), 133.2 (C-1″), 133.2 (C-2″), 130.7 (C-4′), 130.1 (C-5″), 129.1 (C-2′, C-6′), 128.8 (C-3′, C-5′, C-6′′), 122.7 (C-3″), 122.6 (C-5), 118.7 (C-6), 112.0 (C-2), 43.4 (C-7′), 56.0 (OCH3) [21]. HRMS (MALDI) calculated for C20H22ClNO4 [M]+: 495.9927; found 495.9912.
Procedure 3: Preparation of 4-((4-chlorobenzyl) carbamoyl)-2-methoxyphenyl acetate (27). For the acetylation of 15, to a 50 mL flask, equipped with magnetic stirrer was added the chlorinated vanillic amide 15 (0.1000 g, 0.034 mmol), pyridine (0.13 mL, 0.15924 mmol) and acetic anhydride (0.08 mL, 0.8111 mmol). The reaction mixture was subjected to constant magnetic stirring for 24 h. The first step of the reaction product extraction was then carried out by pouring into ice water (30 mL) in a separation funnel using ethyl acetate as extractor solvent (3 × 10 mL). The organic phase was treated with saturated copper sulfate solution (3 × 20 mL). The ethyl acetate phase was washed with water (3 × 30 mL) and dried with anhydrous sodium sulfate (Na2SO4). Subsequently, the organic phase was filtered and concentrated by rotary evaporation [22]. The product was purified by silica gel column chromatography using EtOAc:Hex (65:35) as the mobile phase system to give a crystalline solid; yield: 98% (159 mg), m.p. 117–119 °C, IR νmax (cm−1): 3446 (N-H), 3088 (C-H sp2), 1770 and 1635 (C=O), 1602 and 1421 (aromatic C=C), 1089 (stretching C-Cl). 1H-NMR (CDCl3): 7.43 (d, J = 1.9 Hz, 1H; H-2), 7.33–7.12 (m, 4H; H-2, H-3′, H-5′, H-6′), 7.03–6.86 (m, 1H; H-6), 6.88–6.69 (m, 1H; H-5) ,4.47 (d, J = 5.8 Hz, 2H, H-7′), 3.77 (s, 3H; Me), 2.27 (s, 3H; Me). 13C-NMR (CDCl3): 168.8 (O=C-O), 166.6 (O=C-N), 151.3 (C-3), 142.3 (C-4), 136.6 (C-1′), 133.2 (C-1), 132.9 (C-4′), 129.0 (C-2′, C-6′), 128.7 (C-3′, C-5′), 122.6 (C-5), 118.7 (C-6), 111.8 (C-2), 55.9 (OMe), 43.3 (C-7′), 20.6 (Me) [19]. HRMS (MALDI) calculated for C17H16ClNNaO4 [M + Na]+: 356.0666; found 356.0664.
Procedure 4: Preparation of N-(4-chlorobenzyl)-3-methoxy-4-(4-methylphenetoxy)benzamide (28). In a 100 mL flask equipped with magnetic stirring vanillic amide 15 (0.1000 g, 0.3400 mmol) was added to a solution of acetone (4 mL) together with K2CO3 (0.1388 g, 1.0046 mmol) and 4-methylbenzyl bromide (0.08 mL, 0.6011 mmol) under reflux (60 °C) for 16 h. After the reaction, the solvent was removed under reduced pressure. A solution of CH2Cl2:H2O was poured onto the product, placed in a separation funnel and extracted with three portions of CH2Cl2 (10 mL each). The organic phase was washed 1 N NaOH (3 × 10 mL) and dried with Na2SO4, filtered and concentrated by rotary evaporation. The product was purified by silica gel column chromatography using EtOAc:Hex (65:35) as the mobile phase system to give a white amorphous solid; yield: 30% (42 mg), m.p. 126–129 °C, IR νmax (cm−1): 3290 (N-H), 3001 (C-H sp2), 1629 (C=O), 1600 and 1490 (aromatic C=C), 1029 (stretching C-Cl). 1H-NMR (CDCl3): 7.44 (s, 1H; H-2), 7.32–7.07 (m, 9H; H-6, H-2′, H-3′, H-5′, H-6′, H-2″, H-3″, H-5″, H-6″), 6.80 (d, J = 8.4 Hz, 1H; H-5), 6.49 (t, J = 5.2 Hz, 1H, NH), 4.56 (d, J = 5.8 Hz, 2H; H-7′), 4.18 (t, J = 7.6 Hz, 2H; CH2-O), 3.88 (s, 3H; OCH3), 3.11 (t, J = 7.6 Hz, 2H; CH2-C6H5). 13C-NMR (CDCl3): 167.0 (O=C-N), 151.2 (C-3), 149.3 (C-4), 136.9 (C-1′), 136.2 (C-4″), 134.3 (C-1″), 133.3 (C-4′), 129.2 (C-2′, C-6′), 129.1 (C-3″, C-5″), 128.9 (C-3′, C-5′), 128.8 (C-2″, C-6″), 126.7 (C-1), 119.3 (C-6), 111.6 (C-2), 111.1 (C-5), 69.9 (CH2-O), 56.1 (OMe), 43.3 (C-7′), 35.1 (CH2-C6H5), 21.0 (Me) [23]. HRMS (MALDI) calculated for C24H24ClNNaO3 [M + Na]+: 432.1342; found 432.1328.
Procedure 5: Preparation of N-(4-chlorobenzyl)-4-(4-hydroxyphenoxy)-3-methoxybenzamide (29). In a 100 mL flask equipped with magnetic stirring, vanillic amide 15 (0.1000 g, 0.3400 mmol) was added to a solution of DMF (3.43 mL), K2CO3 (0.0711 g, 0.5142 mmol), and 4-hydroxyphenyl bromide (0.0827 g, 0.4113 mmol), and left stirring for 24 h at room temperature. After the reaction, the product was extracted in a separation funnel with CH2Cl2 (3 × 10 mL). The extraction solution was washed with distilled water (3 × 10 mL), and then with 10% NaOH (10 mL). The solution was treated with Na2SO4, filtered, and concentrated by rotary evaporation [24]. The product was purified by silica gel column chromatography using EtOAc:Hex (65:35) as the mobile phase system to afford a colorless oil; yield: 75% (106 mg), IR νmax (cm−1): 3350 (OH) 3016 (CH sp2), 1645 (C=O), 1600 and 1489 (aromatic C=C), 1014 (stretching C-Cl). 1H-NMR (CDCl3): 7.95 (s, 1H; NH), 7.44 (s, 1H; H-2), 7.30–6.94 (m, 8H; H-5, H-6, H-2′, H-3′, H-5′, H-6′, H-2″, H-6″), 6.79 (dd, J = 8.5, 2.6 Hz, 2H; H-3″, H-5″), 4.54 (d, J = 5.8 Hz, 2H; H-7′), 4.14 (t, J = 7.6 Hz, 2H; CH2-O), 3.82 (s, 3H; OMe), 3.04 (t, J = 7.5 Hz, 2H; CH2-C6H5). 13C-NMR (CDCl3): 167.5 (O=C-N), 155.0 (C-4″), 151.3 (C-4), 149.2 (C-3), 136.7 (C-1′), 133.3 (C-4′), 130.0 (C-2′, C-6′), 129.1 (C-2″, C-6″), 128.8 (C-3′, C-5′), 128.9 (C-1″), 126.4 (C-1), 119.5 (C-6), 115.6 (C-3″, C-5″), 111.6 (C-2), 111.2 (C-5), 70.1 (CH2-O), 56.1 (OCH3), 43.4 (C-7′), 34.8 (CH2-C6H5) [23]. HRMS (MALDI) calculated for C23H22ClNNaO4 [M + Na]+: 434.1135; found 434.1123.
Preparation of N-(4-chlorobenzyl)-3-methoxy-4-propoxybenzamide (30). This product was prepared according to procedure 4, using 1-propyl bromide (0.05 mL, 0.4102 mmol) under reflux (60 °C), for 16 h and finally extracted to give after the silica gel column chromatography a white amorphous solid; yield: 54% (62 mg), m.p. 124–126 °C. νmax IR (cm−1): 3296 (N-H), 1631 (C=O), 1581 to 1450 (aromatic C=C), 1014 (C-Cl stretch). 1H-NMR (CDCl3): 7.40 (d, J = 1.8 Hz, 1H; H-2), 7.31–7.22 (m, 5H; H-6, H-2′, H-3′, H-5′, H-6′), 6.81 (d, J = 8.4 Hz, 1H; H-5), 6.74 (bs, 1H; NH), 4.55 (d, J = 5.8 Hz, 2H; H-7′), 3.98 (t, J = 6.8 Hz, 2H; H-1″), 3.86 (s, 3H; OMe), 2.04–1.76 (m, 2H; H-2″), 1.03 (t, J = 7.4 Hz, 3H; H-3″). 13C-NMR (CDCl3): 167.0 (O=C-N), 151.4 (C-4), 149.2 (C-3), 137.0 (C-1′), 133.1 (C-4′), 129.1 (C-2′, C-6′), 128.7 (C-3′, C-5′), 126.3 (C-1), 119.4 (C-6), 110.9 (C-2), 111.4 (C-5), 70.4 (C-1″), 56.0 (OCH3), 43.3 (C-7′), 22.3 (C-2″), 10.3 (C-3″) [25]. HRMS (MALDI) calculated for C18H20ClNNaO3 [M + Na]+: 356.1029; found 356.1038.
Preparation of N-(4-chlorobenzyl)-4-isopropoxy-3-methoxybenzamide (31). This product was prepared according to procedure 5, using 2-propyl bromide (0.05 mL, 0.4102 mmol), at room temperature and left stirring for 24 h and finally extracted to give, after silica gel column chromatography purification using EtOAc:Hex as the gradient mobile phase system, increasing in polarity and an isocratic EtOAc:Hex (65:35) system after elution of the purified product a white amorphous solid; yield: 54% (62 mg), m.p. 142–147 °C. IV νmax (cm−1): 3284 (N-H), 3078 (C-H sp2), 1631 (C=O), 1598 and 1455 (aromatic C=C), 1031 (stretching C-Cl). 1H-NMR (CDCl3): 7.40 (d, J = 1.8 Hz, 1H; H-6), 7.26–7.18 (m, 5H; H-2, H-2′, H-3′, H-5′, H-6′), 6.79 (d, J = 8.4 Hz, 2H; H-5), 6.79 (bs, J = 8.4 Hz, 2H; NH), 4.51 (sept, J = 6.2 Hz, 1H; H-2″), 4.51 (d, J = 5.7 Hz, 2H; H-7′), 3.81 (s, 3H; OMe), 1.33 (d, J = 6.0 Hz, 6H; H-1″; H-3″). 13C-NMR (CDCl3): 167.1 (O=C-N), 150.3 (C-4), 150.0 (C-3), 137.0 (C-1′), 133.2 (C-4′), 129.1 (C-2′, C-6′), 128.7 (C-3′, C-5′), 126.5 (C-1), 119.4 (C-6), 113.6 (C-5), 111.2 (C-2), 71.2 (C-1″), 56.0 (OCH3), 43.3 (C-7′), 21.9 (Me) [25]. HRMS (MALDI) calculated for C18H20ClNNaO3 [M + Na]+: 356.1059; found 356.1027.
Preparation of N-(4-chlorobenzyl)-4-(decyloxy)-3-methoxybenzamide (32). The product was prepared according to procedure 4, using 1-decyl bromide (0.05 mL, 0.4102 mmol) under reflux (60 °C) for 16 h and finally extracted to give after silica gel column chromatography a white amorphous solid; yield: 81% (120 mg), m.p. 110–105 °C, IR νmax (cm−1): 3305 (N-H), 3000 (C-H sp2), 1627 (C=O), 1602 and 1508 (aromatic C=C), 1035 (stretching C-Cl). 1H-NMR (CDCl3): 7.42 (s, 1H; H-6), 7.26 (s, 5H; H-2, H-2′, H-3′, H-5′, H-6′), 6.54 (s, 1H; H-5), 4.56 (d, J = 5.8 Hz, 2H; H-7′), 4.01 (t, J = 6.8 Hz, 2H; CH2-C6H5), 3.86 (s, 3H; OMe), 1.83 (dt, J = 14.1, 6.9 Hz, 2H; CH2-O), 1.74–1.15 (m, 14H; (CH2)7), 0.86 (t, J = 6.1 Hz, 3H; CH3). 13C-NMR (CDCl3): 167.2 (O=C-N), 151.7 (C-4), 149.4 (C-3), 137.1 (C-1′), 133.4 (C-4′), 129.3 (C-2′, C-6′), 128.9 (C-3′, C-5′), 126.5 (C-1), 119.5 (C-6), 111.6 (C-2), 111.1 (C-5), 69.2 (CH2O), 56.2 (OMe), 43.5 (C-7′), 32.0 (CH2CH2O), 29.7–22.8 ((CH2)7CH2O), 14.3 (CH3). [26]. HRMS (MALDI) calculated for C18H20ClNaNO3 [M + Na]+: 454.2125; found 454.2119.
3.3. Antifungal Activity
3.3.1. Microbiological Strains
The microorganisms used in microbiological tests were strains of Candida albicans (ATCC 76645, LM-106 and LM-23), C. krusei (LM-13 and LM-656) and C. tropicalis (ATCC-13803 and ML-36). The strains were respectively acquired from the Adolfo Lutz Institute in São Paulo (Brazil), and from the Federal University Pharmaceutical Science Mycology Laboratories of São Paulo and Paraiba. The yeast strains were maintained in appropriate medium of Sabouraud Dextrose Broth-SDB prepared and used according to manufacturer’s instructions (Difco Laboratories, MA, USA), and stored at 4 °C and 35 °C. The microorganism suspension was prepared according to McFarland tube 0.5, and adjusted by means of a spectrophotometer (Leitz-Photometer 340-800, Ernst Leitz, Wetzlar, Germany) at 90% T (530 nm) corresponding to approximately 106 CFU mL−1 [29,30,33].
3.3.2. Determination of the Minimum Inhibitory Concentration (MIC)
The MIC value was determined by microdilution method using 96 well “U” shaped micro-titer plates in duplicate. In each well of the plate 100 µL of twice concentrated SDB liquid medium was added. Then, 100 mL of product solution (also doubly concentrated) was placed in the first row of plate wells. Through serial dilution (ratio of two), the concentrations of 1024 µg/mL to 64 µg/mL were obtained, such that in the first line of the plate was the highest concentration and in the latter, the lower concentrations. Finally, 10 µL of inoculum was added to the wells in each plate column that specifically referred to a strain. The same was also done in the culture medium with the fungal drug nystatin (100 IU). The plates were incubated at 37 °C for 24–48 h. For each strain, the MIC was defined as the lowest concentration capable of inhibiting fungal growth in the wells as visually observed compared with the control. All tests were performed in duplicate and the results were expressed as a geometric mean of the MIC values obtained in both tests [34].
3.4. Qualitative-Structure Activity Relationship of Antimicrobial Activity in Amides
3.4.1. Volsurf Descriptors
The molecular structures in three dimensions (3D) were used as input data to the Volsurf + v program. 1.0.7, and subjected to molecular interaction fields (MIC) to generate descriptors using the following probes: N1 (amide hydrogen-nitrogen bond donor probe), O (hydrogen-oxygen carbonyl bond acceptor probe), OH2 (probe water), and DRY (hydrophobic probe). Descriptors not using probes were generated to create a total of 128 descriptors [35].
3.4.2. Models for the Study of Qualitative-Structure Activity Relationship
KNIME 3.1.0 software (3.1.0 KNIME from Konstanz Information Miner, copyright 2003–2014, www.knime.org) [31] was used to perform all of the analyses. For internal validation, cross validation was employed using the “leave-one-out” method. Descriptors were selected, and a model was generated using the training and cross-validation set, using the WEKA nodes [35]. The internal performances of the selected models were analyzed for sensitivity (true positive rate), specificity (true negative rate), and accuracy (overall predictability).
4. Conclusions
A number of N-(4-halobenzyl)amides 1–32 were prepared and their antifungal potential was evaluated in screening experiments carried out with seven fungal strains. Compounds 2, 3, 5, 6, 10, 14, and 15 (MICs = 256 and 512 μg·mL−1) showed considerable antifungal activity against all tested strains of the genus Candida. According to the SAR, it was observed that disubstituted amides, such as meta- and para-hydroxylated or meta-methoxylated/para-hydroxylated or 3,4,5-trihydroxylated N-(4-halobenzyl)amides have better antifungal activity against Candida strains. In addition, the presence of the nitro group in an ortho position in the benzene ring contributes to the antifungal activity. This study with haloamides could help in the development of future therapeutic approaches to the growing problem of microbial pathogens via the discovery of novel antifungal agents.
Acknowledgments
This work was supported by the Brazilian agencies: Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). The authors also thank the Institute CETENE, Pernambuco, for analysis of samples in high resolution mass spectra.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/21/12/1716/s1.
Author Contributions
Ricardo C. Montes and Marianna Oliveira de Araújo prepared the amides. Ana Luiza A.L. Perez and Cássio Ilan S. Medeiros performed the antimicrobial experiments. Edeltrudes de Oliveira Lima was responsible for the analysis of these data. Marcus Tullius Scotti developed the in silico study of the antifungal activity of amides and Damião Pergentino de Sousa planned and coordinated the study.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds are available from the authors.
References
- 1.Barbedo L.S., Sgarbi D.S.G. Candidiase. DST J. Bras. Doenças Sex. Trasm. 2010;22:22–38. [Google Scholar]
- 2.Velikova M., Bankova V., Sorkun K., Houcine S., Tsvetkova I., Kujumgiev A. Propolis from the Mediterranean Region: Chemical Composition and Antimicrobial Activity. Z. Naturforsch. C J. Biosci. 2000;55:790–793. doi: 10.1515/znc-2000-9-1019. [DOI] [PubMed] [Google Scholar]
- 3.De P., Baltas M., Bedos-Belval F. Cinnamic Acid Derivatives as Anticancer Agents—A Review. Curr. Med. Chem. 2011;18:1672–1703. doi: 10.2174/092986711795471347. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt A., Scheel D., Strack D. Elicitor-stimulated biosynthesis of hydroxycinnamoyltyramines in cell suspension cultures of Solanum tuberosum. Planta. 1998;205:51–55. doi: 10.1007/s004250050295. [DOI] [Google Scholar]
- 5.Guzman J.D., Mortazavi P.N., Munshi T., Evangelopoulos D., Mchugh T.D., Gibbons S., Malkinson J., Bhakta S. 2-Hydroxy-substituted cinnamic acids and acetanilides are selective growth inhibitors of Mycobacterium tuberculosis. MedChemComm. 2014;5:47–50. doi: 10.1039/C3MD00251A. [DOI] [Google Scholar]
- 6.Krátký M., Vinsová J., Buchta V. In Vitro Antibacterial and Antifungal Activity of Salicylanilide Benzoates. Sci. World J. 2012;2012:290628. doi: 10.1100/2012/290628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merkl R., Hradkova I., Filip V., Smidrkal J. Antimicrobial and antioxidant properties of phenolic acids alkyl esters. Czech J. Food Sci. 2010;28:275–279. [Google Scholar]
- 8.Zhang M., Lu X., Zhang H., Li N., Xiao Y., Zhu H., Ye Y. Synthesis, structure, and biological assay of cinnamic amides as potential EGFR kinase inhibitors. Med. Chem. Res. 2013;22:986–994. doi: 10.1007/s00044-012-0093-z. [DOI] [Google Scholar]
- 9.Seitz T., Stenzel K. Iminoacetic Acid Amides and Their Use as Pest Control Agents. WO1997014673 A1. Germany Patent. 1997 Apr 24;
- 10.Lo William C., Hunter J.E., Watson G.B., Patny A., Iyer P.S., Boruwa J. Pesticidal Compositions and Processes Related thereto. EP2934117 A4. Patent. 2016 Jul 31;
- 11.Fu J., Cheng K., Zhang Z., Fang R., Zhu H. Synthesis, structure and structure–activity relationship analysis of caffeic acid amides as potential antimicrobials. Eur. J. Med. Chem. 2010;45:2638–2643. doi: 10.1016/j.ejmech.2010.01.066. [DOI] [PubMed] [Google Scholar]
- 12.Waisser K., Pesina M., Holý P., Pour M., Bures O., Kunes J., Klimesová V., Buchta V., Kubanová P., Kaustová J. Antimycobacterial and antifungal isosters of salicylamides. Arch. Pharm. 2003;336:322–335. doi: 10.1002/ardp.200300725. [DOI] [PubMed] [Google Scholar]
- 13.Alves M.J., Ferreira I.C.F.R., Froufe H.J.C., Abreu R.M.V., Martins A., Pintado M. Antimicrobial activity of phenolic compounds identified in wild mushrooms, SAR analysis and docking studies. J. Appl. Microbiol. 2013;115:346–357. doi: 10.1111/jam.12196. [DOI] [PubMed] [Google Scholar]
- 14.Montalbetti C.A.G.N., Falque V. Amide bond formation and peptide coupling. Tetrahedron. 2005;61:10827–10852. doi: 10.1016/j.tet.2005.08.031. [DOI] [Google Scholar]
- 15.Joullié M.M., Lassen K.M. Evolution of amide bond formation. ARKIVOC. 2010;8:189–250. [Google Scholar]
- 16.Montes R.C., de Freitas T.S., Costa M.S., Oliveira F.S., Campina F.F., Ferreira A.R., Ferreira S.O., Melo H.D., Dias C.S., de Sousa D.P. Antimicrobial evaluation of cinnamic and benzoic haloamides. J. Chem. Pharm. Res. 2016;8:311–320. [Google Scholar]
- 17.Malz F., Jancke H. Validation of quantitative NMR. J. Pharm. Biomed. Anal. 2005;38:813–823. doi: 10.1016/j.jpba.2005.01.043. [DOI] [PubMed] [Google Scholar]
- 18.Sadeghian H., Seyedi S.M., Saberi M.R., Arghiania Z., Riazi M. Design and synthesis of eugenol derivatives, as potent 15-lipoxygenase inhibitors. Bioorg. Med. Chem. 2008;16:890–901. doi: 10.1016/j.bmc.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 19.De Morais S.M., Vila-Nova N.S., Bevilaqua C.M.L., Rondon F.C., Lobo C.H., Moura A.A.N., Sales A.D., Rodrigues A.P.R., de Figuereido J.R., Campello C.C. Thymol and eugenol derivatives as potential antileishmanial agents. Bioorg. Med. Chem. 2014;22:6250–6255. doi: 10.1016/j.bmc.2014.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’avila F.M., Oliveira P.S., Dutra F.S.P., Fernandes T.J., de Pereira C.M.P., de Oliveira S.Q., Stefanello F.M., Lencina C.L., Barschak A.G. Eugenol derivatives as potential anti-oxidants: Is phenolic hydroxyl necessary to obtain an effect? J. Pharm. Pharmacol. 2014;66:733–746. doi: 10.1111/jphp.12197. [DOI] [PubMed] [Google Scholar]
- 21.Georgsson J., Hallberg A., Larhed M. Rapid Palladium-Catalyzed Synthesis of Esters from Aryl Halides Utilizing Mo(CO)6 as a Solid Carbon Monoxide Source. J. Comb. Chem. 2003;5:350–352. doi: 10.1021/cc0201086. [DOI] [PubMed] [Google Scholar]
- 22.Andrade L.N., Batista J.S., de Sousa D.P. Spasmolitic activity of p-menthane esters. J. Med. Plants Res. 2011;5:6995–6999. [Google Scholar]
- 23.Leigh W.J., Lathioor E.C., St. Pierre M.J. Photoinduced Hydrogen Abstraction from Phenols by Aromatic Ketones. A New Mechanism for Hydrogen Abstraction by Carbonyl n, π and π, π Triplets. J. Am. Chem. Soc. 1996;118:12339–12348. doi: 10.1021/ja961973v. [DOI] [Google Scholar]
- 24.Xiang Y., He B., Lia X., Zhu Q. The design and synthesis of novel “turn-on” fluorescent probes to visualize monoamine oxidase-B in living cells. RSC Adv. 2013;3:4876–4879. doi: 10.1039/c3ra22789h. [DOI] [Google Scholar]
- 25.Ploypradith P., Cheryklin P., Niyomtham N., Bertoni D.R., Ruchirawat S. Solid-Supported Acids as Mild and Versatile Reagents for the Deprotection of Aromatic Ethers. Org. Lett. 2007;9:2637–2640. doi: 10.1021/ol070781+. [DOI] [PubMed] [Google Scholar]
- 26.Sunitha M.S., Vishnumurthy K.A., Adhikari A.V. Synthesis and two-photon absorption property of new π-conjugated donor-acceptor polymers carrying different heteroaromatics. J. Chem. Sci. 2013;125:29–40. doi: 10.1007/s12039-013-0366-1. [DOI] [Google Scholar]
- 27.Sartoratto A., Machado A.L.M., Delarmelina C., Figueira G.M., Duarte M.C.T., Rehder V.L.G. Composition and antimicrobial activity of essential oils from aromatic plants used in Brazil. Braz. J. Microbiol. 2004;35:275–280. doi: 10.1590/S1517-83822004000300001. [DOI] [Google Scholar]
- 28.Houghton P.J., Howes M.J., Lee C.C., Steventon G. Uses and abuses of in vitro tests in ethnopharmacology: Visualizing an elephant. J. Ethnopharmacol. 2007;110:391–400. doi: 10.1016/j.jep.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 29.Nattional Committee for Clinical Laboratory Standards (NCCLS) Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically. 6th ed. Clinical And Laboratory Standards Institute; Wayne, PA, USA: 2003. p. 19087. Approved Standard. [Google Scholar]
- 30.Hadacek F., Greger H. Testing of antifungal natural products: Methodologies, comparatibility of results and assay choice. Phytochem. Anal. 2000;11:137–147. doi: 10.1002/(SICI)1099-1565(200005/06)11:3<137::AID-PCA514>3.0.CO;2-I. [DOI] [Google Scholar]
- 31.Berthold M.R., Cebron N., Dill F., Gabriel T.R., Kötter T., Meinl T., Ohl P., Sieb C., Thiel K., Wiswedel B. Data Analysis, Machine Learning and Applications. Springer; Berlin, Germany: 2007. pp. 319–326. [Google Scholar]
- 32.Cruciani G., Crivori P., Carrupt P.A., Testa B. Molecular fields in quantitative structure-permeation relationships: The VolSurf approach. J. Mol. Struct. THEOCHEM. 2000;503:17–30. doi: 10.1016/S0166-1280(99)00360-7. [DOI] [Google Scholar]
- 33.Eloff J.N. A sensitive and quick microplate method to determine the minimal inhibitory concentration of plant extracts for bactéria. Planta Med. 1978;64:711–713. doi: 10.1055/s-2006-957563. [DOI] [PubMed] [Google Scholar]
- 34.Souza E.L., Stamford T.L.M., Lima E.O., Trajano V.N. Effectiveness of Origanum vulgare L. essential oil to inhibit the growth of food spoiling yeasts. Food Control. 2007;18:409–413. doi: 10.1016/j.foodcont.2005.11.008. [DOI] [Google Scholar]
- 35.Hall M., Frank E., Holmes G., Pfahringer B., Reutemann P., Witten I.H. The WEKA Data Mining Software: An Update. SIGKDD Explor. 2009;11:10–18. doi: 10.1145/1656274.1656278. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.