

Abstract
Herein we present, an exhaustive docking analysis considering the case of autotaxin (ATX). HA155, a small molecule inhibitor of ATX, is co-crystallized. In order to further extract conclusions on the nature of the bond formed between the ligands and the amino acid residues of the active site, density functional theory (DFT) calculations were undertaken. However, docking does not provide reproducible results when screening boronic acid derivatives and their binding orientations to protein drug targets. Based on natural bond orbital (NBO) calculations, the formed bond between Ser/Thr residues is characterized more accurately as a polar covalent bond instead of a simple nonpolar covalent one. The presented results are acceptable and could be used in screening as an active negative filter for boron compounds. The hydroxyl groups of amino acids are bonded with the inhibitor’s boron atom, converting its hybridization to sp3.
Keywords: virtual screening, autotaxin, boronic acid, organoboron, drug design, warhead
1. Introduction
Boronic acid is considered as a bioisostere for carboxylic acids bearing lower acidity [1]. It emerged as a pharmacophore group after the clinical approval of the drug bortezomib [2] in 2003 as an anticancer agent. Boron is located before carbon in the periodic table of elements. Both elements present similarities in behavior when participating in organic molecules [3]. In nature, boron-containing compounds can be found in plants, algae, microorganisms [4], and in certain natural products derived from bacteria. [5] Boron contains an empty p orbital in its sp2 hybridization state, presenting a trigonal planar geometry. Upon covalent bond formation, sp3 hybridization takes place, leading to a tetrahedral geometry [6].
Several recent reviews pointed to the significance of boron-containing compounds and to their medicinal interest. Boron derivatives were found to present various biological activities: Neuroprotection [7], antifungal [8], anti-inflammatory [9,10,11,12,13,14], anti-oxidant [15], anti-trypanosomial [16], antidiabetic [17], anticancer [2,9,10,11,12,18], antibacterial [19,20] and/or antiviral [21]. Recently, a second boron compound, tavaborole [8], gained approval as a topical antifungal agent. Thus, boronic acid derivatives continuously attract researchers’ attention for medicinal applicability as novel “lead compounds”. However, their biological mechanism of action and their mutagenic activity in mammals are still under study and are not well defined. Thus, several researchers have tested various boronic acid derivatives as mutagenic risk factors using the Ames assay as a reference protocol of mutagenicity. Bortezomib and the tested compounds were registered as Ames-negative [22].
Based on our previous 2D-QSAR and 3D in silico studies on boronic acid derivatives [23,24], we present herein a docking analysis in order to define the typical steps and variables that are important to incorporate in a library’s virtual screening when organoboron derivatives are included. The enzyme autotaxin (ATX) was used as a biological target in our investigation. The enzyme is located extracellularly and participates in lipid metabolism, converting lysophosphatidylcholine (LPC) to lysophosphatidic acid (LPA). Its active site is located within its catalytic phosphodiesterase (PDE) domain with known recorded XRD data (PDB entry 2XRG) in the presence of a small molecule inhibitor HA155 [25], presenting a threonine-mediated covalent bond between the enzyme and the inhibitor. Moreover, we performed DFT calculations in order to explore the nature of the covalent bond formed between the inhibitor HA155 and two residues similar like Ser/Thr. Additionally, boron compounds are known to form covalent bonds with His [26] and/or Asp [18] residues. The use of covalent “warhead” groups in Medicinal Chemistry are an emerging strategy to achieve irreversible protein inhibition [27,28]. Hence, designed molecules bearing a covalent warhead can be used in targeting shallow pockets or clefts at the surface of proteins to selectively disrupt protein–protein interactions (PPIs) [29] in vivo and in other contexts.
2. Results and Discussion
The usage of typical constraints in the screening of boronic acid derivative libraries could lead to a false negative presentation with regard to binding poses and scoring function values, failing to replicate possible covalent bonds among boron and the amino acids Ser/Thr existing in the active site. Thus, alternate steps must be taken into account when we are dealing with hit compounds bearing boron in their structures producing plausible bindng poses. In particular, using standard hydrogen bond (HB) or contact constraints for Thr209 provided HA155 solutions with 100% off poses based only on the inhibitor’s shape score with minimum HB network scores. Alternatively, the introduction of custom constraints incorporating a smart pattern-like option is needed in order to better simulate the original HA155 query in our training sets (see Scheme 1). Additionally, smart patterns I and II reproduce a number of binding modes with other neighboring residues bearing an oxygen atom, such as Asp171/Tyr306 (and excepting Thr209). The statistics for our study groups are presented in Table 1.
Scheme 1.

General smart pattern representation structures (I, II, III, IV) used for the performed constrained docking simulations.
Table 1.
Summary statistics for our study groups.
| Best Poses | General Score (%) | HBs Formed (%) | Chemgauss4 Score |
|---|---|---|---|
| # Training set_1 | 68–93 | 87–95 | −5.64 |
| Training set_2 | 40–46 | 82–90 | −9.48 |
| Training set_3 | 20 | 94 | −8.67 |
| Training set_4 | 77–82 | 92–98 | −9.35 |
| Training set_5 | 82–85 | 71–95 | −9.21 |
| Training set_6 | 66–92 | 92–93 | −9.05 |
| * Training set_7 | 7–30 | 55–80 | −12.18 (off position) |
# Smart pattern constraint I; * no active constraints. HB: hydrogen bond.
Training set_1 used custom constraint I and provided the worst results. On the other hand, Training set_7 (with no active constraints) resulted in impressively high scoring functions, but entirely lacked the correct binding mode. Training set_3 had as active constraint patterns I, II, and III, and provided improved results. For the study of the remaining cases, all of the above-mentioned constraints (I–IV) were used (differentiated on the included distance). The distance was presented as a sphere with descending radius centered on the Thr209 hydroxyl group varying from preset 3.5 to 1.25 Angstroms (Å). The presented results were acceptable and could be used in screening as an active negative filter for boron compounds. Among Training sets_2, _4, _5, and _6 (based on their statistics), the most enriched proved to be Training set_4, with a cut-off radius of 2.5 Å. Figure 1 provides a detailed 2D map analysis of the ATX active site.
Figure 1.

Schematic representation of the covalent bond as orange dashed lines between compound HA155 and neighboring residues of the autotaxin (ATX) active site.
Images of the successful case studies are given in Figure 2, both in overlay with the original query compound HA155 and in detailed 3D representations inside the protein’s active site.
Figure 2.


Overlay representation of query compound HA155 as gold sticks with Thr209 residue present and the predicted solutions as colored lines (top); best scoring poses of modeled compound HA155 within the active site of ATX (bottom). The figure was obtained by PyMol version 1.4.1 [30].
The optimized geometries of serine and threonine amino acids are the global minima (as shown in Figure 3). Two hydrogen bonds are observed in serine. The first is located between the carboxylic hydrogen and the nitrogen of the backbone amine (1.929 Å), and the second HB is between the carboxylic oxygen and the residual hydroxyl group (2.040 Å). In threonine, the HBs can be observed between the carboxylic hydrogen and the amine group (1.899 Å).
Figure 3.

Optimized geometries of Serine and Threonine amino acids at B3LYP/6-31+G(d,p) level of theory with their respective intramolecular HBs as dashed lines.
The optimized geometries of the inhibitor HA155 are depicted in Figure 4. The HA155 (2) structure is the optimized crystal structure as experimentally isolated. This conformation of the inhibitor is more stable than the HA155 (1) structure, which is another local minimum conformation in the potential energy surface, varying by 2.02 kcal/mol, including the zero point correction energy.
Figure 4.

Optimized geometries of inhibitor HA155, a local minima conformer (1) and a global minima crystal structure conformer (2), at the B3LYP/6-31+G(d,p) level of theory.
The optimized geometries of the bonded serine and threonine with boronic acid (HA155) are presented in Figure 5. In both structures, the hydroxyl groups of the amino acids are bonded with the inhibitor’s boron atom, converting its hybridization to sp3. The geometric parameters are shown in Table 2. The highest occupied molecular orbitals (HOMO) of the two complexes are shown in Figure 6, depicting the electron density concentrated between the boron atom of compound HA155 and the amino acids. The natural bond orbital (NBO) calculations—both in HA155-Ser and HA155-Thr complexes—showed that the bond between boron and oxygen (B-O3, Figure 5) more correctly refers to a polar covalent bond. Based on these calculations, it seems that the occupation number in oxygen is approximately 1.65 electrons, and in boron 0.40 electrons, respectively. Accordingly, the degree of polarity for our models was calculated as 1.770 for HA155-Thr and 1.821 for HA155-Ser, with a threshold of 1.700 for the covalent bond.
Figure 5.
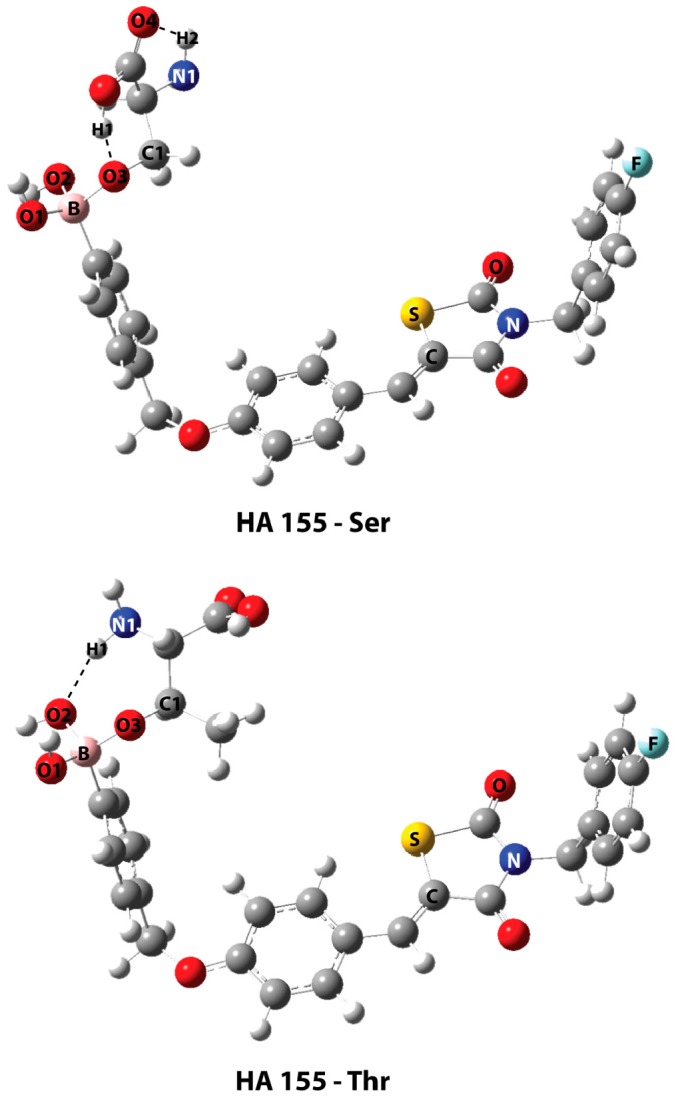
Optimized geometries of the bonded structures of the inhibitor HA155 with serine (top) and threonine (bottom) at the B3LYP/6-31+G(d,p) level of theory, with their respective intramolecular HBs as dashed lines.
Table 2.
Geometrical parameters of the HA155-Ser and HA155-Thr. The bonds are given in Å and the angles in degrees.
| HA155-Ser | HA155-Thr | |
| Bonds * | ||
| B-O1 | 1.460 | 1.465 |
| B-O2 | 1.484 | 1.496 |
| B-O3 | 1.521 | 1.495 |
| Angles | ||
| B-O3-C1 | 119.6 | 122.8 |
| Intermolecular Hydrogen Bond | ||
| H1…O3 | 1.568 | - |
| H2…O4 | 2.210 | - |
| H1…O2 | - | 1.955 |
* The atom numbering follows the same format as represented in Figure 4.
Figure 6.
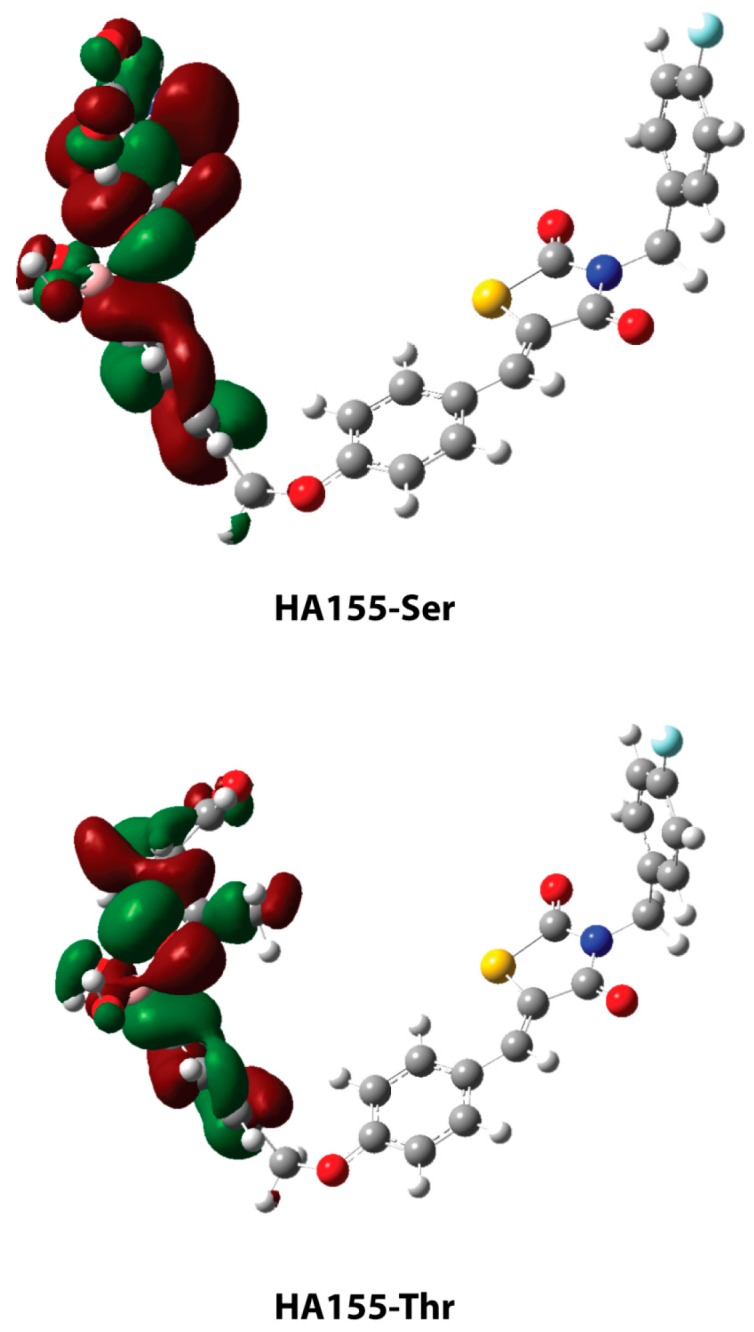
Highest occupied molecular orbitals (HOMOs) of the HA155-Ser (top) and HA155-Thr (bottom) at the B3LYP/6-31+G(d,p) level of theory.
The binding energies of the HA155-Ser complex were calculated both in the gas phase as well as in water solution, providing 311.18 and 300.13 kcal/mol, respectively, whereas the energies for HA155-Thr were 326.49 and 309.52 kcal/mol. The high binding energies found here support the formation of a weak covalent bond between compound HA155 and both Ser/Thr residues. These results provided extra evidence of the reversible nature of this bond, as it was also previously experimentally documented [13,15].
3. Computational Details
Docking studies. The target enzyme ATX (PDB entry 2XRG) co-crystallized with the small molecule inhibitor HA155 was used in our study [25]. All simulations were reproduced on a typical desktop PC running a Windows 7 64-bit operating system (Dual Core Intel Pentium 3.2 GHz CPU processors, RAM 8 GB), using OEDocking suite v. 3.0.1 programs (OpenEye Scientific Software, Inc., Santa Fe, NM, USA; www.eyesopen.com) [31,32,33] using Exhaustive Search Algorithm. Visualization of the docking solutions was performed with PyMol v. 1.4.1 software [30].
The PDB data were used to evaluate our results, and the HA155 molecule was represented in smile format compiled in a simple *.txt file, where with the use of Open Babel [34] we transformed it to the respective *.smi file. Consequently, we acquired its conformer library file with the use of Omega v.2.5.1.4 software (OpenEye Scientific Software, Inc., Santa Fe, NM, USA; www.eyesopen.com) [35,36] in *.oeb.gz format. Preparation of the respective *.pdb-formatted protein was done using the OEDocking v. 3.0.1 suite program MAKE RECEPTOR (OpenEye Scientific Software, Inc., Santa Fe, NM, USA; www.eyesopen.com) [31,32,33]. All water molecules were removed. The docking box was centered on the protein active site which included the co-crystallized HA155 molecule implementing a balanced site-shape potential by an outer contour docking space. No residue modifications were presented unless otherwise specified. Several different constraint options were utilized for residue Thr209 in an effort to better reproduce the original data, and the protein was then saved as a *.oeb-formatted file for further use. Command line-based docking was then performed, specifying the above prepared files as input for the OEDocking v. 3.0.1 suite software (OpenEye Scientific Software, Inc., Santa Fe, NM, USA; www.eyesopen.com) [31,32,33], generating 60 final poses for each model. All of the results were further refined by the use of OEDocking suite program FRED rescore v. 3.0.1 (OpenEye Scientific Software, Inc., Santa Fe, NM, USA; www.eyesopen.com) [31,32,33], providing the sorting of poses using default parameters. Scoring functions were based on Chemgauss4 values, and the solutions were saved as *.sdf files, offering the possibility of being used further for visualization purposes.
Density Functional Theory (DFT) calculations. All geometry optimization calculations were carried out on a typical desktop PC running a Windows 7 64-bit operating system (Intel i7 2.9 GHz CPU processors, RAM 4 GB), in gas phase, using the G09W [37] software package. The hybrid DFT method with Becke’s [38] three-parameter functional and the nonlocal correlation provided by the Lee, Yang, and Parr expression [39] (B3LYP) was used for optimization, employing the 6-31+G(d,p) basis set [40,41,42]. Frequency calculations for all optimized geometries ensured that the calculated structures were the global minima of the potential energy surface of the molecules. Single point calculations starting from the optimized geometry of all structures were also carried out in water solvent using the PCM [43,44,45] model as implemented in G09W software. Natural bond orbital analysis was performed using NBO 3.1 software package as implemented in the G09W package [37], in order to evaluate the covalent bond nature between the studied compounds.
4. Conclusions
Virtual screening of large libraries of boronic acid derivatives fail to dock in a natural mode, since they commonly form covalent bonds with Ser and/or Thr residues when they are presented in the protein active site. Hence, they are left out as false negatives both in regard to their binding poses and their scoring function values. This issue demands programs that can incorporate custom constraints such as smart pattern options or pinpoint selection.
Based on NBO calculations (performed originally herein), the formed bond between Ser/Thr residues is characterized more accurately as a polar covalent bond instead of a simple nonpolar covalent one. The presented results are acceptable, and could be used in screening as an active negative filter for boron compounds. The hydroxyl groups of amino acids are bonded with the inhibitor’s boron atom, converting its hybridization to sp3. Furthermore, the extremely high binding energies of the HA155-Ser/Thr complexes revealed that this reaction is not spontaneous. These theoretical data are also in accordance with the experimentally witnessed reversible binding of the inhibitor HA155 inside the ATX catalytic domain active site.
The findings described above highlight general options that need to be considered when large libraries of boron compounds are virtually screened to identify novel hits in drug design.
Acknowledgments
The authors would like to thank Openeye Scientific Software, Inc., Santa Fe, NM, USA; www.eyesopen.com, for providing an academic license of their programs. Furthermore the authors are grateful to the Aristotle’s University of Thessaloniki Scientific Computing Center (SCC) for providing access to their computing facilities. S.K. is also thankful to the Aristotle University of Thessaloniki Research Committee for financial support through an excellence Post-doctoral Scholarship.
Abbreviations
The following abbreviations are used in this manuscripts
| 2D | two-dimensional |
| 3D | three-dimensional |
| ATX | autotaxin |
| B3LYP | Becke three-parameter Lee-Yang-Par |
| CPU | central processing unit |
| DFT | density functional theory |
| HB | hydrogen bond |
| HOMO | highest occupied molecular orbital |
| HTS | high-throughput screening |
| LPA | lysophosphatidic acid |
| LPC | lysophosphatidylcholine |
| NBO | natural bond orbital |
| PDB | protein data bank |
| PDE | phosphodiesterase |
| PPI | protein-protein interaction |
| RAM | random access memory |
| QSAR | quantitative structure-activity relationship |
| XRD | X-ray diffraction |
Author Contributions
S.K. and D.H.-L. conceived and designed the experiments; S.K. performed the docking experiments; A.G.P. performed the DFT calculations; S.K. and D.H.-L. analyzed and discussed the data; All the authors contributed to the final writing of this paper. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Not available.
References
- 1.Ballatore C., Huryn D.M., Smith A.B., 3rd Carboxylic acid (bio)isosteres in drug design. ChemMedChem. 2013;8:385–395. doi: 10.1002/cmdc.201200585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams J., Behnke M., Chen S., Cruickshank A.A., Dick L.R., Grenier L., Klunder J.M., Ma Y.T., Plamondon L., Stein R.L. Potent and selective inhibitors of the proteasome: Dipeptidyl boronic acids. Bioorg. Med. Chem. Lett. 1998;8:333–338. doi: 10.1016/S0960-894X(98)00029-8. [DOI] [PubMed] [Google Scholar]
- 3.Lesnikowski Z.J. Recent developments with boron as a platform for novel drug design. Expert Opin. Drug. Discov. 2016;11:569–578. doi: 10.1080/17460441.2016.1174687. [DOI] [PubMed] [Google Scholar]
- 4.Dembitsky V.M., Smoum R., Al-Quntar A.A., Abu Ali H., Pergament I., Srebnik M. Natural occurrence of boron-containing compounds in plants, algae and microorganisms. Plant Sci. 2002;163:931–942. doi: 10.1016/S0168-9452(02)00174-7. [DOI] [Google Scholar]
- 5.Trippier P.C., McGuigan C. Boronic acids in medicinal chemistry: Anticancer, antibacterial and antiviral applications. MedChemComm. 2010;1:183–198. doi: 10.1039/c0md00119h. [DOI] [Google Scholar]
- 6.Das B.C., Thapa P., Karki R., Schinke C., Das S., Kambhampati S., Banerjee S.K., Van Veldhuizen P., Verma A., Weiss L.M., et al. Boron chemicals in diagnosis and therapeutics. Future Med. Chem. 2013;5:653–676. doi: 10.4155/fmc.13.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jimenez-Aligaga K., Bermejo-Bescos P., Martin-Aragon S., Csaky A.G. Discovery of alkenylboronic acids as neuroprotective agents affecting multiple biological targets involved in Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2013;23:426–429. doi: 10.1016/j.bmcl.2012.11.068. [DOI] [PubMed] [Google Scholar]
- 8.Rock F.L., Mao W., Yaremchuk A., Tukalo M., Crepin T., Zhou H., Zhang Y.K., Hernandez V., Akama T., Baker S.J., et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759–1761. doi: 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- 9.Albers H.M., Ovaa H. Chemical evolution of autotaxin inhibitors. Chem. Rev. 2012;112:2593–2603. doi: 10.1021/cr2003213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Albers H.M., Hendrickx L.J., van Tol R.J., Hausmann J., Perrakis A., Ovaa H. Structure-based design of novel boronic acid-based inhibitors of autotaxin. J. Med. Chem. 2011;54:4619–4626. doi: 10.1021/jm200310q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albers H.M., Dong A., van Meeteren L.A., Egan D.A., Sunkara M., van Tilburg E.W., Schuurman K., van Tellingen O., Morris A.J., Smyth S.S., et al. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc. Natl. Acad. Sci. USA. 2010;107:7257–7262. doi: 10.1073/pnas.1001529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albers H.M., van Meeteren L.A., Egan D.A., van Tilburg E.W., Moolenaar W.H., Ovaa H. Discovery and optimization of boronic acid based inhibitors of autotaxin. J. Med. Chem. 2010;53:4958–4967. doi: 10.1021/jm1005012. [DOI] [PubMed] [Google Scholar]
- 13.Akama T., Baker S.J., Zhang Y.K., Hernandez V., Zhou H., Sanders V., Freund Y., Kimura R., Maples K.R., Plattner J.J. Discovery and structure-activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis. Bioorg. Med. Chem. Lett. 2009;19:2129–2132. doi: 10.1016/j.bmcl.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 14.Minkkila A., Saario S.M., Kasnanen H., Leppanen J., Poso A., Nevalainen T. Discovery of boronic acids as novel and potent inhibitors of fatty acid amide hydrolase. J. Med. Chem. 2008;51:7057–7060. doi: 10.1021/jm801051t. [DOI] [PubMed] [Google Scholar]
- 15.Knott K., Fishovitz J., Thorpe S.B., Lee I., Santos W.L. N-Terminal peptidic boronic acids selectively inhibit human ClpXP. Org. Biomol. Chem. 2010;8:3451–3456. doi: 10.1039/c004247a. [DOI] [PubMed] [Google Scholar]
- 16.Jacobs R.T., Nare B., Wring S.A., Orr M.D., Chen D., Sligar J.M., Jenks M.X., Noe R.A., Bowling T.S., Mercer L.T., et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop. Dis. 2011;5:e1151. doi: 10.1371/journal.pntd.0001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia-Soria G., Gonzalez-Galvez G., Argoud G.M., Gerstman M., Littlejohn T.W., 3rd, Schwartz S.L., O’Farrell A.M., Li X., Cherrington J.M., Bennett C., et al. The dipeptidyl peptidase-4 inhibitor PHX1149 improves blood glucose control in patients with type 2 diabetes mellitus. Diabetes Obes. Metab. 2008;10:293–300. doi: 10.1111/j.1463-1326.2008.00868.x. [DOI] [PubMed] [Google Scholar]
- 18.Ban H.S., Usui T., Nabeyama W., Morita H., Fukuzawa K., Nakamura H. Discovery of boron-conjugated 4-anilinoquinazoline as a prolonged inhibitor of EGFR tyrosine kinase. Org. Biomol. Chem. 2009;7:4415–4427. doi: 10.1039/b909504g. [DOI] [PubMed] [Google Scholar]
- 19.Bandyopadhyay A., McCarthy K.A., Kelly M.A., Gao J. Targeting bacteria via iminoboronate chemistry of amine-presenting lipids. Nat. Commun. 2015;6:6561. doi: 10.1038/ncomms7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mendes R.E., Alley M.R., Sader H.S., Biedenbach D.J., Jones R.N. Potency and spectrum of activity of AN3365, a novel boron-containing protein synthesis inhibitor, tested against clinical isolates of Enterobacteriaceae and nonfermentative Gram-negative bacilli. Antimicrob. Agents Chemother. 2013;57:2849–2857. doi: 10.1128/AAC.00160-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramkumar K., Tambov K.V., Gundla R., Manaev A.V., Yarovenko V., Traven V.F., Neamati N. Discovery of 3-acetyl-4-hydroxy-2-pyranone derivatives and their difluoridoborate complexes as a novel class of HIV-1 integrase inhibitors. Bioorg. Med. Chem. 2008;16:8988–8998. doi: 10.1016/j.bmc.2008.08.067. [DOI] [PubMed] [Google Scholar]
- 22.Hansen M.M., Jolly R.A., Linder R.J. Boronic Acids and Derivatives-Probing the Structure-Activity Relationships for Mutagenicity. Org. Process Res. Dev. 2015;19:1507–1516. doi: 10.1021/acs.oprd.5b00150. [DOI] [Google Scholar]
- 23.Katsamakas S., Bermperoglou E., Hadjipavlou-Litina D. Considering Autotaxin Inhibitors in Terms of 2D-QSAR and 3D-Mapping- Review and Evaluation. Curr. Med. Chem. 2015;22:1428–1461. doi: 10.2174/0929867322666150227154253. [DOI] [PubMed] [Google Scholar]
- 24.Katsamakas S., Hadjipavlou-Litina D. Boronic Acid Based Inhibitors of Autotaxin: Understanding their Biological Role in Terms of Quantitative Structure Activity Relationships (QSAR) Lett. Drug Des. Discov. 2013;10:11–18. doi: 10.2174/157018013804142483. [DOI] [Google Scholar]
- 25.Hausmann J., Kamtekar S., Christodoulou E., Day J.E., Wu T., Fulkerson Z., Albers H.M., van Meeteren L.A., Houben A.J., van Zeijl L., et al. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011;18:198–204. doi: 10.1038/nsmb.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsilikounas E., Kettner C.A., Bachovchin W.W. Identification of serine and histidine adducts in complexes of trypsin and trypsinogen with peptide and nonpeptide boronic acid inhibitors by proton NMR spectroscopy. Biochemistry. 1992;31:12839–12846. doi: 10.1021/bi00166a019. [DOI] [PubMed] [Google Scholar]
- 27.Adeniyi A.A., Muthusamy R., Soliman M.E. New drug design with covalent modifiers. Expert Opin. Drug Discov. 2016;11:79–90. doi: 10.1517/17460441.2016.1115478. [DOI] [PubMed] [Google Scholar]
- 28.Mah R., Thomas J.R., Shafer C.M. Drug discovery considerations in the development of covalent inhibitors. Bioorg. Med. Chem. Lett. 2014;24:33–39. doi: 10.1016/j.bmcl.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 29.Smith M.C., Gestwicki J.E. Features of protein-protein interactions that translate into potent inhibitors: Topology, surface area and affinity. Expert Rev. Mol. Med. 2012;14:e16. doi: 10.1017/erm.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The PyMOL Molecular Graphics System. Schrödinger, LLC; Cambridge, MA, USA: [(accessed on 6 September 2016)]. Version 1.4.1. Available online: https://www.pymol.org. [Google Scholar]
- 31.McGann M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011;51:578–596. doi: 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- 32.McGaughey G.B., Sheridan R.P., Bayly C.I., Culberson J.C., Kreatsoulas C., Lindsley S., Maiorov V., Truchon J.F., Cornell W.D. Comparison of topological, shape, and docking methods in virtual screening. J. Chem. Inf. Model. 2007;47:1504–1519. doi: 10.1021/ci700052x. [DOI] [PubMed] [Google Scholar]
- 33.McGann M.R., Almond H.R., Nicholls A., Grant J.A., Brown F.K. Gaussian docking functions. Biopolymers. 2003;68:76–90. doi: 10.1002/bip.10207. [DOI] [PubMed] [Google Scholar]
- 34.O’Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hawkins P.C., Nicholls A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012;52:2919–2936. doi: 10.1021/ci300314k. [DOI] [PubMed] [Google Scholar]
- 36.Hawkins P.C., Skillman A.G., Warren G.L., Ellingson B.A., Stahl M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., et al. Gaussian 09, Revision A. 02. Gaussian, Inc.; Wallingford, CT, USA: 2009. [Google Scholar]
- 38.Becke A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- 39.Lee C.T., Yang W.T., Parr R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 40.Clark T., Chandrasekhar J., Spitznagel G.W., Schleyer P.V. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li-F. J. Comput. Chem. 1983;4:294–301. doi: 10.1002/jcc.540040303. [DOI] [Google Scholar]
- 41.Krishnan R., Binkley J.S., Seeger R., Pople J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980;72:650–654. doi: 10.1063/1.438955. [DOI] [Google Scholar]
- 42.McLean A.D., Chandler G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980;72:5639–5648. doi: 10.1063/1.438980. [DOI] [Google Scholar]
- 43.Amovilli C., Barone V., Cammi R., Cancès E., Cossi M., Mennucci B., Pomelli C.S., Tomasi J. Advances in Quantum Chemistry. Volume 32. Academic Press; San Diego, CA, USA: 1998. Recent Advances in the Description of Solvent Effects with the Polarizable Continuum Model; pp. 227–261. [Google Scholar]
- 44.Tomasi J., Persico M. Molecular-Interactions in Solution—An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994;94:2027–2094. doi: 10.1021/cr00031a013. [DOI] [Google Scholar]
- 45.Miertuš S., Scrocco E., Tomasi J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981;55:117–129. doi: 10.1016/0301-0104(81)85090-2. [DOI] [Google Scholar]