

Abstract
Introduction:
The protein disulfide isomerase (PDI) family of thiol isomerases are intracellular enzymes known to catalyze the oxidation, reduction and isomerization of disulfide bonds during protein synthesis in the endoplasmic reticulum. PDI and related members of the thiol isomerase family are known to localize extracellularly where they possess various functions. Among these, the role of PDI in the initiation of thrombus formation is best characterized. PDI is secreted within seconds from activated platelets and endothelial cells at the site of vascular injury and accumulates in the developing platelet-fibrin thrombus. Inhibition of PDI by antibodies or small molecule inhibitors blocks thrombus formation. Efforts are underway to identify extracellular substrates of PDI that participate in the network pathways linking thiol isomerases to thrombus formation. ERp57, ERp5 and ERp72 also play a role in initiation of thrombus formation but their specific extracellular substrates are unknown.
Areas covered:
The following review gives an overview of biochemistry of vascular thiol isomerases followed by a detailed description of their role in thrombosis and its clinical implications.
Expert commentary:
The thiol isomerase system, by controlling the initiation of thrombus formation, provides the regulatory switch by which the normal vasculature is protected under physiologic conditions from thrombi generation.
Keywords: anti-thrombotic drugs, disulfide, endothelium, isomerase, oxidase, platelet, protein disulfide, reductase, thiol isomerases, thrombus formation
1. Introduction
Thiol isomerases belong to a large group of multidomain, multifunctional proteins, of which protein disulfide isomerase (PDI) is the prototype [1]. These proteins, now 21 in total (Fig 1), have evolved from an ancestral thioredoxin gene found in primitive unicellular organisms. The thioredoxin-like domain consists of 4-stranded antiparallel β sheet surrounded by 3 ∝ helices (Figure 2A) [2]. The active site of PDI family thiol isomerases contain Cysteine-X-X-Cysteine (CXXC) motif, where X is any amino acid. Duplication and evolution of the thioredoxin-like fold resulted in multidomain proteins with both active or catalytic domains (termed a, a’ or a⁰) and substrate-binding domains (b or b’) [3]. The catalytic domains contain the CXXC motif and carry out the oxidoreductase function of these enzymes, whereas the substrate-binding domains have developed particular features, such as hydrophobic pockets, to facilitate protein-protein interaction [4, 5].
Figure 1. PDI family of thiol isomerases.
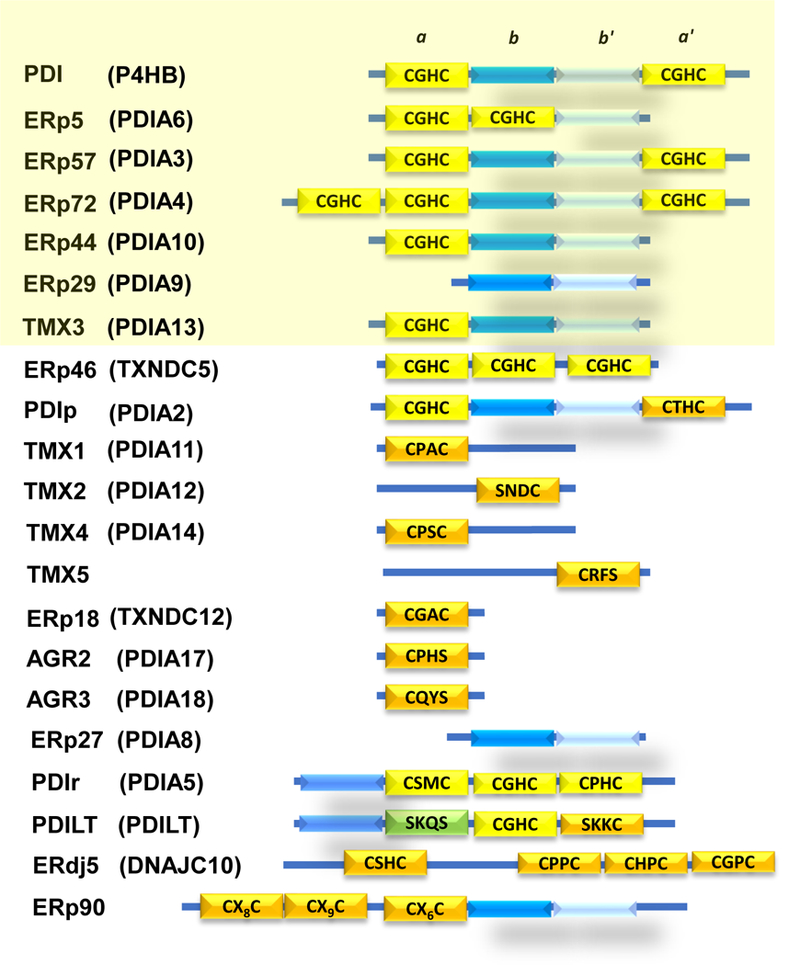
PDI is the archetypal member of the thiol isomerase family and contains four domains: a, b, b’, a’. The a and a’ domains are catalytically active thioredoxin-like domains containing the CGHC motif (yellow), whereas b and b’ are catalytically inactive thioredoxin-like domains (blue and light blue, respectively). Other vascular thiol isomerases are highlighted in yellow. PDI, ERp57, ERp5 and ERp72 are implicated in thrombus formation. Adapted from [88].
Figure 2. X-ray crystal structure of PDI.
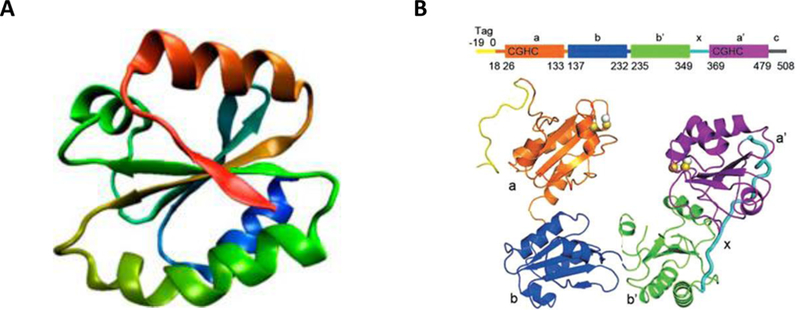
A.The structure of thioredoxin-like domain [106] B. The tertiary structure of PDI is U-shaped, composed of four thioredoxin-like domains: a, b, b’ and a’ [107]. The active site motifs CGHC are found in the a and a’ domains. b and b’ are catalytically inactive substrate binding domains with b’ thought to be important for substrate specificity. The b’ and a’ domains are connected via a short 19-amino acid peptide x-linker. Adapted from [88].
Thiol isomerases are multifunctional enzymes [1]. Their primary function is to catalyze oxidation, reduction and isomerization reactions related to disulfide bonds. Interacting with a protein substrate with lower redox potential, thiol isomerases function as oxidases by forming disulfide bonds from free sulhydryl groups within the substrate. Conversely, acting as reductases on substrates with higher redox potential, they cleave disulfide bonds. They can also act as isomerases by modifying disulfide bond formation within the substrate protein without net change in free thiol content, resulting in alteration in substrate protein structure. In addition, by facilitating protein folding in the absence of catalytic activity, they act as chaperones [6, 7]. These functions are essential in nascent protein folding in the endoplasmic reticulum (ER). Thiol isomerases can also act as nitrosylases/denitrosylases upon modification of the catalytic cysteines by transfer of nitric oxide (NO) to or from substrates [8, 9]. Thus, thiol isomerases are multifunctional proteins involved in multiple catalytic as well as non-catalytic processes.
Although PDI family isomerases contain classical C-terminal ER retention sequence, e.g. KDEL in PDI, a subpopulation is transported extracellularly during cell activation, particularly in platelets and endothelium, where they support a variety of processes [10]. These include viral entry, triglyceride transport, maintenance of surface redox potential, antigen presentation, tumor micro-invasion, and, best characterized, thrombus formation [10–15]. The release of these proteins is regulated and occurs upon injury or chemical stimuli, but the mechanism of release remains poorly understood. The identification of the critical role that PDI plays in thrombus formation was unanticipated, and has expanded our understanding of proximal molecular events which regulate the initiation of the hemostatic process upon vascular injury. This review focuses on the biochemistry and recent advances in our understanding of the role of vascular thiol isomerases in thrombus formation.
2. Vascular thiol isomerases
2.1. Protein Disulfide Isomerase
PDI is the archetypal and most abundant thiol isomerase in the ER and was originally identified by Anfinsen as the microsomal enzyme capable of oxidative folding of ribonuclease A [16]. A polypeptide with molecular weight of 57,000, encoded by the P4HB gene, PDI has 4 domains: a, b, b’ and a’, with a linker peptide, “x”, C-terminal to b’ and a short “c” segment C-terminal to the a’ domain (Figure 2B.) [17, 18]. The a and a’ domains contain the catalytic active sites containing Cys-Gly-His-Cys (CGHC) motif. The two catalytic domains are not redundant in function as demonstrated by the critical role of a’ domain of PDI in thrombus formation [4].
The a and a’ domain CGHC motifs of PDI appear to be predominantly reduced in vivo, although the redox state of these thiol isomerases is dynamically regulated by sulfhydryl oxidases such as Ero1 [19, 20, 21]. The CGHC motif active sites enable PDI to cycle between the reduced thiol and disulfide-bonded state, key to its participation as an oxidoreductase. It can directly catalyze the formation of disulfide bridge formation in a number of substrates and is dependent on sulfhydryl oxidases to be oxidized itself prior to acting on its substrate. A sulfhydryl oxidase couples the oxidation of a CXXC motif in PDI to reduction of a small molecule cofactor, such as flavin adenine nucleotide (FAD). The reaction proceeds through a mixed disulfide intermediate between the sulfhydryl oxidase and active domain of a PDI family protein. Understanding of this linear electron transfer pathway with the sulfhydryl enzyme first oxidizing CXXC motif in a thiol isomerase, which subsequently donates its disulfide to a nascent secretory protein, is essential in understanding in vivo trapping of substrates, as discussed below [22, 23]. A number of oxidative substrates (e.g. procollagen, thyroglobulin, γ-gliadin) of PDI have been identified but many remain elusive [24, 25, 26].
The essential function of PDI in cell growth may lie in its chaperone activity and not its catalytic activity. The complete deletion of PDI in yeast is lethal, and yet yeast cells that only express mutant PDI with inactivated catalytic sites (AXXA) are viable [27]. Consistently, PDI knockout mice are embryonic lethal. Unlike oxidoreductase activity, the structural determinants of PDI chaperone activity remain unclear [28]. A mutant PDI with the 51 C-terminal amino acids deleted (abb’xa’), maintained its catalytic activity but lost its chaperone function, although this may have resulted from the loss of substrate-binding by the b’ domain secondary to destabilization of the a’ domain [29]. Examples of chaperone activity of PDI, which occurs in complex with other enzymes, include hydroxylation of proline and membrane transport of triglycerides [12].
PDI undergoes S-nitrosylation following exposure to nitric oxide (NO) donors [30]. S-nitrosylation of catalytic cysteines within PDI enables transfer of NO to other proteins and into cells. Knockdown of PDI in HEL cells inhibits delivery of NO from a S-NO donor into cytosol [9]. Similarly, inhibition of PDI blocks transfer of NO from a physiologic NO donor, S-nitrosoglutathione, into both megakaryocytes and into platelets [31]. NO can also be transferred from the heme moiety in guanylyl cyclase, as evidenced by its activation and marked increase in cGMP levels [9, 32]. However, the significance of this mechanism in vivo has not yet been determined.
PDI is abundant in the ER and is often used as an ER marker. However, it is also located in small secretory granules, including the GRO-∝ granule of the endothelial cells and T-granule of the platelet, which may be specific compartments of the dense tubular system [33, 34]. PDI and other thiol isomerases contain C-terminal ER retention sequence, KDEL. But despite this, subpopulations are transported extracellularly [35]. It is this subpopulation that carries out its extracellular function by acting on its extracellular substrates during thrombus formation. Stimulation of platelets and endothelial cells by secretagogues in vitro results in rapid exocytosis of PDI [10]. Similarly, in vivo endothelial injury models reveal rapid appearance of PDI on endothelial surface followed by platelet surface in a growing thrombus [33]. The dynamics of released PDI upon endothelial injury, its sequestration at the site of growing thrombus, and its potential substrates, are discussed in greater detail below. The route by which PDI is released extracellularly remains unresolved. Some data suggests that it follows the classical secretory mechanism, that is, ER to Golgi to plasma membrane transport [36]. This occurs in association with the KDEL receptor. The KDEL receptor was initially thought to function in recycling of proteins with a KDEL motif back to the ER. Now, it is known to also exist in the trans-Golgi network (TGN) as well as in plasma membrane. PDI and KDEL receptors colocalize in TGN and on plasma membranes. Additionally, the knockdown of the KDEL receptor in endothelial cells results in reduced agonist-induced exocytosis of PDI [36]. More recently, it has been suggested that PDI release follows unconventional protein secretion pathways, as used by other ‘leaderless’ proteins [37]. It has also been shown that there is a basal constitutive release of small quantities of PDI from the endothelium.
2.2. ERp57
Erp57 is expressed in all eukaryotic cells and is widely distributed in mammalian tissues [38]. A polypeptide with molecular weight of 57,000, encoded by the gene PDIA3, ERp57 shares a common domain structure with PDI. It also has four thioredoxin-like domains, a-b-b’-a’, with a and a’ domains carrying the catalytically active site with the CGHC motifs. Despite this similarity, these enzymes are functionally non-redundant as ERp57 cannot rescue PDI deficiency in yeast [39]. This is likely related to the distinct substrate-binding domains b’ in ERp57 and PDI accounting for different substrate specificities. For example, the b’ subunit of ERp57 interacts with lectin chaperones calnexin and calreticulin, whereas PDI does not [40, 41]. Thus, intracellular substrates of ERp57 likely includes glycoproteins that enter the calnexin cycle [42]. ERp57 and PDI also possess similar reduction potentials, which are substantially more oxidizing than those of the substrate thiols, but differ in their capacities for redox modulation by endogenous sulfhydryl oxidases such as Ero1∝ [28]. ERp57 knockout mice are embryonic lethal [43].
ERp57 is primarily located in the ER and like PDI contains the ER retention sequence, KDEL. Unlike, PDI, extra-ER sites of ERp57 storage are not known, although it is found in the membrane fraction of the endothelial cells in addition to the ER [33]. A subpopulation of ERp57 is transported to the plasma membrane and the extracellular space via an undetermined route, where it carries out its extracellular functions by acting on its extracellular substrates. Agonist-stimulation of platelets and endothelial cells leads to ERp57 release and it is this extracellular subpopulation of ERp57 that participates in thrombosis [44, 45, 46].
2.3. ERp5
ERp5 is a single polypeptide of molecular weight 48,000, encoded by the gene PDIA6. Its domain structure is a unique a-a’-b arrangement, with a and a’ carrying active sites with the CGHC motif [47]. ERp5 is thought to carry out oxidoreductase, isomerase and chaperone activities in the ER, although it is known to be a less efficient thiol isomerase than PDI in vitro [47]. Few substrates of ERp5 are known. Similar to PDI and ERp57, a subpopulation of ERp5 is transported to the cell membrane and extracellularly, where it carries out its extracellular functions, including thrombus formation [15, 48].
2.4. ERp72
ERp72, encoded by the gene PDIA4 is one of the largest thiol isomerases with 5 thioredoxin-like domains due to an additional N-terminal domain a⁰ to yield an a⁰-a-b-b’-a’ architecture [49]. The a⁰ domain also contains the CGHC active site, in addition to those present in the a and a’ domains. The non-catalytic b and b’ domains of ERp72 are similar to ERp57, but despite this similarity, ERp72 does not bind calnexin [50]. While the b’ domain in PDI is the primary substrate binding site, unlike PDI, the b’ domain in ERp72 is not hydrophobic, and does not bind scrambled RNase or small peptides. Instead, there are hydrophobic patches in the active site domains a⁰, a and a’ which likely participate in substrate recognition and binding as well [51]. These differences in the structural recognition sites of these enzymes can be extrapolated to its extracellular substrates, particularly those involved in thrombus formation.
3. Thiol isomerases and thrombus formation
3.1. PDI
PDI has been known to present on the platelet membrane surface and released from activated platelets [52–54]. Inhibitors of thiol isomerases were also known to impair platelet aggregation, and, in addition, PDI catalyzed the formation of thrombin-antithrombin-thrombospondin 1 and thrombin-antithrombin-vitronectin complexes in vitro. Despite this knowledge, the critical role of PDI in thrombus formation was not appreciated until development of intravital microscopy to study thrombus formation in a live mouse [23, 44, 55, 56]. This was also actively pursued in quest for the oxidoreductase that catalytically deencrypted inactive tissue factor, as proposed by Hogg et al [57, 58]. The mechanism of deencryption of tissue factor remains controversial. However the finding of controlled release of PDI from platelets and endothelial cells within seconds of vessel wall injury, preceding tissue factor activity, and that inhibition of PDI inhibited thrombus formation, was unanticipated. Thrombus formation in the mouse cremaster arterioles was visualized after laser injury and the dynamics of released PDI monitored [10]. PDI was released within seconds of the endothelial injury and incorporated in the growing thrombus. Inhibition of PDI activity by the non-specific thiol isomerase inhibitor, bacitracin, as well as by an inhibitory anti-PDI antibody, blocked platelet thrombus formation and fibrin generation. Using a carotid artery injury and ligation models of thrombosis with intravital live microscopy, significant impairment of fibrin generation by an inhibitory anti-PDI antibody was observed [59]. Platelet-released PDI was also shown to promote fibrin generation in the ligation model of common carotid artery in mice expressing human tissue factor [59]. Subsequently, this was also demonstrated in other models of vascular injury [4, 60, 61]. The conclusion of this body of work was that fibrin generation is dependent on the release of PDI at the site of vessel wall injury.
The dynamics of PDI release observed at the site of vessel wall injury, with PDI lining the injured vasculature prior to platelet accumulation, suggested that endothelium was the primary source of PDI. By blocking platelets from participating in thrombus formation by inhibiting platelet ∝IIbβ3 with eptifibatide confirmed that endothelium is the initial as well as the principal source of released PDI at the site of vessel wall injury [33]. It was demonstrated that endothelium-derived PDI, in the absence of platelet participation in thrombus formation, is sufficient for fibrin generation. Subsequently, a study of conditional platelet-specific PDI knockout mice, corroborated these findings [60]. In this model, platelet PDI was only required for late thrombus growth, but not for initial platelet adhesion or fibrin generation. PDI inhibition impairs platelet aggregation, in vitro, but PDI is not required for P-selectin surface expression, calcium mobilization, β3-talin interaction or platelet spreading [60]. Par4−/− mice, with platelets that cannot be activated by thrombin, generate normal levels of fibrin despite the absence of platelet thrombus formation at the site of vessel wall injury [62]. This is consistent with the observation that endothelial PDI is sufficient for supporting fibrin clot formation by itself. β3−/− mice failed to generate fibrin [63]. This is likely due to absence of endothelium ∝vβ3 in β3−/− mice, which is likely a critical intermediate for PDI-dependent fibrin generation.
How is endothelial and platelet-derived PDI retained at the site of vessel wall injury? This occurs via interaction of PDI with β3 integrin, both platelet ∝IIbβ3 and endothelial ∝vβ3 [63]. Retention of PDI favors the activated conformer of β3 integrin in vitro. PDI binds to β3 with a Kd of 1–2 μM. Interaction of PDI with β3 raised the possibility of β3 being a substrate of PDI in its role in thrombus formation. It has been known that thiol isomerase inhibitors and specific PDI inhibitors inhibit platelet ∝IIbβ3 and ∝vβ1 activation [64, 65]. Whether this is related to direct action of PDI on β3, or indirect, through impairment of platelet activation and altered inside-out signaling, is unclear. β3 has a total 46 cysteines forming 23 disulfide bonds, but none are free [66]. On the other hand, mutagenesis studies revealed that mutation of specific cysteine residues on β3 rendered the molecule constitutively active [67, 68]. Also, recombinant ∝IIbβ3 has intrinsic thiol isomerase activity from 9 intrinsic C-X-X-C residues [69]. The activation of β3 and whether PDI, or other thiol isomerases, play a role remains elusive [70].
Recently, platelet polyphosphates, which are contained in platelet dense granules, have been implicated in activation of zymogens of the contact pathway of coagulation, particularly factor XII and factor V, bypassing the tissue factor pathway [71]. To study the role of platelet dense granules in PDI-mediated thrombus formation, Hermansky-Pudlak Syndrome 6 (HPS6) mice that lack platelet dense granules were evaluated. In the laser injury model, these mice not only demonstrated defective platelet thrombus formation but significant impairment in fibrin accumulation [72]. This implied that there existed an endothelial defect in this syndrome. Evaluation of HPS6 platelets as well as human umbilical vein endothelial cells (HUVEC) following siRNA-mediated HPS6 depletion led to the finding of defective PDI release from both platelets and endothelium in this syndrome, in vitro. Furthermore, supplementation of recombinant PDI corrected thrombus formation in HPS6−/− mice (S.H. Kim and B. Furie, unpublished observations, 2013). This work linked a human bleeding disorder with deficient PDI release.
3.1.1. PDI substrates
With the requirement of PDI in initiation of thrombus formation firmly established, the biggest question that arises is, What are the extracellular substrates of PDI? A mechanism-based kinetic trapping has been developed for identifying extracellular substrates of PDI. This is based on the linear electron transfer pathway between a thiol isomerase and its substrate, modified from what has been employed previously to study the electron transport pathway in the ER and on the cell surfaces [73, 74]. When the PDI CGHC motif is reduced, a substrate disulfide can be reduced at the expense of the PDI active site. This reaction occurs through transient formation of a mixed disulfide between the N-terminal Cys in the PDI CGHC motif and a Cys in the substrate. Resolution of the mixed disulfide requires the C-terminal Cys of the CGHC motif. Mutation of the C-terminal Cys to alanine (Ala) in the PDI active site (PDI-CGHA) leads to the accumulation of the PDI-substrate mixed disulfide intermediate complex, which can then be isolated and analyzed (Figure 3).
Figure 3. Mechanism-based trapping of PDI substrates.
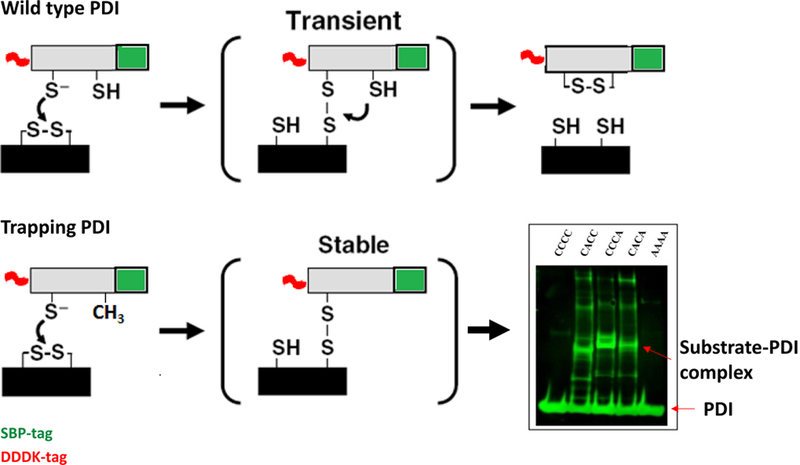
Reduction of a substrate disulfide by PDI occurs through transient formation of a mixed disulfide between the N-terminal Cys in the PDI CGHC motif and a Cys in the substrate, as depicted in the top panel. Resolution of the mixed disulfide requires the C-terminal Cys of the CGHC motif. Mutation of the C-terminal Cys to alanine (Ala) in the PDI active site (CGHC to CGHA) makes this mixed PDI-substrate disulfide stable, as depicted in the lower panel. These PDI-substrate complexes can then be isolated and analyzed. The Western blot on the right shows one such reaction where no substrate is isolated with the use of wild-type PDI (CCCC) or inactive PDI (AAAA), but only with PDI trapping mutants with C-terminal Cys modified to Ala in one (CACC and CCCA) or both (CACA) catalytically active domains.
Using this technique, multiple protein substrates of PDI have been identified in platelet-rich plasma stimulated with collagen and thrombin. These include vitronectin, complement factor 3, complement factor 5, ∝2-macroglobulin, protein S, histidine-rich glycoprotein, thrombospondin 1, prothrombin and CD5 antigen-like binding protein [75]. Among these, vitronectin, histidine rich glycoprotein, protein S and prothrombin are redox substrates of PDI. The molecular basis of PDI-vitronectin interaction and its physiological significance both in vitro and in vivo has also been characterized. Vitronectin is an abundant plasma protein with a concentration of 300 μg ml−1, present in both monomeric and multimeric forms depending on its association with other molecular species and activity of the proteolytic enzymes [76]. A smaller quantity is also present in platelet ∝-granules and released upon platelet activation [77]. Vitronectin binds to ∝vβ3 and ∝IIbβ3, plasminogen activator I, urokinase receptor and collagen, and complement C5b-7, and is involved in thrombus formation, as demonstrated in vitronectin−/− mice, which have absent platelet thrombus formation and significantly reduced fibrin generation [78, 79]. Vitronectin has multiple disulfide bonds, and PDI reduces Cys 137- Cys 161, an intradomain linkage, and Cys 274- Cys 453, an interdomain linkage in the hemipexin-like domain of vitronectin. It remains unclear whether one or both disulfides require reduction for vitronectin activation. Reduction of these thiols correlates with β3 binding, as native vitronectin does not bind ∝vβ3, but only after treatment with reduced PDI. Binding of vitronectin to ∝vβ3 on human umbilical vein endothelial cells (HUVEC) is enhanced by PDI, which can be blocked by either Arg-Gly-Asp (RGD) peptide or ∝vβ3 inhibitory antibody, suggesting that PDI reduces a disulfide bond in vitronectin that exposes the RGD site, facilitating its binding to ∝vβ3. Moreover, inhibition of PDI by either small molecule inhibitor quercetin-3-rutinoside or inhibitory antibodies blocks vitronectin accumulation at the site of thrombus formation in vivo. This is the first demonstration of molecular interaction of PDI with its substrate and its relevance to thrombus formation.
The major drawback of the above described technique is that it can only identify substrates that are subject to disulfide reduction, as CXXA variants of the active motif can never exist in an oxidized state. To identify substrate proteins of PDI which require oxidation using kinetic substrate trapping, a PDI variant must retain both active site cysteine residues, but instead be modified to perform disulfide exchange much more slowly that the wild-type PDI, so that the PDI-substrate intermediates formed during the oxidoreductate reaction can be ‘frozen’ using alkylating agents, and subsequently isolated and identified. Using two such variants where His of CGHC motif was modified to either Pro (CGPC) or Arg (CGAC) in both a and a’ domains, multiple other substrates released from activated platelets have now been identified [80]. These include factor V, annexin V, heparanase, ERp57, kallekrein 14, serpin B6, tetranectin, and collagen VI, which show a bias for reduction, whereas cathepsin G, glutaredoxin-1, thioredoxin, GPIb, and fibrinogen show a bias for oxidation by PDI. Cathepsin G is an ∝-granule protein known to play a role in thrombosis [81, 82]. Treating washed platelet releasate with oxidized PDI increases the activity of native cathepsin G, confirming the physiological importance of PDI-cathepsin G interaction. Similarly, inhibition of PDI activity with quercetin-3-rutinoside significantly reduced platelet factor Va generation following platelet activation, which paralleled the reduction in platelet-dependent thrombin generation, implying that PDI is involved in conversion of platelet factor V to Va, which in turn is involved in thrombin generation [83].
Factor XI has also been implicated as a redox substrate of PDI [84]. Reduction of factor XI by purified PDI enhanced cleavage of its chromogenic substrate and factor IX in vitro and resulted in opening of Cys362-Cys482 disulfide bond. Disruption of this disulfide bond resulted in enhanced accessibility of thrombin to factor XI secondary to broader binding-site [84]. Previously, purified platelet PDI was also shown to catalyze formation of disulfide-linked complexes of thrombospondin-thrombin-antithrombin in vitro [23, 85]. The physiologic relevance of interaction of PDI with factor XI or thrombospondin remains unknown.
3.1.2. PDI and tissue factor
Oxidation of the Cys 186-Cys 209 allosteric bond in tissue factor is one of the suggested mechanisms by which its encrypted inactive form becomes active [57]. Cys 209 in tissue factor has been reported to be constitutively S-glutathionylated or S-nitrosylated [58]. This modification was hypothesized to prevent formation of Cys 186-Cys 209 disulfide. PDI has been suggested to deencrypt tissue factor, whereas inhibition of PDI has been suggested to increase tissue factor activity via expression of phosphatidylserine [86]. PDI also regulates a P2X7 receptor-dependent signaling pathway that is associated with deencryption of tissue factor [87]. The role of PDI in tissue factor activation remains controversial.
3.2. ERp57
ERp57 was first identified in profiling studies designed to identify thiol isomerases present in mouse and human platelets, which revealed that stimulated platelets released ERp57 and ERp57-containing microparticles [44]. Subsequently, it was shown that anti-ERp57 inhibitory antibodies inhibited CRP-XL-induced platelet aggregation and ATP release, but not PF4 or P-selectin release, implying that ERp57 was involved in dense but not ∝-granule release [45]. Calcium mobilization, an early event in platelet activation, and ∝IIbβ3 stimulation, as measured by fibrinogen binding, were altered as well. These antibodies also inhibited platelet thrombus formation in collagen glass fibers in vitro. ERp57 accumulates in the growing thrombus formation in the mouse laser injury model of thrombus formation, where anti-ERp57 antibodies inhibited platelet thrombus formation [45]. These data established a role of ERp57 in thrombus formation.
Inhibitory monoclonal antibodies to ERp57 confirmed that ERp57 is essential for platelet aggregation [46]. Recombinant wildtype ERp57 enhanced platelet aggregation whereas a mutant inactive form altered platelet aggregation. The activation of platelet ∝IIbβ3 but also P-selectin release is inhibited by this antibody, unlike the prior work. In mice treated with Vivo-ERp57 morpholino to deplete the total ERp57, as ERp57−/− are embryonic lethal, platelet aggregation and β3 activation was abnormal ex vivo [88]. Additionally, both platelet thrombus formation and fibrin accumulation were significantly reduced in the laser injury model of thrombus formation in Vivo-ERp57 morpholino mice as compared to the control mice. Platelet-specific ERp57 conditional knockout mice demonstrated increased tail bleeding time and occlusion times in mouse model of ferric chloride-induced carotid and mesenteric arteriolar injury [89]. Platelet incorporation in the growing thrombus in this mouse model is dependent on the presence of β3 integrin. ERp57-null platelets had reduced platelet aggregation and ∝IIbβ3 stimulation. ERp57 bound to β3 in thrombin-activated platelets but not in platelet lacking β3. Fibrin generation is reduced in the laser injury model. Although studies performed by our group show that ERp57fl/flPF4Cre-positive mice that do not express ERp57 in their platelets have normal fibrin generation, similar to the results obtained for PDI [88]. This remains to be resolved. Endothelium-specific ERp57 conditional knockout mice have also been generated, and show that the fibrin generation is further reduced in the laser injury model. These data suggest that ERp57 is critical for platelet activation and fibrin generation in vivo [90].
Unlike PDI, the extracellular substrates of ERp57 in plasma or those released from activated platelets have not been identified although preliminary studies indicate that ERp57 targets the components of the lectin pathway of complement activation (Eriksson and Furie, 2018) Vitronectin is not a substrate of ERp57 [75].
3.3. ERp5
Like PDI and ERp57, ERp5 is secreted from platelets upon cell activation, and inhibition of ERp5 activity using anti-ERp5 antibody prevents platelet aggregation and fibrinogen binding to the activated platelets in vitro [91]. ∝IIbβ3 is a binding partner of ERp5 demonstrated by co-immunoprecipitation studies with β3 integrin [91]. ERp5 binds to β3 with or without Mn2+, implying that β3 activation is not critical for this interaction, and the cysteine residues in the ERp5 active sites are not required for β3 binding [48]. Inhibitory anti-ERp5 antibodies inhibit ERp5-dependent platelet and endothelial cell disulfide reductase activity in vitro. Using the laser injury model of thrombus formation in the live mouse, ERp5 accumulates at the site of vessel wall injury in the growing thrombus, and inhibition of ERp5 with specific antibodies reduce platelet thrombus formation and fibrin accumulation [48]. These data support a novel role of ERp5 in thrombus formation.
3.4. ERp72
Like ERp5, the release of ERp72 by activated platelets was reported some time ago, but only recently was its role in thrombus formation confirmed in vivo [44, 92, 93]. Platelet-specific conditional ERp72-knockout mice that do not express ERp72 in platelets, but retain normal levels of PDI, ERp57 and ERp5 [92]. These mice show decreased thrombin-induced activation of ∝IIbβ3 and P-selectin, and thrombin and convulxin-induced aggregation and ATP release. ERp72 binding to thrombin-activated or Mn2+-treated β3-null platelets is substantially reduced, implying that ERp72 binds to platelet ∝IIbβ3. Double active site mutant ERp72(ss-oo-oo) with a and a’ active sites inactivated, failed to rescue aggregation the defect in ERp72-null platelets, while ERp72(oo-ss-oo) and ERp72(oo-oo-ss), with a0030 and a or a0 and a’ sites inactivated, respectively, partially recovered the aggregation defect in these platelets in vitro. This implies that active sites in the a and a’ domains of ERp72 but not the a0 domain support platelet aggregation. In addition, PDI or ERp57 did not rescue aggregation defect in ERp72-null platelets, supporting a distinct role of ERp72 in platelet function.
ERp72 accumulates at the site of vessel wall injury in the laser injury model of thrombus formation in vivo and a humanized monoclonal inhibitory anti-ERp72 antibody inhibited thrombus formation in this model [92]Holbrook, 2017 #94). [93]. As with PDI, endothelial-specific conditional ERp72-knockout mice with absent endothelial ERp72, have both reduced platelet thrombus formation and fibrin accumulation, whereas platelet-specific endothelial knockout mice only had reduced platelet thrombus formation in the laser injury model. Extracellular substrates of ERp72 that are involved in thrombus formation are not known.
4. Clinical implications
The modification of disulfide bonds by thiol isomerases represents a previously unrecognized layer of control of thrombus formation. Despite the similarity between modification of disulfide bonds by thiol isomerases and cleavage of bonds between amino acids by classic serine proteases of the coagulation cascade, there is a major distinction between these two mechanisms. Unlike proteases of the coagulation cascade, thiol isomerases do not directly initiate blood coagulation, but only relieve the suppression of thrombus formation. Thus, inhibition of thiol isomerases could achieve control of thrombosis without increasing bleeding risks. Conversely, thiol isomerases may serve as hemostatic agents in coagulopathies without increasing the risk of thrombosis. In fact, genetic deletion of PDI in mice megakaryocytes/platelets, or inhibition of PDI and other thiol isomerases by inhibitory antibodies, impairs thrombus formation in vivo without prolonging tail bleeding times [10, 60]. Chronically elevated plasma thiol isomerase levels, as may occur in conditions with increased cell turnover and/or cell death, such as cancers or inflammatory disorders, may predispose individuals with these conditions to thrombotic tendencies. Thiol isomerase-targeted therapy to prevent thromboembolism may be a strategy in these conditions and plasma levels of PDI and thiol isomerases may serve as biomarkers of thromboembolic risk.
4.1. Targeting PDI as antithrombotic agents
Using high-throughput screening of a small molecule library, flavonoid quercetins (e.g. quercetin-3-rutinoside and isoquercetin) were discovered as inhibitors of PDI [94]. Quercetin-3-rutinoside demonstrated the most potent inhibitory activity of all compounds tested with an IC50 of 6 uM. When tested in mice, doses as low as 0.1 mg/kg of quercetin-3-rutinoside inhibited thrombus formation, without increasing tail bleeding times. Infusion of recombinant PDI reversed this inhibitory effect [94], indicating that PDI is the specific target of quercetin-3-rutinoside in this model. Orally administered quercetin-3-rutinoside was also antithrombotic in doses that are commonly used as dietary supplements and are not known to have any significant adverse effects. These findings provided preliminary proof-of-principle that inhibition of PDI to block thrombus formation could be accomplished safely. Epidemiologic data support that consumption of flavonoids is associated with reduced risk of cardiovascular death and strokes [95, 96].
Due to higher bioavailability, isoquercetin is currently being evaluated as an antithrombotic agent in the Cancer Associated Thrombosis and Isoquercetin Trial (CAT-IQ) [97]. This is a 600-patient randomized phase 2/3 trial designed to determine the safety and efficacy of isoquercetin in preventing cancer-associated thrombosis. The efficacy outcome measurements include thrombosis at 60 days and plasma D-dimer levels, whereas bleeding remains the primary safety outcome. To assess adequate plasma concentrations of isoquercetin and its active metabolites, PDI inhibitory activity in patient plasma was determined. Inhibition of PDI does not alter standard coagulation tests such as the prothrombin time or partial thromboplastin time, but does affect platelet aggregation studies and platelet-dependent thrombin generation assays [83, 94].
The PDI binding site of quercetin-3-rutinoside has been suggested. Using fluorescence enhancement-based assay and isothermal calorimetry, quercetin-3-rutinoside binds to the b’x domain of PDI with a 1:1 stoichiometry [98]. Furthermore, infusion of b’x domain of PDI rescued thrombus formation that was inhibited by quercetin-3-rutinoside in the laser injury model. Due to the lack of active site motif CGHC, the b’ domain lacks reductase activity, and has the potential as an antidote to reverse the antithrombotic effect of quercetin-3-rutinoside.
Another class of compounds termed bepristats have recently been identified as inhibitors of PDI via high-throughput screening of another small molecule repository. These compounds bind to the b’ domain of PDI and reversibly inhibit platelet aggregation and thrombus formation in vivo. These compounds paradoxically enhance the catalytic activity of a and a’ domains by displacing the x-linker, which acts as an allosteric switch to increase the reductase activity in the catalytic domains [99].
Bacitracin is a topical antibiotic used as an inhibitor of thiol isomerases in the laboratory for decades [100]. It is a non-specific inhibitor of thiol isomerases, low in potency, and highly nephrotoxic, precluding its clinical use as an antithrombotic agent. Similarly, aminoglycosides bind and inhibit PDI but only in toxic doses. Several synthetic PDI antagonists have also been synthesized in recent years. To give an example, ML359 was identified as a potent, selective inhibitor of PDI by high throughput screening of another small molecule library after discovery of flavonoids as PDI inhibitors [101]. Other examples include juniferdin, PACMA-31, adenanthin, phenyl vinyl sulfonate and RB-11-ca that have been tested for other indications but not thrombosis [102]. Inhibitors of other non-PDI thiol isomerases as antithrombotic agents have not been tested.
4.2. Thiol isomerases in coagulopathy
Deficient release of PDI at the site of vessel wall injury in the laser injury model has been reported in HPS6 [72]. In addition, supplementation of recombinant PDI to these mice rescues the defective thrombus formation. But, whether or not recombinant PDI shortens bleeding time or platelet aggregation defect in these mice has not yet been determined. Upregulation of platelet PDI has been reported in hemophilia A, but it is not known whether infusion of recombinant thiol isomerase can mitigate bleeding in hemophilia and other coagulopathies [103].
4.3. PDI as a biomarker
Currently, there are little data on circulating plasma levels of vascular thiol isomerases. Autoantibodies to PDI have been identified in cancers, SLE and infections [104]. Similarly, autoantibodies to ERp57 have been detected in cancers and infections [105]. This could indicate that these thiol isomerases circulate at a higher level in these disorders, but whether this is associated with a prothrombotic phenotype is unknown. Plasma levels of soluble and microparticle-associated PDI are currently under evaluation in many disorders including cancers and cardiovascular disease to see if elevated levels correlate with a higher risk of thromboembolic disease.
5. Conclusion
Thiol isomerases were originally described as ER proteins critical for protein folding. However, over the recent years, our understanding of these enzymes has expanded to include their role as extracellular regulators of thrombus formation. PDI and the other thiol isomerases are now implicated at multiple steps in the thrombus formation, including their effect on platelet function and the initiation of coagulation and fibrin formation. More recently, substrates of PDI have been discovered. These comprise of both soluble plasma proteins, including those of the coagulation and complement cascades, as well as membrane receptors. Similar analyses for other thiol isomerases implicated in thrombus formation are underway. inhibitors of PDI are being tested clinically as novel antithrombotic agents.
6. Expert Commentary
A large body of data now suggests that vascular thiol isomerases, released from activated endothelial cells and platelets, regulate the initiation of the hemostatic process following vessel wall injury by redox modulation of structures of proteins involved in thrombus formation. This is in contrast to the classical belief that hemostasis begins with exposure of tissue factor and collagen, normally sequestered spatially in the absence of vessel wall injury. Once initiated, the process of thrombus formation is modulated mainly by natural anticoagulant systems: tissue factor pathway inhibitor, the protein C system, and antithrombin. But, what regulates the initiation of thrombus formation? We suggest that absence of extracellular thiol isomerases in the circulation suppresses thrombus formation. Upon vascular injury, this suppression is relieved by the local release of thiol isomerases by injured endothelium and bound platelets. The released thiol isomerases, via modification of disulfide bonds then covalently modify their substrates from inactive to active state, which can now participate in thrombus formation. The thiol isomerase system, therefore, provides the regulatory switch by which the normal vasculature is protected under physiologic conditions from generation of thrombi.
Despite agreement in the critical role that PDI family thiol isomerases in regulation of thrombus formation, several key questions remain unanswered. While PDI, ERp57, ERp5 and ERp72 have already been implicated in thrombus formation, the full repertoire of thiol isomerases that participate in thrombosis remains to be identified. Secondly, how extracellular isomerases interact with one another in the context of thrombosis is unknown. Are these pathways parallel and redundant, to maximize redox capacity ensuring fidelity of extracellular redox reactions, or separate and discriminatory, to enable selective activation of specific substrates, or hybrid, with elements of both? Several lines of evidence suggest that thiol isomerases may have distinct substrates. The best characterized substrate of PDI to date, vitronectin, does not bind ERp57. The substrates of PDI and ERp57 in the ER are unique, with the latter having propensity to modify disulfide bonds in glycoproteins. Additionally, inhibitory anti-ERp57 antibodies further impair platelet aggregation, ATP release and ∝IIbβ3 activation in PDI-null mouse platelets. In fact, a mechanism for the participation of both these thiol isomerases in ∝IIbβ3 activation is suggested by a study of polyomavirus infection showing that three thiol isomerases coordinately catalyze the unfolding of the C-terminal arm of VP1, the major coat protein of the virus, to facilitate infection.
Another important question that remains unexplained is how thiol isomerases are released by platelets and endothelium. There is limited data to suggest that this may be through unconventional protein secretion routes, but the protein machinery involved in this, and whether there are separate storage granules for PDI and the other vascular thiol isomerases, remains unresolved. Understanding how and under what conditions thiol isomerases are released is central to understanding their function, development of thiol isomerase-targeted antithrombotic therapy, and their potential utility as biomarkers in various prothrombotic conditions. Furthermore, identifying the whole range of substrates of thiol isomerases and the sequence in which these undergo oxidoreduction during thrombus formation will be critical in understanding activation pathways that rely on disulfide bond rearrangements.
7. Five-year view
The next five years will see a major development in both basic and clinical elements in the role of thiol isomerases in hemostasis and thrombosis. With development of biochemical techniques to identify and evaluate substrates of thiol isomerases, other substrates of PDI as well as substrates of other vascular thiol isomerases will be identified and characterized. Simultaneously, completion of original clinical trials of PDI inhibitors will lay the ground for development and testing of other inhibitors of vascular thiol isomerases. Lastly, disease states associated with elevated circulating PDI and other vascular thiol isomerases will be identified and its utility as a biomarker evaluated.
Figure 4. Vitronectin is a substrate of PDI.
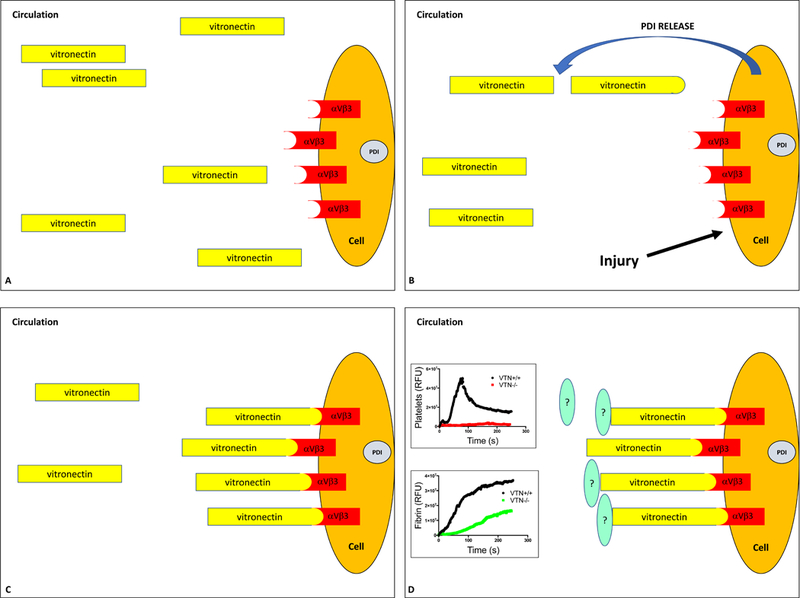
A.Vitronectin, a major plasma protein, does not bind to its receptors, endothelial ∝vβ3 and platelet ∝IIbβ3, under normal physiologic conditions. B. Vascular injury results in endothelial activation and release of PDI at the site of injury. Extracellular PDI reduces one or two disulfide bonds in vitronectin resulting in a structural change. C and D. ‘Activated’ vitronectin can now engage with its receptors ∝vβ3 and ∝IIbβ3 to support platelet aggregation and fibrin generation. The graphs show impaired platelet accumulation and fibrin generation at the site of vessel wall injury in the laser injury model of thrombus formation in vitronectin−/− mice. The downstream substrates of vitronectin in thrombus formation remain elusive.
8. Key Issues.
PDI family of thiol isomerases are ER resident proteins that also play an important role in a variety of extracellular processes
The critical role of PDI in initiation of thrombus formation is best characterized
PDI appears within seconds at the site of vessel wall injury and precedes platelet-fibrin thrombus formation; Inhibition of PDI activity in the vasculature inhibits thrombus formation
Vitronectin is a specific extracelluar substrate of PDI. Reduction of vitronectin by PDI enhances its binding to endothelial ∝vβ3 integrin supporting thrombus formation.
ERp5, ERp57 and ERp72 also support thrombus formation but their extracellular substrates have not been identified
Acknowledgments
Funding
This work was supported by grants from the National Institutes of Health (U54 HL112302; R01 HL136394).
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1.Ellgaard L, Ruddock LW. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 2005; 6:28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qin J, Clore GM, Gronenborn AM. The high-resolution three-dimensional solution structures of the oxidized and reduced states of human thioredoxin. Structure. 1994;2:503–22. [DOI] [PubMed] [Google Scholar]
- 3.McArthur AG, Knodler LA, Silberman JD, et al. The evolutionary origins of eukaryotic protein disulfide isomerase domains: new evidence from the Amitochondriate protist Giardia lamblia. Mol Biol Evol. 2001;18:1455–63. [DOI] [PubMed] [Google Scholar]
- 4.Zhou J, Wu Y, Wang L, et al. The C-terminal CGHC motif of protein disulfide isomerase supports thrombosis. J Clin Invest. 2015125:4391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tian G, Xiang S, Noiva R, et al. The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell. 2006;124:61–73. [DOI] [PubMed] [Google Scholar]
- 6.Song JL, Wang CC. Chaperone-like activity of protein disulfide-isomerase in the refolding of rhodanese. Eur J Biochem. 1995;231:312–6.. [DOI] [PubMed] [Google Scholar]
- 7.Yao Y, Zhou Y, Wang C. Both the isomerase and chaperone activities of protein disulfide isomerase are required for the reactivation of reduced and denatured acidic phospholipase A2. EMBO J. 1997;16:651–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sliskovic I, Raturi A, Mutus B. Characterization of the S-denitrosation activity of protein disulfide isomerase. J Biol Chem. 2005;280:8733–41. [DOI] [PubMed] [Google Scholar]
- 9.Zai A, Rudd MA, Scribner AW, et al. Cell-surface protein disulfide isomerase catalyzes transnitrosation and regulates intracellular transfer of nitric oxide. J Clin Invest. 1999;103:393–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho J, Furie BC, Coughlin SR, et al. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest. 2008;118:1123–31.*First demonstration of important role of extracellular PDI in inititiation of thrombus formation in a mouse model of thrombosis
- 11.Walczak CP, Tsai B. A PDI family network acts distinctly and coordinately with ERp29 to facilitate polyomavirus infection. J Virol. 2011;5:2386–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wetterau JR, Combs KA, McLean LR, et al. Protein disulfide isomerase appears necessary to maintain the catalytically active structure of the microsomal triglyceride transfer protein. Biochemistry. 1991;30:9728–35. [DOI] [PubMed] [Google Scholar]
- 13.Jiang XM, Fitzgerald M, Grant CM, et al. Redox control of exofacial protein thiols/disulfides by protein disulfide isomerase. J Biol Chem. 1999;274:2416–23. . [DOI] [PubMed] [Google Scholar]
- 14.Lindquist JA, Hammerling GJ, Trowsdale J. ER60/ERp57 forms disulfide-bonded intermediates with MHC class I heavy chain. FASEB J. 2001;15:1448–50. [DOI] [PubMed] [Google Scholar]
- 15.Kaiser BK, Yim D, Chow IT, et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature. 2007;447:482–6. [DOI] [PubMed] [Google Scholar]
- 16.Sela M, White FH Jr., Anfinsen CB Reductive cleavage of disulfide bridges in ribonuclease. Science. 1957;125:691–2. [DOI] [PubMed] [Google Scholar]
- 17.Goldberger RF, Epstein CJ, Anfinsen CB. Acceleration of reactivation of reduced bovine pancreatic ribonuclease by a microsomal system from rat liver. J Biol Chem. 1963;238:628–35. [PubMed] [Google Scholar]
- 18.Laboissiere MC, Sturley SL, Raines RT. The essential function of protein-disulfide isomerase is to unscramble non-native disulfide bonds. J Biol Chem. 1995;270:28006–9.. [DOI] [PubMed] [Google Scholar]
- 19.Appenzeller-Herzog C, Ellgaard L. The human PDI family: versatility packed into a single fold. Biochim Biophys Acta. 2008;1783:535–48 [DOI] [PubMed] [Google Scholar]
- 20.Frand AR, Kaiser CA. Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol Cell. 1999:469–77. [DOI] [PubMed] [Google Scholar]
- 21.Lundstrom J, Holmgren A. Determination of the reduction-oxidation potential of the thioredoxin-like domains of protein disulfide-isomerase from the equilibrium with glutathione and thioredoxin. Biochemistry. 1993;32:6649–55. [DOI] [PubMed] [Google Scholar]
- 22.Alon A, Heckler EJ, Thorpe C, et al. QSOX contains a pseudo-dimer of functional and degenerate sulfhydryl oxidase domains. FEBS Lett. 2010;584:1521–5. [DOI] [PubMed] [Google Scholar]
- 23.Milev Y, Essex DW. Protein disulfide isomerase catalyzes the formation of disulfide-linked complexes of thrombospondin-1 with thrombin-antithrombin III. Arch Biochem Biophys. 1999;361:120–6. [DOI] [PubMed] [Google Scholar]
- 24.Forster SJ, Freedman RB. Catalysis by protein disulphide-isomerase of the assembly of trimeric procollagen from procollagen polypeptide chains. Biosci Rep. 1984;4:223–9. [DOI] [PubMed] [Google Scholar]
- 25.Di Jeso B, Park YN, Ulianich L, et al. Mixed-disulfide folding intermediates between thyroglobulin and endoplasmic reticulum resident oxidoreductases ERp57 and protein disulfide isomerase. Mol Cell Biol. 2005;25:9793–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bulleid NJ, Freedman RB. Defective co-translational formation of disulphide bonds in protein disulphide-isomerase-deficient microsomes. Nature. 1988;335:649–51. [DOI] [PubMed] [Google Scholar]
- 27.LaMantia ML, Lennarz WJ. The essential function of yeast protein disulfide isomerase does not reside in its isomerase activity. Cell. 1993;74:899–908. [DOI] [PubMed] [Google Scholar]
- 28.Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–50.. [DOI] [PubMed] [Google Scholar]
- 29.Dai Y, Wang C. A mutant truncated protein disulfide isomerase with no chaperone activity. J Biol Chem. 1997;272:27572–6. [DOI] [PubMed] [Google Scholar]
- 30.Uehara T, Nakamura T, Yao D, et al. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–7. [DOI] [PubMed] [Google Scholar]
- 31.Shah CM, Bell SE, Locke IC, et al. Interactions between cell surface protein disulphide isomerase and S-nitrosoglutathione during nitric oxide delivery. Nitric Oxide. 2007;16:135–42. [DOI] [PubMed] [Google Scholar]
- 32.Bell SE, Shah CM, Gordge MP. Protein disulfide-isomerase mediates delivery of nitric oxide redox derivatives into platelets. Biochem J. 2007;403:283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jasuja R, Furie B, Furie BC. Endothelium-derived but not platelet-derived protein disulfide isomerase is required for thrombus formation in vivo. Blood. 2010;116:4665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thon JN, Peters CG, Machlus KR, et al. T granules in human platelets function in TLR9 organization and signaling. J Cell Biol. 2012;198:561–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaux D, Tooze J, Fuller S. Identification by anti-idiotype antibodies of an intracellular membrane protein that recognizes a mammalian endoplasmic reticulum retention signal. Nature. 1990;345:495–502. [DOI] [PubMed] [Google Scholar]
- 36.Wan SW, Lin CF, Lu YT, et al. Endothelial cell surface expression of protein disulfide isomerase activates beta1 and beta3 integrins and facilitates dengue virus infection. J Cell Biochem. 2012;113:1681–91. [DOI] [PubMed] [Google Scholar]
- 37.Araujo TL, Zeidler JD, Oliveira PV, et al. Protein disulfide isomerase externalization in endothelial cells follows classical and unconventional routes. Free Radic Biol Med. 2017;103:199–208 [DOI] [PubMed] [Google Scholar]
- 38.Marcus N, Shaffer D, Farrar P, et al. Tissue distribution of three members of the murine protein disulfide isomerase (PDI) family. Biochim Biophys Acta. 1996;1309:253–60. [DOI] [PubMed] [Google Scholar]
- 39.Gunther R, Srinivasan M, Haugejorden S, et al. Functional replacement of the Saccharomyces cerevisiae Trg1/Pdi1 protein by members of the mammalian protein disulfide isomerase family. J Biol Chem. 1993;268:7728–32. [PubMed] [Google Scholar]
- 40.High S, Lecomte FJ, Russell SJ, et al. Glycoprotein folding in the endoplasmic reticulum: a tale of three chaperones? FEBS Lett. 2000;476:38–41. [DOI] [PubMed] [Google Scholar]
- 41.Klappa P, Stromer T, Zimmermann R, et al. A pancreas-specific glycosylated protein disulphide-isomerase binds to misfolded proteins and peptides with an interaction inhibited by oestrogens. Eur J Biochem. 1998;254:63–9. [DOI] [PubMed] [Google Scholar]
- 42.Oliver JD, van der Wal FJ, Bulleid NJ, et al. Interaction of the thiol-dependent reductase ERp57 with nascent glycoproteins. Science. 1997;275:86–8. [DOI] [PubMed] [Google Scholar]
- 43.Garbi N, Tanaka S, Momburg F, et al. Impaired assembly of the major histocompatibility complex class I peptide-loading complex in mice deficient in the oxidoreductase ERp57. Nat Immunol. 2006;7:93–102. [DOI] [PubMed] [Google Scholar]
- 44.Holbrook LM, Watkins NA, Simmonds AD, et al. Platelets release novel thiol isomerase enzymes which are recruited to the cell surface following activation. Br J Haematol. 2010;148:627–37. [DOI] [PubMed] [Google Scholar]
- 45.Holbrook LM, Sasikumar P, Stanley RG, et al. The platelet-surface thiol isomerase enzyme ERp57 modulates platelet function. J Thromb Haemost. 2012;10:278–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu Y, Ahmad SS, Zhou J, et al. The disulfide isomerase ERp57 mediates platelet aggregation, hemostasis, and thrombosis. Blood. 2012;119:1737–46.*Demonstration of the role of ERp57 in thrombus formation
- 47.Kikuchi M, Doi E, Tsujimoto I, et al. Functional analysis of human P5, a protein disulfide isomerase homologue. J Biochem. 2002;132:451–5. [DOI] [PubMed] [Google Scholar]
- 48.Passam FH, Lin L, Gopal S, et al. Both platelet- and endothelial cell-derived ERp5 support thrombus formation in a laser-induced mouse model of thrombosis. Blood. 2015;125:2276–85.*Demonstration of the role of extracellular ERp5 in thrombus formation
- 49.Mazzarella RA, Srinivasan M, Haugejorden SM, et al. ERp72, an abundant luminal endoplasmic reticulum protein, contains three copies of the active site sequences of protein disulfide isomerase. J Biol Chem. 1990;265:1094–101. [PubMed] [Google Scholar]
- 50.Kozlov G, Maattanen P, Schrag JD, et al. Structure of the noncatalytic domains and global fold of the protein disulfide isomerase ERp72. Structure. 2009;17:651–9.. [DOI] [PubMed] [Google Scholar]
- 51.Kramer B, Ferrari DM, Klappa P, et al. Functional roles and efficiencies of the thioredoxin boxes of calcium-binding proteins 1 and 2 in protein folding. Biochem J. 2001;357:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen K, Lin Y, Detwiler TC. Protein disulfide isomerase activity is released by activated platelets. Blood. 1992;79:2226–8. [PubMed] [Google Scholar]
- 53.Essex DW, Chen K, Swiatkowska M. Localization of protein disulfide isomerase to the external surface of the platelet plasma membrane. Blood. 1995;86:2168–73.. [PubMed] [Google Scholar]
- 54.Chen K, Detwiler TC, Essex DW. Characterization of protein disulphide isomerase released from activated platelets. Br J Haematol. 1995;90:425–31. [DOI] [PubMed] [Google Scholar]
- 55.Essex DW, Miller A, Swiatkowska M, et al. Protein disulfide isomerase catalyzes the formation of disulfide-linked complexes of vitronectin with thrombin-antithrombin. Biochemistry. 1999;38:10398–405. [DOI] [PubMed] [Google Scholar]
- 56.Falati S, Gross P, Merrill-Skoloff G, et al. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med. 2002;8:1175–81.. [DOI] [PubMed] [Google Scholar]
- 57.Chen VM, Ahamed J, Versteeg HH, et al. Evidence for activation of tissue factor by an allosteric disulfide bond. Biochemistry. 2006;45:12020–8. [DOI] [PubMed] [Google Scholar]
- 58.Ahamed J, Versteeg HH, Kerver M, et al. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci U S A. 2006;103:13932–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reinhardt C, von Bruhl ML, Manukyan D, et al. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Invest. 2008;118:1110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim K, Hahm E, Li J, et al. Platelet protein disulfide isomerase is required for thrombus formation but not for hemostasis in mice. Blood. 2013;122:1052–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou J, May L, Liao P, et al. Inferior vena cava ligation rapidly induces tissue factor expression and venous thrombosis in rats. Arterioscler Thromb Vasc Biol. 2009;29:863–9. [DOI] [PubMed] [Google Scholar]
- 62.Vandendries ER, Hamilton JR, Coughlin SR, et al. Par4 is required for platelet thrombus propagation but not fibrin generation in a mouse model of thrombosis. Proc Natl Acad Sci U S A. 2007;104:288–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cho J, Kennedy DR, Lin L, et al. Protein disulfide isomerase capture during thrombus formation in vivo depends on the presence of beta3 integrins. Blood. 2012;120:647–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lahav J, Jurk K, Hess O, et al. Sustained integrin ligation involves extracellular free sulfhydryls and enzymatically catalyzed disulfide exchange. Blood. 2002. October 1;100(7):2472–8. doi: 10.1182/blood-2001-12-0339 PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 65.Lahav J, Wijnen EM, Hess O, et al. Enzymatically catalyzed disulfide exchange is required for platelet adhesion to collagen via integrin alpha2beta1. Blood. 2003;102:2085–92. [DOI] [PubMed] [Google Scholar]
- 66.Xiao T, Takagi J, Coller BS, et al. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mor-Cohen R, Rosenberg N, Landau M, et al. Specific cysteines in beta3 are involved in disulfide bond exchange-dependent and -independent activation of alphaIIbbeta3. J Biol Chem. 2008;283:19235–44. [DOI] [PubMed] [Google Scholar]
- 68.Mor-Cohen R, Rosenberg N, Einav Y, et al. Unique disulfide bonds in epidermal growth factor (EGF) domains of beta3 affect structure and function of alphaIIbbeta3 and alphavbeta3 integrins in different manner. J Biol Chem. 2012;287:8879–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O’Neill S, Robinson A, Deering A, et al. The platelet integrin alpha IIbbeta 3 has an endogenous thiol isomerase activity. J Biol Chem. 2000;275:36984–90. [DOI] [PubMed] [Google Scholar]
- 70.Ye F, Kim C, Ginsberg MH. Reconstruction of integrin activation. Blood. 2012;119:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morrissey JH, Choi SH, Smith SA. Polyphosphate: an ancient molecule that links platelets, coagulation, and inflammation. Blood. 2012;119:5972–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharda A, Kim SH, Jasuja R, et al. Defective PDI release from platelets and endothelial cells impairs thrombus formation in Hermansky-Pudlak syndrome. Blood. 2015;125:1633–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wynn R, Cocco MJ, Richards FM. Mixed disulfide intermediates during the reduction of disulfides by Escherichia coli thioredoxin. Biochemistry. 1995;34:11807–13. [DOI] [PubMed] [Google Scholar]
- 74.Qin J, Clore GM, Kennedy WM, et al. Solution structure of human thioredoxin in a mixed disulfide intermediate complex with its target peptide from the transcription factor NF kappa B. Structure. 1995;3:289–97. [DOI] [PubMed] [Google Scholar]
- 75.Bowley SR, Fang C, Merrill-Skoloff G, et al. Protein disulfide isomerase secretion following vascular injury initiates a regulatory pathway for thrombus formation. Nat Commun. 2017;8:14151.**Identification of vitronectin as a redox substrate of PDI in its regulatory role in thrombus formation.
- 76.Barnes DW, Silnutzer J, See C, et al. Characterization of human serum spreading factor with monoclonal antibody. Proc Natl Acad Sci U S A. 1983;80:1362–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Preissner KT, Reuning U. Vitronectin in vascular context: facets of a multitalented matricellular protein. Semin Thromb Hemost. 2011;37:408–24. [DOI] [PubMed] [Google Scholar]
- 78.Seiffert D, Smith JW. The cell adhesion domain in plasma vitronectin is cryptic. J Biol Chem. 1997;272:13705–10. [DOI] [PubMed] [Google Scholar]
- 79.Reheman A, Gross P, Yang H, et al. Vitronectin stabilizes thrombi and vessel occlusion but plays a dual role in platelet aggregation. J Thromb Haemost. 2005;3:875–83.. [DOI] [PubMed] [Google Scholar]
- 80.Stopa JD, Baker KM, Grover SP, et al. Kinetic-based trapping by intervening sequence variants of the active sites of protein-disulfide isomerase identifies platelet protein substrates. J Biol Chem. 2017;292:9063–74.*Identification of other putative extracellular substrates of PDI
- 81.Sambrano GR, Huang W, Faruqi T, et al. Cathepsin G activates protease-activated receptor-4 in human platelets. J Biol Chem. 2000;275:6819–23. [DOI] [PubMed] [Google Scholar]
- 82.Korkmaz B, Moreau T, Gauthier F. Neutrophil elastase, proteinase 3 and cathepsin G: physicochemical properties, activity and physiopathological functions. Biochimie. 2008;90:227–42. [DOI] [PubMed] [Google Scholar]
- 83.Stopa JD, Neuberg D, Puligandla M, et al. Protein disulfide isomerase inhibition blocks thrombin generation in humans by interfering with platelet factor V activation. JCI Insight. 2017;2:e89373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zucker M, Seligsohn U, Yeheskel A, et al. An allosteric disulfide bond is involved in enhanced activation of factor XI by protein disulfide isomerase. J Thromb Haemost. 2016;14:2202–2211. [DOI] [PubMed] [Google Scholar]
- 85.Danishefsky KJ, Alexander RJ, Detwiler TC. Formation of a stable complex of thrombin and the secreted platelet protein glycoprotein G (thrombin-sensitive protein, thrombospondin) by thiol-disulfide exchange. Biochemistry. 1984;23:4984–90. [DOI] [PubMed] [Google Scholar]
- 86.Popescu NI, Lupu C, Lupu F. Extracellular protein disulfide isomerase regulates coagulation on endothelial cells through modulation of phosphatidylserine exposure. Blood. 2010;116:993–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Furlan-Freguia C, Marchese P, Gruber A, et al. P2X7 receptor signaling contributes to tissue factor-dependent thrombosis in mice. J Clin Invest. 2011;121:2932–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schulman S, Bendapudi P, Sharda A, et al. Extracellular Thiol Isomerases and Their Role in Thrombus Formation. Antioxid Redox Signal. 2016;24:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang L, Wu Y, Zhou J, et al. Platelet-derived ERp57 mediates platelet incorporation into a growing thrombus by regulation of the alphaIIbbeta3 integrin. Blood. 2013;122:3642–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhou J, Wu Y, Wang L, et al. The disulfide isomerase ERp57 is required for fibrin deposition in vivo. J Thromb Haemost. 2014;12:1890–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jordan PA, Stevens JM, Hubbard GP, et al. A role for the thiol isomerase protein ERP5 in platelet function. Blood. 2005;105:1500–7. [DOI] [PubMed] [Google Scholar]
- 92.Zhou J, Wu Y, Chen F, et al. The disulfide isomerase ERp72 supports arterial thrombosis in mice. Blood. 2017;130:817–828*Extracellular ERp72 supports thrombus formation
- 93.Holbrook L, Sandhar GK, Sasikumar P, et al. A humanized monoclonal antibody that inhibits platelet-surface ERp72 reveals a role for ERp72 in thrombosis. J Thromb Haemost. 2017. 16:367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jasuja R, Passam FH, Kennedy DR, et al. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J Clin Invest. 2012;122:2104–13.**Identification of flavanoids as inhibitors of PDI activity; Preclinical study of rutin as an antithrombotic.
- 95.Geleijnse JM, Launer LJ, Van der Kuip DA, et al. Inverse association of tea and flavonoid intakes with incident myocardial infarction: the Rotterdam Study. Am J Clin Nutr. 2002;75:880–6.. [DOI] [PubMed] [Google Scholar]
- 96.McCullough ML, Peterson JJ, Patel R, et al. Flavonoid intake and cardiovascular disease mortality in a prospective cohort of US adults. Am J Clin Nutr. 2012;95:454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Flaumenhaft R, Furie B, Zwicker JI. Therapeutic implications of protein disulfide isomerase inhibition in thrombotic disease. Arterioscler Thromb Vasc Biol. 2015;35:16–23.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lin L, Gopal S, Sharda A, et al. Quercetin-3-rutinoside Inhibits Protein Disulfide Isomerase by Binding to Its b’x Domain. J Biol Chem. 2015;290:23543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bekendam RH, Bendapudi PK, Lin L, et al. A substrate-driven allosteric switch that enhances PDI catalytic activity. Nat Commun. 2016;7:12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mandel R, Ryser HJ, Ghani F, et al. Inhibition of a reductive function of the plasma membrane by bacitracin and antibodies against protein disulfide-isomerase. Proc Natl Acad Sci U S A. 1993;90:4112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Khodier C, VerPlank L, Nag PP, et al. Identification of ML359 as a Small Molecule Inhibitor of Protein Disulfide Isomerase. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD) 2010. [PubMed] [Google Scholar]
- 102.Xu S, Sankar S, Neamati N. Protein disulfide isomerase: a promising target for cancer therapy. Drug Discov Today. 2014;19:222–40. [DOI] [PubMed] [Google Scholar]
- 103.Voigtlaender M, Holstein K, Spath B, et al. Expression and release of platelet protein disulphide isomerase in patients with haemophilia A. Haemophilia. 2016;22:e537–e544. [DOI] [PubMed] [Google Scholar]
- 104.Nagayama S, Yokoi T, Tanaka H, et al. Occurrence of autoantibody to protein disulfide isomerase in patients with hepatic disorder. J Toxicol Sci. 1994;19:163–9. [DOI] [PubMed] [Google Scholar]
- 105.Caorsi C, Niccolai E, Capello M, et al. Protein disulfide isomerase A3-specific Th1 effector cells infiltrate colon cancer tissue of patients with circulating anti-protein disulfide isomerase A3 autoantibodies. Transl Res. 2016;171:17–28 e1–2. [DOI] [PubMed] [Google Scholar]
- 106.Weichsel A, Gasdaska JR, Powis G, et al. Crystal structures of reduced, oxidized, and mutated human thioredoxins: evidence for a regulatory homodimer. Structure. 1996. 15;4:735–51.. [DOI] [PubMed] [Google Scholar]
- 107.Wang C, Li W, Ren J, et al. Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid Redox Signal. 2013;19:36–45. [DOI] [PubMed] [Google Scholar]