

Abstract
Circulating Tumor Cells (CTCs) are commonly detected in the systemic blood of cancer patients with metastatic tumors. However, the mechanisms controlling the viability of cancer cells in blood and length of time spent in circulation, as well as their potential for generating additional tumors are still undefined. Here, it is demonstrated that CX3CR1, a chemokine receptor, drives reseeding of breast CTCs to multiple organs. Antagonizing this receptor dramatically impairs the progression of breast cancer cells in a relevant model of human metastatic disease, by affecting both tumor growth and numerical expansion. Notably, therapeutic targeting of CX3CR1 prolongs CTC permanence in the blood, both promoting their spontaneous demise by apoptosis and counteracting metastatic reseeding. These effects lead to containment of metastatic progression and extended survival. Finally, targeting CX3CR1 improves blood exposure of CTCs to doxorubicin and in combination with docetaxel shows synergistic effects in containing overall tumor burden.
INTRODUCTION
The seeding of CTCs departing from primary tumors, such as breast adenocarcinoma, in distant organs is a necessary event for the emergence of metastatic disease. Secondary tumors are by far the major cause of death for patients, but motivation and efforts to avert their occurrence have been historically thwarted. This is likely due to the long-held belief that distant dissemination predominantly occurs prior the detection of the primary tumor. Indeed, early spreading from primary site, prolonged dormancy and return to a proliferative state characterize a commonly accepted paradigm for cancer cells. However, an additional scenario in which cancer cells depart from established metastases to seed either new lesions (reseeding) or existing lesions (cross-seeding) (1) (2) (3) has been recently confirmed (4) (5). Despite the fact that metastatic expansion by reseeding likely hastens clinical progression towards a lethal outcome (1) measures to prevent or contains this occurrence are only minimally pursued in the clinic (6) (7). In part, this is due to the paucity of information concerning (1) the longevity of cancer cells in the blood, (2) the mechanistic basis and chronological dynamics of their reseeding and (3) uncertainty about the true potential of CTCs to initiate new metastatic lesions after being shed from secondary tumors (8).
This pre-clinical study was pursued to gain a better understanding of the cellular dynamics and molecular underpinning of metastatic reseeding. Combining this new knowledge with effective therapeutics for counteracting tumor spreading from existing metastases, will improve management of cancer patients by increasing the likelihood of either stabilizing early metastatic disease or delaying further widespread dissemination of multiple secondary lesions.
We have previously shown that CX3CR1, the receptor for the chemokine Fractalkine, plays an instrumental role in guiding breast CTCs to the skeleton (9). Recently, we generated JMS-17–2, a potent small-molecule antagonist for this receptor, which impaired the homing of breast CTCs to the skeleton and reduced tumor burden in pre-clinical models of metastatic disease (10). Here we employed FX-68, an improved CX3CR1 antagonist, to examine the timing and kinetics of breast CTCs re-entry and disappearance from circulation as well as to ascertain how counteracting reseeding affects CTCs viability, regulates the emergence of new lesions and impacts overall survival. Finally, we asked whether combining CX3CR1 antagonists with doxorubicin would affect metastatic progression and exposure of CTCs to chemotherapeutic drugs in the blood.
MATERIALS AND METHODS
Cell lines and cell cultures
MDA-MB-231 (MDA-231) human and 4T-1 murine breast cancer cell lines were purchased from ATCC and cultured in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen) or RPMI-1640, respectively, supplemented with 10% fetal bovine serum (Hyclone) and 0.1% gentamicin (Invitrogen). All cell lines were cultured at 37° C and 5% CO2. Starting from the original vials from ATCC, each cell line was expanded and frozen in different aliquots that were used for not more than 10 passages and not longer than 2 months following resuscitation. Control for Mycoplasma contamination and cell authentication by Single Tandem Repeat were performed by IDEXX Radil. Each cell line was genetically engineered to stably express Green Fluorescent Protein (GFP) by transduction with a pLenti CMV Blast vector (Addgene) in DMEM for 24 hours.
SDS-PAGE and Western Blotting
Cell lysates were collected with RIPA lysis and extraction buffer (#89900 Thermo Fisher) containing a phosphatase inhibitor cocktail (Calbiochem), protease inhibitor cocktail (Calbiochem), 10% glycerol and 0.5 M EDTA (Thermo Fisher). A BCA protein assay (Pierce) was used to determine protein concentrations and 50 μg of proteins were loaded onto 10% polyacrylamide gels and then transferred onto Immobilon PVDF membranes (Millipore Corporation). Membranes were blocked for one hour at room temperature with 0.1% Tween-20/TBS with 5% (w/v) powdered milk. CX3CR1 was detected using a primary antibody (AB8021, Abcam) used at 0.5μg/ml, diluted in 0.1% Tween-20, 5% dry milk in TBS and incubated overnight at 4°C. A secondary, HRP-conjugated antibody (Pierce) was used at 10ng/ml. Blotted membranes were processed with SuperSignal Femto chemiluminescence substrates (Pierce) and visualized using a FluorChem imaging system (ProteinSimple).
For quantitative Western blotting, membranes were blocked for one hour at room temperature with Odyssey Blocking Buffer (P/N 927–50000 LI-COR Biosciences; Lincoln, NE). The activation of the ERK signaling pathway was detected with rabbit phospho-ERK and mouse total-ERK antibodies (Cell Signaling Technology) used at 10ng/ml. Both primary antibodies were combined in the Odyssey Blocking Buffer with 0.2% Tween-20 and incubated overnight at 4°C. Two-color detection was achieved by multiplexing the IRDye 800CW Goat anti-Rabbit IgG (1:15000 P/N 925–32211 LI-COR Biosciences) and IRDye 680RD Goat anti-Mouse IgG (1:15000 P/N 925–68070 LI-COR Biosciences; Lincoln, NE) secondary antibodies in the Odyssey Blocking Buffer with 0.2% Tween-20 and 0.01% SDS. Blotted membranes were visualized using the Odyssey Fc Imaging System (LI-COR Biosciences) and densitometry analysis was performed using the Image Studio software (v. 5.2)
Pharmacokinetic studies for FX-68
The FX-68 small-molecule compound is a newly synthesized antagonist of CX3CR1, (US Provisional Patent Application No. 62/500,134) that showed high selectivity towards 33 plasma membrane receptors (CEREP 44 screen, outsourced to Eurofins Pharma (www.eurofins.com) and with 55.3% inhibition of adrenergic α1A and 56.1% inhibition of dopaminergic D2b receptors only at concentrations equal or higher than 1μM. FX-68 also tested negative for inhibition of 364 different kinases in an assay outsourced to Reaction Biology (www.reactionbiology.com). The functional IC50 for FX-68 was established by evaluating the inhibition of MAPK activation in cancer cells stimulated with 50nM Fractalkine, as assessed by quantitative Western Blotting (see methods section). We tested eight different concentrations of FX-68, ranging from 0.05nM to 100nM, in three different experiments; the percentages of inhibition for each dose were plotted using Prism v5.0 (GraphPad Software) and the IC50 was calculated as 4.4 nM (Suppl. Fig. 1).
For pharmacokinetic studies, mice were administered via intraperitoneal (i.p.) route with 10mg/kg of FX-68 in 10% dimethylacetamide (DMAC), 10% tetraethylene glycol and 10% Solutol HS15 in sterile ddH2O. Animals were then anesthetized as described above and 300μl of blood samples were collected by cardiac puncture at the designated time points and transferred in K2EDTA tubes. Blood samples were placed on ice and tested after dilution. The measurement of FX-68 concentrations in blood was outsourced to Alliance Pharma (www.alliancepharmaco.com). The results showed that this compound reached a plasma concentration of ~200ng/ml one hour after administration, which declined to 60ng/ml at six hours, corresponding to 460nM and 140nM, respectively (FX-68 m.w.= 434.90). These concentrations are several folds higher than the IC50 measured in vitro for this compound but far below the 1μM concentration at which FX-68 showed still very high selectivity towards 33 plasma membrane receptors and lack of inhibitory activity on 364 different kinases (see above).
Animal models of metastasis
Female SCID mice (Taconic CB17-SCRF, ~ 20g body weight) were housed in a germ-free barrier. At 6 weeks of age, mice were anesthetized with a mixture of ketamine (80mg/kg) and xylazine (10mg/kg). Tumor cells (2.5×104 MDA-231 or 5×103 4T-1) were delivered as a 100 μl suspension of serum-free culture medium by carefully accessing the left ventricle of the heart through the chest with an insulin syringe mounting a 30-gauge needle. After intracardiac injection, the experimenter distributed each animal in unlabeled cages. Each cage was randomly assigned to different treatment groups by a second experimenter.
Model of tumor seeding.
Animals were treated i.p. with FX-68 dissolved in 4% DMSO, 4% Cremophor EL (Kolliphor EL, Sigma-Aldrich, MO) in sterile PBS or vehicle one-hour prior and three hours after being injected with cancer cells. The dosing regimen was selected based on results from pharmacokinetic analyses. Mice were sacrificed 24 hours post-injection. Validation of the intracardiac delivery of cancer cells was performed as previously described (10). For the detection and enumeration of single cancer cells, the experimenter was not blinded but was assisted by the Nuance FX multiplex imaging software (see below). A fluorescence image was acquired for each tissue section analyzed, approximately 36 serial sections spanning the entire width of each knee-joint. The identification and enumeration of single cells was obtained by three different experimenters, including the Principal Investigator, and occasional discrepancies reconciled prior to compiling the final data. All cells detected in both knew-joints for each animal on the study were averaged and expressed as DTCs in the respective figure legends.
Model of metastatic disease.
One week after intracardiac injection, animals were randomly assigned to control and treated group and then imaged for tumors in the skeleton and soft-tissue organs. Vehicle or FX-68 (10mg/kg) were administered i.p. twice/day, for the entire duration of the study and animals were imaged weekly. Doxorubicin was purchased from LEK Laboratories (D5794), dissolved in PBS at 1mg/ml and stored at −20 °C before use.
All experiments were performed in agreement with NIH guidelines for the humane use of animals. The use of animals for this study was approved by the Institutional Animal Care and Use Committee (IACUC) at Drexel University College of Medicine Committee and by the Animal Care and Use Review Office (ACURO) at the U.S. Army Medical Research and Materiel Command.
Labeling of cancer cells with CM-Dil
The CellTracker™ CM-Dil (ThermoFisher Scientific) was diluted as 50μg in 50 μl of DMSO to prepare a stock solution. Immediately before use, the stock solution was diluted with D-PBS to a 1μM working solution. Cells were exposed to the working solution at 37° C for 5 minutes and then at 4° C for 15 minutes and then washed with PBS followed by F-12 cell culture medium.
In vivo bioluminescence Imaging
MDA-231 cells were stably transduced with the pLeGo-IG2-Luc2 vector. Prior to imaging, mice were injected i.p. with 150 mg/kg of D-luciferin (ViVoGlo, Promega) and anesthetized using 3% isoflurane. Animals were then transferred to the chamber of an IVIS Lumina XR (PerkinElmer) where they received 2% isoflurane throughout the image acquisition. Fifteen minutes after injection of the substrate, five-minute exposures of both dorsal and ventral views were obtained and quantification and analysis of bioluminescence was performed using the Living Image software.
Processing of animal tissues.
Bones and soft-tissue organs were collected and fixed in 4% paraformaldehyde solution (Electron Microscopy Sciences) for 24 hours and then transferred into fresh formaldehyde for additional 24 hours. Soft-tissue organs were then placed either in 30% sucrose for cryoprotection or 1% paraformaldehyde for long-term storage. Bones were decalcified in 0.5M EDTA (Fisher Scientific) for 7 days followed by incubation in 30% sucrose. Tissues were maintained at 4°C for all aforementioned steps and frozen in O.C.T. medium (Sakura Finetek) by placement over dry-ice chilled 2-methyl butane. Soft-tissue organs as well as knee joints (femora and tibiae) were processed to obtain serial frozen sections, 80¼m in thickness, using a Microm HM550 cryostat (11). All sections spanning the entire bone width of femur and tibia were inspected to obtain either accurate enumeration of DTCs or visualization and measurement of tumor foci in the inoculated animals.
Fluorescence microscopy and morphometric analysis of metastases
Fluorescent images of tumor cells and skeletal metastases were acquired using an Axio Scope.A1 microscope (Zeiss) connected to a Nuance Multispectral Imaging System (PerkinElmer). Digital images were analyzed and processed using the Nuance Software (v. 2.4). Microscope and software calibration for size measurement is regularly performed using a TS-M2 stage micrometer (Oplenic Optronics) (12)
Enumeration and collection of CTCs from mouse blood
We use blood withdrawn by cardiac puncture, as this route has been previously shown to be the most effective and reliable among all alternatives (13). AMD-3100 was purchased from Selleckchem (S8030) dissolved in PBS at 1mg/ml and at −20 °C before use. Tumor-bearing animals were anesthetized as described above and 200–250μl of systemic blood was collected at the designated time points and transferred in K2EDTA tubes. Blood samples were placed on ice, diluted using a saline buffer (0.5% BSA, 2% EDTA in PBS) at a 1:8 ratio and then filtered through a 70μm cell strainer immediately prior to achieve a label-free, microfluidic isolation of CTCs. For enumeration, we used the Captor® system (Clearbridge Biomedics, Singapore), which allows visualization of individual CTCs trapped into crescent-shape microwells (14). Following complete flowing of the diluted blood, the CTChip® was transferred to the stage of an inverted fluorescent microscope and all trapped CTCs were counted. For the recovery of fully viable CTCs we used the ClearCell® FX system (Clearbridge Biomedics, Singapore), which employs the CTChip® FR1 to isolate cancer cells from blood cells based on size, deformability and inertia through a Dean Flow Fractionation process (15).
Collection of CTCs from human blood and immunofluorescence staining.
Breast cancer patients were enrolled in a prospective biomarker study. All subjects gave informed consent; the study was approved by the Institutional Review Board at Feinberg School of Medicine. The clinical trial enrolled patients with stage III and IV disease irrespective of subtype, about to start next systemic therapy. Blood form a breast cancer patient diagnosed with ER+/PR+/HER2− Intraductal Carcinoma (IDC), staging T4N1M1, was first processed for enrichment using the Parsortix™ system (Angle plc, Surrey, UK) and then CTCs were collected using the Captor® system (Clearbridge Biomedics, Singapore). Detection of CX3CR1 was achieved by on-chip immunofluorescence staining using a primary antibody (ab8021, 1ng/ml) from Abcam (Cambridge, MA) followed by Tyramide Signal Amplification with AlexaFluor™ 594 (B40944, ThermoFisher Scientific, Waltham, MA). For fluorescence microscopy and imaging we used an EVOS FL system (ThermoFisher Scientific, Waltham, MA).
qRT-PCR for viability assessment of CTCs
CTCs were lysed on the same microfluidic chip used for capture using buffer RLT and total RNA was isolated using an RNeasy Mini Kit (Qiagen). CTCs were collected from one mouse - for each time point and treatment - for RNA isolation and qRT-PCR (two mice were pooled together if necessary to recover enough RNA). TaqMan RNA-to-Ct 1-step kit (cat. 4392653) was used with an Applied Biosystem 7900HT Fast Real-Time PCR system. TaqMan Gene Expression Assays were purchased from ThermoFisher Scientific for BAX (Assay ID Hs00180269_m1) and Bcl2 (Assay ID Hs00608023_m1).
Statistical analysis
One-way Anova with either Dunnett’s or Newman-Keuls post-tests was used to compare multiple experimental groups. Student’s t-test with Welch’s correction (GraphPad Prism 5.0) was used to compare two experimental groups assuming no equal variance. Log-rank (Mantel-Cox) test was used for comparison of survival curves.
RESULTS
Targeting CX3CR1 reduces tumor seeding and progression, and extends survival.
We initially sought to validate our novel compound FX-68 for its ability to interfere with the seeding of circulating breast cancer cells by antagonizing CX3CR1. To this end, human (MDA-MB-231) or murine (4T-1) breast cancer cells – both expressing CX3CR1 when compared to THP-1 human monocytic cell line as control (Fig. 1A, top and (10)) were grafted via the left cardiac ventricle into immuno-compromised (SCID) or immuno-competent (BALB-c) mice, respectively, to allow unbiased tumor spreading to different organs (16). Animals were administered with FX-68 (10mg/kg) or vehicle via intraperitoneal route (i.p.) one hour before and 3 hours after the inoculation of cancer cells, based on previously conducted pharmacokinetic studies and as described before (10). The effects of FX-68 confirmed the results previously shown by the closely related compound JMS-17–2 (10). Treatment with FX-68 severely impaired both MDA-231 and 4T-1 cell types from seeding the skeleton in treated animals, as shown by the dramatic reduction in the total number of Disseminated Tumor Cells (DTCs) detected in distal femur and proximal tibia of both knee-joints examined for each animal in the study (Fig. 1A,B). Interestingly, when the same experiment was conducted by administering animals with AMD-3100, a well characterized and widely used small-molecule inhibitor of the chemokine receptor CXCR4 (17), the seeding of breast cancer cells was unaffected (Fig.1B). In animals grafted with MDA-231 cells as described above, small tumors were detected by bioluminescence imaging in the skeleton and soft-tissues at one-week post-inoculation, providing a valuable pre-clinical model of early metastatic disease (Fig.1C). Following randomization of inoculated animals into control and treated groups, we found that targeting CX3CR1 both limited the progressive expansion in tumor numbers typically observed in untreated animals (Fig. 1D) and contained the overall tumor burden below 50% of the levels assessed in the control group (Fig.1E), an effect that we have previously reported as likely due to the inhibition of a set of CX3CR1-regulated tumor-associated genes and signaling pathways (10). Notably, these effects translated into a dramatic extension of overall survival induced by the CX3CR1 antagonist as compared to control animals treated with vehicle (Fig.1F). It is important to emphasize that the effects observed with FX-68 are matching - for temporal kinetic as well as extent and type tumor inhibition – previous results we obtained either administering JMS-17–2 to tumor-bearing animals or inhibiting CX3CR1 expression by CRISPRi (10). Taken together, these observations further corroborated the implication of the chemokine receptor CX3CR1 in the metastatic behavior of breast cancer cells, and most importantly indicate that the numerical expansion of early tumors into additional metastatic sites could be effectively counteracted by targeting this chemokine receptor with pharmacological means.
Figure 1. Impaired metastatic seeding and progression by pharmacological antagonism of CX3CR1.
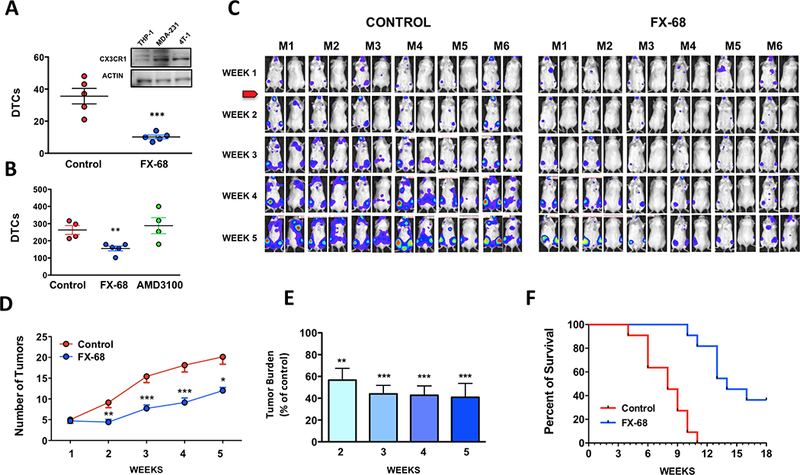
(A) Murine 4T-1 and (B) human MDA-231 breast cancer cells, both expressing CX3CR1 (a, inset) were significantly impaired in seeding the skeleton of mice treated with FX-68. In contrast, the CXCR4 antagonists AMD-3100 had no effect on tumor seeding in similar experiments (** P=0.0058; *** P=0.0009, unpaired Student’s t-test). (C) Mice were inoculated with MDA-231 cells, monitored weekly by bioluminescence imaging and allowed to develop small tumors for one week, prior to administration of either vehicle or FX-68 for the entire duration of the experiment. (D) The number of tumors and (E) total tumor burden in FX-68 treated animals were both strongly reduced by the CX3CR1 antagonist as compared to controls (7 mice /group; Tumor numbers: *P=0.014, **P=0.009, ***P=0.002, paired t-test; tumor burden: ***P= 0.0002, One-way Anova with Dunnett’s post-test. (F) Kaplan-Meier curve showing increase in overall survival with FX-68 treatment (11 mice/group, Log-rank test: χ2 = 19.95, P<0.0001).
Number of CTCs in blood and their viability are affected by CX3CR1 antagonist.
Our next aim was to determine whether targeting CX3CR1 contained the emergence of additional tumors by directly preventing CTCs from reseeding skeleton and soft-tissues. It is widely recognized that tracking the movements and fate of CTCs is inherently challenging. Cancer cells in the blood are a dynamic pool that constantly fluctuates as cells enter the circulation upon being dislodged from tumors, reseed additional sites or perish to anoikis if they fail to do so (18). Indeed, determining the time spent by cancer cells in the blood has been characteristically problematic, both in humans (19,20) and animals (21) . The potential for CTCs dislodged from existing metastatic lesions to spread to other organ sites was first inferred by mathematical models (22) and then recently revealed by genetic studies in humans (4) (5); however, hard evidence from cell-tracking experiments has been lacking. Understandably, a major hurdle for these types of studies is the limited ability to monitor the timing of CTCs’ entry into and egress from the blood during steady-state conditions. To circumvent these issues, we proceeded by acutely mobilizing cancer cells from tumors in the skeleton and soft-tissues in mice – above steady-state counts – and then thereafter sampling the blood at specific intervals, reasoning that a time-dependent decline of the number of CTCs in the blood would result from a combination of cancer cells seeding back to tissues and dying in circulation. Mice were grafted via the left cardiac ventricle with MDA-231 cells to generate unbiased tumor spreading and reproduce the clinical scenario of metastatic disease; after three weeks animals developed large tumors in skeleton and soft-tissues (refer to Fig.1) and were treated with a single dose (5mg/kg) of AMD3100 (23), an antagonist of the CXCR4 receptor (24). This compound mobilizes hematopoietic stem cells (25) and multiple myeloma cells (26) from the bone marrow of humans and mice (23) and has been also used to dislodge DTCs from the skeleton of mice (27). Following AMD-3100, we isolated CTCs from the blood of tumor-bearing mice and found that administration of the CXCR4 antagonist induced a rapid increase in the number of CTCs. The number of CTCs post-treatment peaked at 3 hours and subsequently declined, and after 48 hrs returned to the steady-state counts routinely observed in untreated animals (Fig. 2A,C). In contrast to AMD-3100, FX-68 does not mobilize cancer cells back in the blood (Fig.2B). We surmised that by targeting CX3CR1 this compound would hold CTCs longer in circulation by preventing reseeding to the skeleton and possibly soft-tissues. Indeed, when FX-68 was co-administered with AMD-3100, and then alone twice/day thereafter (Fig.2D), animals showed a rapid increase in CTCs counts that occurred at the same time-point as with AMD-3100 alone, but of a magnitude more than two-fold higher than without the CX3CR1 antagonist. Furthermore, return of CTC counts to steady-state levels was clearly prolonged when FX-68 was combined with AMD-3100 (AUC=731) as compared to AMD-3100 alone (AUC=345) (Fig.2C), suggesting that targeting CX3CR1 delays the egress of CTCs from the blood circulation.
Figure 2. CX3CR1 antagonism retains CTCs in the blood and promotes apoptosis.
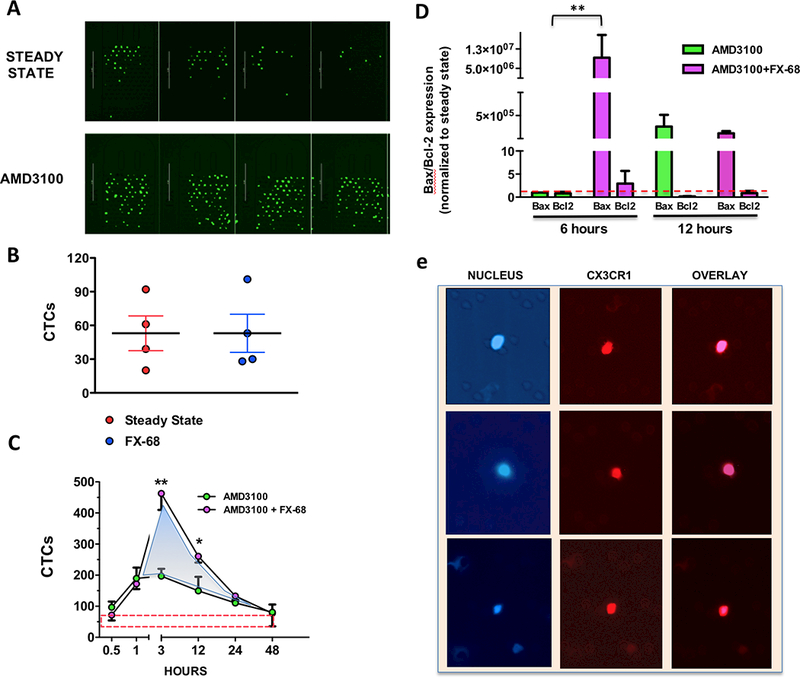
(A) Fluorescent CTCs collected on a microfluidic chip of the Captor Instrument from the blood of mice bearing disseminated tumors. AMD-3100 forcefully mobilized additional CTCs above the steady-state levels in the absence of treatment. (B) The CX3CR1 antagonist FX-68 does not mobilize cancer cells back into circulation, as shown by comparable numbers of CTCs detected at steady-state and 3 hours after administration of FX-68 (C) CTCs enumerated at different time points following administration of AMD-3100 alone or combined with FX-68. The area under the curve was measured as 485 for AMD-3100 alone and 852 for AMD3100+FX-68, which equates to a 75% increase induced by the CX3CR1 antagonist (shaded area). The red-dotted box indicates the numerical range of CTCs detected at steady-state, i.e in the absence of any treatment (3 mice/group; *P=0.04 paired Student’s t-test; **P=0.02 One-way Anova with Dunnett’ post-test. (D) CTCs collected upon treatment with AMD3100 alone or AMD3100+FX-68 were collected at 6 hours and 12 hours (refer also to c) and levels of Bax and Bcl2 were measured as indication of the extent of apoptotic cells for each time-point and treatment. Bax expression was found to be dramatically increased at 6 hours when the re-seeding of CTCs was impaired by FX-68; at 12 hours the apoptotic fractions were comparable between CTCs mobilized by AMD-3100 in the presence or absence of FX-68. All results were normalized to Bax and Bcl2 expression measured in CTCs collected at steady state (red dotted line) **P=0.01 One-way Anova with Dunnett’ post-test. (e) Human CTCs collected from a patient with metastatic breast cancer, showing blue nuclear staining (DAPI) and red fluorescent staining for CX3CR1.
When spreading via the systemic circulation cancer cells become susceptible to anoikis (28,29) and extending the time spent in the blood should increase their odds of incurring into this form of apoptotic cell death. Thus, we speculated that counteracting reseeding by targeting CX3CR1 would also affect the viability of cancer cells. To test this hypothesis, CTCs were recovered from mice harboring 3-week tumors at 6 and 12 hours following a dose of AMD-3100 alone or in combination with FX-68, which was successively administered alone after 3 and 9 hours, respectively, prior to CTC collection. The fraction of apoptotic CTCs in the two treated groups was assessed and compared by measuring the relative expression of Bax and Bcl-2 (30) (31). As shown in Fig.2D, targeting CX3CR1 increased the percentage of apoptotic CTCs collected at 6 hours as compared to cells mobilized by AMD-3100 in the absence of FX-68. Interestingly, 12 hours following mobilization, the percentage of apoptotic cancer cells in the blood was found to be equivalent in the presence or absence of CX3CR1 targeting, suggesting that blocking a relatively fast re-seeding of CTCs has the highest impact on their viability. Finally, to corroborate the translational significance of our study, we analyzed CX3CR1 expression in a blood sample collected from a patient with metastatic breast cancer (ER+, PR+, HER2−) and identified CTCs with a strong expression of this chemokine receptor as shown by immunofluorescence staining (Fig. 2E). While this initial evidence is indeed compelling, we are fully aware that additional and extended studies will be necessary to fully characterize CX3CR1 expression in CTCs from human breast cancer patients and eventually correlate the intensity and frequency of CX3CR1 expression with molecular subtypes, occurrence and progression of metastatic disease, and overall survival as clinical endpoints, among others.
Higher numbers of CTCs in blood corresponds to a reduced number of reseeded cancer cells.
Next, we sought to confirm that the protracted permanence of CTCs in the blood and consequent impairment of their viability observed upon targeting CX3CR1 were truly the result of a direct inhibition of CTCs reseeding. To this end, we labeled green-fluorescent MDA-231 cells with the cell-tracker CM-Dil, a widely validated red-fluorescent dye that, while being stably retained by non-dividing cells, is transferred to daughter cells upon mitosis and eventually becomes too diluted and undetectable in rapidly dividing cell populations (32,33). Based on these features, we predicted that GFP/CM-Dil double-labeled cancer cells, when reseeding from established tumors would emit green fluorescence but lack red fluorescence, as the results of high proliferation during tumor growth and consequential dilution of the CM-Dil dye. On the other hand, cancer cells that became dormant upon seeding different tissues following intracardiac injection would retain the CM-Dil dye, thereby emitting signals in both green and red fluorescent spectra (Fig.3A). For these experiments, mice were grafted with double-labeled cancer cells in the left cardiac ventricle and allowed to develop tumors for two weeks. At this time, animals were then treated with AMD-3100 with or without FX-68 (10mg/Kg, i.p., 1 hr prior and 3 hrs after AMD-3100) and killed 12 or 24 hours later. Lung and knee-joints were harvested and inspected for fluorescent cancer cells using a Multispectral Imaging System. Fig. 3B shows that cancer cells with features of reseeded cells were identified at both time points and their numbers were dramatically reduced by the systemic administration of the CX3CR1 antagonist as compared to untreated animals.
Figure 3. Compromised reseeding of CTCs to bone and lungs.
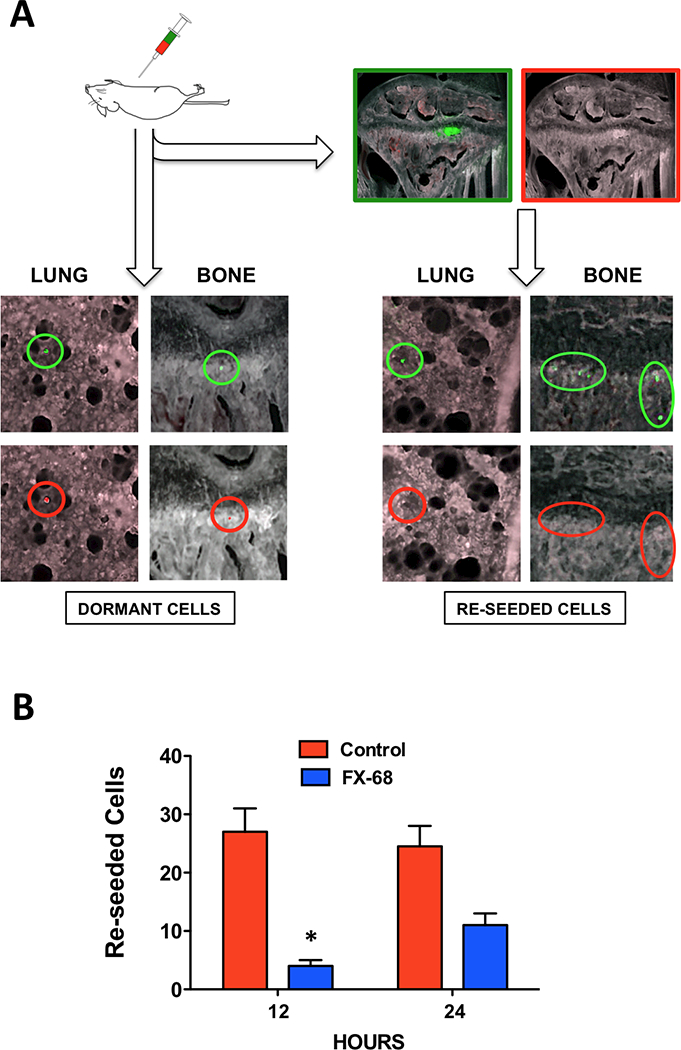
(A) MDA-231 cells, stably expressing GFP, were labeled with the red dye CM-Dil to distinguish re-seeded cells from dormant cells in mice harboring skeletal and soft-tissue tumors. Highly proliferating cancer cells in tumors and re-seeded cancer cells lacked red fluorescence, which was detected in non-proliferating, dormant cancer cells. (B) Re-seeded cells detected in bone and lungs of animals treated with AMD-3100 in combination with FX-68 were significantly fewer as compared to AMD-3100 alone (2 mice/data point). *P=0.03, Student’s t-test).
CTCs retain metastatic potential
The results reported above indicate that cancer cells can effectively seed different tissues upon departing from existing disseminated tumors and this process can be countered by the pharmacological targeting of CX3CR1. Thus, it was reasonable to hypothesize that obstructing tumor reseeding by interfering with this chemokine receptor would curb the emergence of additional lesions originated by existing metastases. Indeed, this paradigm would support our in vivo imaging and survival data from mice with early metastatic disease that were treated with FX-68 (Fig.1d-f).
To test this hypothesis, we first asked whether CTCs – in addition to reseeding – retained full metastatic behavior as defined by the ability to colonize and grow into detectable tumor foci. Indeed, this issue is still debated (8) and it is only starting to be investigated in animal models (21). Thus, we used a label-free microfluidic system to isolate CTCs at steady state from tumor-bearing mice grafted three weeks earlier via intracardiac route with 4T-1 cells, which generated multiple tumors in skeleton and soft-tissues. For re-inoculation experiments, CTCs from four mice were collected, counted, pooled into one cell suspension and re-inoculated into a single mouse at ~300 CTCs/animal. Despite the very low number of inoculated cancer cells, three mice inoculated as described above showed multiple lesions by bioluminescence in vivo imaging at 2 weeks post-grafting and larger tumors at the fourth week (Fig.4A). This is the first available pre-clinical evidence of full metastatic potential retained by CTCs departed from multiple tumors reproducing metastatic disease in humans. Experiments with GFP-labeled 4T-1 cells also stained with the DM-Cil dye demonstrated that CTCs isolated from three-week tumor-bearing animals were mostly cells shed in circulation by high-proliferating tumors, since >70% had lost the red dye, as expected and in contrast to >90% of red fluorescent CTCs isolated from the blood of mice inoculated only three days earlier (Fig.4B).
Figure 4. CTCs from metastases have tumor-forming ability and can reseed to generate additional lesions.
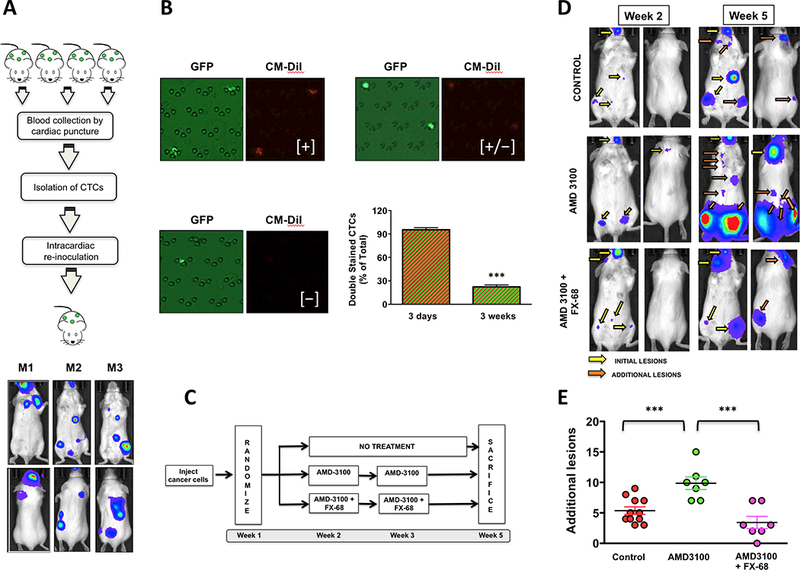
(A) CTCs collected from three mice harboring multiple tumors in skeleton and soft-tissues were collected and re-inoculated. Tumors were detected in three out of five recipient mice within 2 weeks. (B) Mice were grafted via intracardiac route with GFP-expressing 4T-1 cells also labeled with the CM-Dil red-fluorescent dye. Three days or three weeks following grafting, CTCs were collected and the percentage of cells detected as positive to GFP-only (shed from proliferating tumors) or GFP/DM-Dil (quiescent) was assessed for each time point. Cells showing only moderate red fluorescence [+/−] were still considered positive to DM-Dil. The vast majority of CTCs collected from animals three weeks post-grafting were GFP-only, indicating their shedding into the blood circulation from highly proliferating tumors. 3 mice/time point, ***P=0.0001, unpaired t-test. (C) Schematic of the experiment aimed to assess the metastatic potential of CTCs and the protective effect of CX3CR1 antagonism (D) Mice harboring disseminated tumors reproducing early metastatic disease and treated as shown in (b) were monitored by in vivo bioluminescence imaging at two weeks for initial lesions and three weeks later for additional lesions. (E) AMD-3100 doubled the number of additional lesions as compared to control animals, an effect that was fully blunted by co-treatment with FX-68. (Control 11 mice, Treated 7 mice/group; ***P=0.0002, One-way Anova with Dunnett’ post-test).
We then conducted complementary studies in mice harboring experimental metastases, as detected at one-week post-grafting, and left either untreated or treated at the second and third week with AMD-3100, alone or in combination with FX-68 (Fig.4C). All lesions identified at the fifth week that had not been previously detected at the second week of the experiment were counted and compared among treatment groups. We found that mice receiving AMD-3100 showed a doubled number of additional lesions as compared to untreated animals, an effect that was ablated by co-administration of the CX3CR1 antagonist (Fig.4D, E). These results provide compelling evidence that cancer cells departing from secondary tumors in bone and soft-tissues retain the potential to colonize different tissues and generate additional metastases, incidentally explaining why prolonged treatment with AMD-3100 did not extend overall survival of mice with experimental lung metastases (34).
Preventing CTCs from reseeding prolongs their exposure to doxorubicin
Next, we sought to determine whether reducing the number of reseeded tumor cells by targeting CX3CR1 would compare to chemotherapeutic drugs in containing the overall expansion of tumor load. We assumed that impairing reseeding would extend the time that CTCs are exposed to cytotoxic drugs in the blood. This paradigm was tested by treating mice harboring 3-week disseminated tumors with the anthracycline antibiotic Doxorubicin (5mg/Kg, i.p.), alone or in combination with FX-68 (10mg/Kg, i.p.). Our goal was to determine whether retaining cancer cells in the blood circulation by targeting CX3CR1 could extend their exposure to chemotherapeutics, thus inferring a potential impact of applying this approach to the clinic. We took advantage of the fluorescent properties of Doxorubicin (35) and examined the red-spectrum emitted fluorescence of CTCs collected at steady state from mice harboring disseminated tumors and treated with this drug alone, one hour after or three hours prior administration of FX-68. CTCs were collected - independently of the type of treatment - four hours after Doxorubicin administration, based on published pharmacokinetic studies for this drug (36). The uptake of anthracycline was assessed by fluorescence microscopy (Fig.5A) and the analysis showed that a prior administration of the CX3CR1 antagonist caused a significant increase in the number of CTCs positive for intracellular Doxorubicin as compared to mice exposed to the drug either alone or followed by FX-68 (Fig.5B). These results strongly imply that impairing reseeding also increases the exposure of CTCs to drugs, which could thereby improve the efficacy of cytotoxic and targeted therapeutics by increasing their cellular bioavailability. This concept was further investigated by treating animals harboring 1-week disseminated tumors with FX-68 (10mg/Kg, i.p., b.i.d.) in combination with the cytotoxic drug Docetaxel (2mg/Kg, i.p.,b.i.w.), which is standard of care for metastatic breast cancer (37). Animals were monitored by in vivo imaging and both compounds showed dramatic reduction in overall tumor burden, starting from the 5th week of treatment (Fig.5C), indicating that interfering with CX3CR1 functioning compares with the effects exerted by a widely used chemotherapeutic drug. Furthermore, at the 11th week of treatment, the combination of FX-68 with Docetaxel showed an evident synergistic anti-tumor effect, further supporting the potential use of CX3CR1 antagonists in the clinic not only as standalone approach but also in combinatorial strategies.
Figure 5. Obstructing the reseeding of CTCs improves drug exposure in blood and combination of CX3CR1 antagonist with Docetaxel shows synergistic anti-tumor effects.
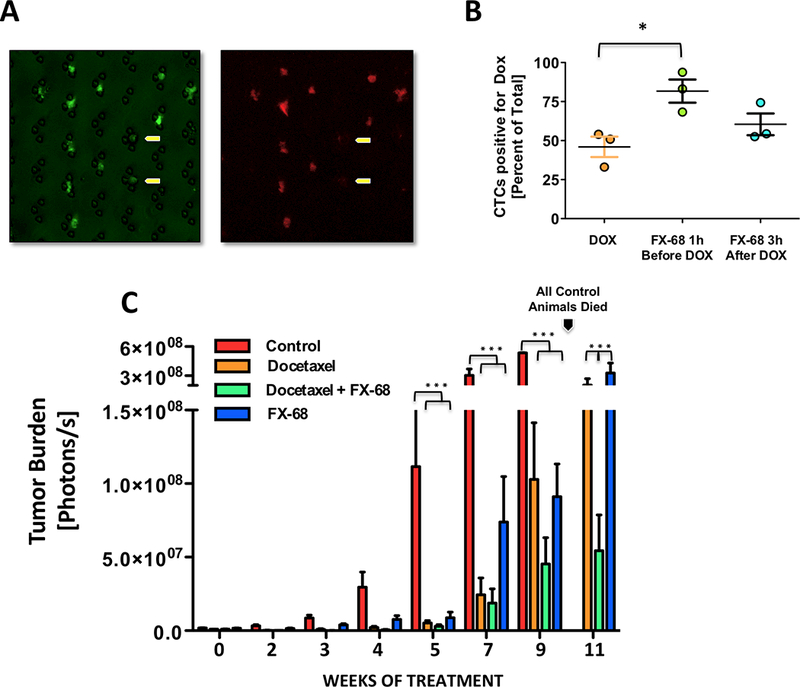
(A, B) Retaining CTCs in the blood by administering FX-68 increased the exposure to Doxorubicin, as measured by the percentage of cells showing red fluorescence emitted by the drug. Yellow arrows show two cancer cells that did not incorporate Doxorubicin (3 mice/group; *P=0.03, One-way Anova with Dunnett’s post-test. (C). Both FX-68 and Docetaxel were highly effective in controlling tumor burden when used separately and in combination, as compared to control animals receiving vehicle. At 11 weeks of treatment, when all control animals had died (see arrow), combination of FX-68 with Docetaxel significantly improved the control of tumor burden as compared to the effects shown by each of these two compounds when used alone. (Control and FX-68 alone 4 mice, Docetaxel alone and Docetaxel/FX-68 in combination, 7 mice; ***P=0.0001, One-way Anova with Newman- Keuls post-test.
DISCUSSION
We have discovered a new functional role for CX3CR1 in metastasis, employing a novel small molecule antagonist of this chemokine receptor and utilizing unique experimental approaches to prove the reseeding and metastatic potential of CTCs.
Furthermore, this study radically improves our knowledge of the spatial and temporal dynamics involved in cancer cells drifting in and out of the blood circulation following their mobilization from metastatic tumors. Indeed, here we show that metastatic disease relies on reseeding for expansion to additional organ sites, providing experimental support to this idea as previously inferred by genomic analyses of metastatic tumors in patients (4) (5).
The re-inoculation of CTC harvested from experimental metastatic tumors and the long-term studies investigating the emergence of additional lesions following mobilization of cancer cells back into circulation make a strong argument in favor of tumor-initiating potential as a feature of at least sub-populations of CTCs. Our finding are in line with similar work previously conducted by others in which CTCs isolated from patients with metastatic breast cancer generated lesions in bone and soft-tissues when grafted in mice (38) (39). These studies should provide impetus to molecular and genetic analyses, in both pre-clinical and clinical spaces, aiming to define highly metastatic CTC phenotypes with the intent of developing predictive biomarkers for patients with the highest risk of tumor spreading from existing metastases.
Our study also demonstrates that preventing CTCs from leaving circulation compromises their viability, in line with the concept of anoikis - the result of a loss of contact with either the extracellular matrix or neighboring cells (40). Cancer cells are expected to acquire anoikis resistance in order to develop a metastatic behavior (28); interestingly, we show that a significant percentage of the human breast cancer cells tested in our model perished when forcefully retained in the blood despite their proven metastatic features. Indeed, this could be due to the fact that in these experiments a relevant fraction of CTCs were not spontaneously mobilized but rather dislodged by a pharmacologic agent. Therefore, some of these CTCs may have not yet acquired anoikis resistance. Nevertheless, the evidence that provoking a surge of CTCs upon conditions preventing their subsequent reseeding exerts a cytotoxic effect makes a captivating case for pursuing similar approaches for therapeutic purposes.
The concept of mobilizing breast cancer cells from micrometastases has been recently proposed also with the intent of excising dormant cancer cells from potential sites of late tumor recurrence (41). Here we provide experimental support to this paradigm by testing whether mobilizing – and also retaining – CTCs in blood could boost exposure to Doxorubicin, which is commonly used for metastatic breast cancer (42), and found that more than ~80% of CTCs had been exposed to the drug as compared to ~50% observed at steady state (i.e. not mobilized by AMD-3100).
Finally, we found that targeting CXCR4 did not prevent breast CTCs from seeding the skeleton, in line with similar findings from others (41), and despite the widely reported association of this receptor with the homing of breast cancer cells to bone (43–45) (46). Indeed, these latter studies drew their conclusions from the detection of small tumor foci or macroscopic tumors as indicators of tumor seeding, instead of imaging and enumerating individual disseminated cancer cells. Thus, homing abilities could not be directly assessed as they were in our study. It is highly plausible that CXCR4 – rather than promoting CTC seeding – supports cancer cells after their homing to bone, for instance during initial colonization and/or progression. Indeed, this scenario is supported by a number of reports (47) (41,48).
On the other hand, the pre-clinical efficacy of the small-molecule antagonist FX-68 in our model of metastatic disease reaffirms the idea that interfering with CX3CR1 dramatically limits the seeding of skeleton and soft-tissues by breast CTCs. Notably, interfering with the functioning of CX3CR1 directly on cancer cells could be combined with a blockade of the same receptor on immune cells such as monocytes (49,50), which have been implicated in the metastatic behavior of different tumor types. Finally, the pharmacological antagonism of CX3CR1 by FX-68 compares to Docetaxel in controlling tumor burden and shows synergistic anti-tumor effects when used in combination with it. Thus, inhibitory targeting of CX3CR1, as a standalone or in combinatorial approaches, should be developed with the ultimate goal of establishing treatment strategies to contain further propagation of metastatic lesions and improve exposure of cancer cells to a range of therapeutics.
Supplementary Material
Implications:
The current findings shed light on CTCs reseeding dynamics and support the development of CX3CR1 antagonism as a viable strategy to counteract metastatic progression.
ACKNOWLEDGEMENTS
The authors wish to thank Dr. Kenneth J. Pienta at the Brady Urological Institute of Johns Hopkins University, Baltimore MD, and members of his laboratory for valuable discussion. The FX-68 small-molecule compound was synthesized in the laboratory of Dr. Joseph Salvino (current address: The Wistar Institute, Philadelphia, PA), which the authors wish to thank also for coordinating the efforts of Eurofins Pharma and Reaction Biology toward this project.
This work was funded by grants from the National Institutes of Health (CA202929), Wallace H. Coulter Foundation and Breast Cancer Alliance to A.F. and O.M. and from the Department of Defense, Breast Cancer Program Breakthrough Award Level 2 (BC150659) to A.F.
Footnotes
DISCLOSURE OF POTENTIAL CONFLICT OF INTEREST
A. Fatatis and O. Meucci have ownership interests (including patents) in Kerberos Biopharma Inc. No competing financial interests were disclosed by the other authors.
REFERENCES
- 1.Labelle M, Hynes RO. The Initial Hours of Metastasis: The Importance of Cooperative Host-Tumor Cell Interactions during Hematogenous Dissemination. Cancer Discovery. 2012;2:1091–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–64. [DOI] [PubMed] [Google Scholar]
- 3.Marsden CG, Wright MJ, Carrier L, Moroz K, Rowan BG. Disseminated breast cancer cells acquire a highly malignant and aggressive metastatic phenotype during metastatic latency in the bone. Rameshwar P, editor. PLoS ONE. 2012;7:e47587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hong MKH, Macintyre G, Wedge DC, Van Loo P, Patel K, Lunke S, et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun. 2015;6:6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones SE. Metastatic breast cancer: the treatment challenge. Clinical Breast Cancer. 2008;8:224–33. [DOI] [PubMed] [Google Scholar]
- 7.Faltas B Cornering metastases: therapeutic targeting of circulating tumor cells and stem cells. Front Oncol. 2012;2:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Toom EE, Verdone JE, Pienta KJ. Disseminated tumor cells and dormancy in prostate cancer metastasis. Curr Opin Biotechnol. 2016;40:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jamieson-Gladney WL, Zhang Y, Fong AM, Meucci O, Fatatis A. The chemokine receptor CX3CR1 is directly involved in the arrest of breast cancer cells to the skeleton. Breast Cancer Research 2010 12:307. BioMed Central Ltd. BioMed Central Ltd; 2011;13:R91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen F, Zhang Y, Jernigan DL, Feng X, Yan J, Garcia FU, et al. Novel Small-Molecule CX3CR1 Antagonist Impairs Metastatic Seeding and Colonization of Breast Cancer Cells. Mol Cancer Res. 2016;14:518–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Russell MR, Liu Q, Fatatis A. Targeting the {alpha} receptor for platelet-derived growth factor as a primary or combination therapy in a preclinical model of prostate cancer skeletal metastasis. Clin Cancer Res. 2010;16:5002–10. [DOI] [PubMed] [Google Scholar]
- 12.Liu Q, Russell MR, Shahriari K, Jernigan DL, Lioni MI, Garcia FU, et al. Interleukin-1β promotes skeletal colonization and progression of metastatic prostate cancer cells with neuroendocrine features. Cancer Res. 2013;73:3297–305. [DOI] [PubMed] [Google Scholar]
- 13.Eliane J-P, Repollet M, Luker KE, Brown M, Rae JM, Dontu G, et al. Monitoring serial changes in circulating human breast cancer cells in murine xenograft models. Cancer Res. 2008;68:5529–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan SJ, Yobas L, Lee GYH, Ong CN, Lim CT. Microdevice for the isolation and enumeration of cancer cells from blood. Biomed Microdevices. 2009;11:883–92. [DOI] [PubMed] [Google Scholar]
- 15.Bhagat A, Hou HW, Li LD, Lim CT, Han J. Dean flow fractionation (DFF) isolation of circulating tumor cells (CTCs) from blood. 2012.
- 16.Eckhardt BL. Strategies for the discovery and development of therapies for metastatic breast cancer. Nature reviews Drug discovery. 2012;11:479–97. [DOI] [PubMed] [Google Scholar]
- 17.Hatse S, Princen K, Bridger G, De Clercq E, Schols D. Chemokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Letters. 2002;527:255–62. [DOI] [PubMed] [Google Scholar]
- 18.Joosse SA, Gorges TM, Pantel K . Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol Med. 2015;7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10:8152–62. [DOI] [PubMed] [Google Scholar]
- 20.Stott SL, Lee RJ, Nagrath S, Yu M, Miyamoto DT, Ulkus L, et al. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci Transl Med. 2010;2:25ra23–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carvalho FLF, Simons BW, Antonarakis ES, Rasheed Z, Douglas N, Villegas D, et al. Tumorigenic potential of circulating prostate tumor cells. Oncotarget. 2013;4:413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newton PK, Mason J, Bethel K, Bazhenova L, Nieva J, Norton L, et al. Spreaders and sponges define metastasis in lung cancer: a Markov chain Monte Carlo mathematical model. Cancer Res. 2013;73:2760–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hendrix CW, Flexner C, MacFarland RT, Giandomenico C, Fuchs EJ, Redpath E, et al. Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrobial Agents and Chemotherapy. 2000;44:1667–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uy GL, Rettig MP, Cashen AF. Plerixafor, a CXCR4 antagonist for the mobilization of hematopoietic stem cells. Expert Opin Biol Ther. 2008;8:1797–804. [DOI] [PubMed] [Google Scholar]
- 26.Azab AK, Runnels JM, Pitsillides C, Moreau A-S, Azab F, Leleu X, et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113:4341–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011;121:1298–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim Y-N, Koo KH, Sung JY, Yun U-J, Kim H. Anoikis resistance: an essential prerequisite for tumor metastasis. International Journal of Cell Biology. 2012;2012:306879–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McInnes LM, Jacobson N, Redfern A, Dowling A, Thompson EW, Saunders CM. Clinical implications of circulating tumor cells of breast cancer patients: role of epithelial-mesenchymal plasticity. Front Oncol. 2015;5:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perlman H, Zhang X, Chen MW, Walsh K, Buttyan R. An elevated bax/bcl-2 ratio corresponds with the onset of prostate epithelial cell apoptosis. Cell Death Differ. 1999;6:48–54. [DOI] [PubMed] [Google Scholar]
- 31.Del Poeta G, Venditti A, Del Principe MI, Maurillo L, Buccisano F, Tamburini A, et al. Amount of spontaneous apoptosis detected by Bax/Bcl-2 ratio predicts outcome in acute myeloid leukemia (AML). Blood. 2003;101:2125–31. [DOI] [PubMed] [Google Scholar]
- 32.Andrade W, Seabrook TJ, Johnston MG, Hay JB. The use of the lipophilic fluorochrome CM-DiI for tracking the migration of lymphocytes. J Immunol Methods. 1996;194:181–9. [DOI] [PubMed] [Google Scholar]
- 33.Schormann W, Hammersen FJ, Brulport M. Tracking of human cells in mice. Histochemistry and cell …. 2008;130:329–38. [DOI] [PubMed] [Google Scholar]
- 34.Smith MCP, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, et al. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64:8604–12. [DOI] [PubMed] [Google Scholar]
- 35.Motlagh NSH, Parvin P, Ghasemi F, Atyabi F. Fluorescence properties of several chemotherapy drugs: doxorubicin, paclitaxel and bleomycin. Biomed Opt Express. 2016;7:2400–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddy LH, Murthy RSR. Pharmacokinetics and biodistribution studies of Doxorubicin loaded poly(butyl cyanoacrylate) nanoparticles synthesized by two different techniques. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2004;148:161–6. [DOI] [PubMed] [Google Scholar]
- 37.Palmeri L, Vaglica M, Palmeri S. Weekly docetaxel in the treatment of metastatic breast cancer. Ther Clin Risk Manag. 2008;4:1047–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L, Ridgway LD, Wetzel MD, Ngo J, Yin W, Kumar D, et al. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci Transl Med. 2013;5:180ra48–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol. 2013;31:539–44. [DOI] [PubMed] [Google Scholar]
- 40.Valentijn AJ, Zouq N, Gilmore AP. Anoikis. Biochem Soc Trans 2004;32:421–5. [DOI] [PubMed] [Google Scholar]
- 41.Price TT, Burness ML, Sivan A, Warner MJ, Cheng R, Lee CH, et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci Transl Med. 2016;8:340ra73–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Brien MER, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15:440–9. [DOI] [PubMed] [Google Scholar]
- 43.Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. [DOI] [PubMed] [Google Scholar]
- 44.Liang Z, Wu T, Lou H, Yu X, Taichman RS, Lau SK, et al. Inhibition of breast cancer metastasis by selective synthetic polypeptide against CXCR4. Cancer Res. 2004;64:4302–8. [DOI] [PubMed] [Google Scholar]
- 45.Liang Z, Yoon Y, Votaw J, Goodman MM, Williams L, Shim H. Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res. 2005;65:967–71. [PMC free article] [PubMed] [Google Scholar]
- 46.Peng S-B, Zhang X, Paul D, Kays LM, Gough W, Stewart J, et al. Identification of LY2510924, a novel cyclic peptide CXCR4 antagonist that exhibits antitumor activities in solid tumor and breast cancer metastatic models. Molecular Cancer Therapeutics. 2015;14:480–90. [DOI] [PubMed] [Google Scholar]
- 47.Darash-Yahana M, Pikarsky E, Abramovitch R, Zeira E, Pal B, Karplus R, et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 2004;18:1240–2. [DOI] [PubMed] [Google Scholar]
- 48.Sauvé K, Lepage J, Sanchez M, Heveker N, Tremblay A. Positive feedback activation of estrogen receptors by the CXCL12-CXCR4 pathway. Cancer Res. 2009;69:5793–800. [DOI] [PubMed] [Google Scholar]
- 49.Zheng J, Yang M, Shao J, Miao Y, Han J, Du J. Chemokine receptor CX3CR1 contributes to macrophage survival in tumor metastasis. Mol Cancer. 2013;12:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanna RN, Cekic C, Sag D, Tacke R, Thomas GD, Nowyhed H, et al. Patrolling monocytes control tumor metastasis to the lung. Science. 2015;350:985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.