

Abstract
Junctional Epidermolysis Bullosa (JEB) is a severe, often lethal genetic disease caused by mutations in genes encoding the basement membrane component laminin-332. Surviving JEB patients develop chronic skin and mucosa wounds, which impair their quality of life and lead to skin cancer. Here we show that autologous transgenic keratinocyte cultures regenerated an entire, fully functional epidermis on a 7-year-old child suffering from a devastating, life-threatening form of JEB. The proviral integration pattern was maintained in vivo and epidermal renewal did not cause any clonal selection. Clonal tracing showed that human epidermis is not sustained by equipotent progenitors, but by a limited number of long-lived stem cells, detected as holoclones, able to extensively self-renew in vitro and in vivo and to produce progenitors that replenish terminally differentiated keratinocytes. This study provides a blueprint that can be applied to other stem cell-mediated combined ex vivo cell and gene therapies.
Generalized Junctional Epidermolysis Bullosa (JEB) is a severe, often lethal genetic disease characterized by structural and mechanical fragility of the integuments. Skin and mucosal blisters and erosions occur within the lamina lucida of the basement membrane upon minor trauma. Massive chronic skin wounds greatly impair the patients’ quality of life, lead to recurrent infections and scars and are predisposing to skin cancer. JEB is caused by mutations in LAMA3, LAMB3 or LAMC2 genes, which jointly encode laminin-332 (a heterotrimeric protein, also known as laminin 5, consisting of α3, β3, and γ2 chains) and in genes encoding collagen XVII and α6β4 integrins1. Deleterious mutations causing absence of laminin-332 are usually early lethal. In nonlethal JEB, laminin-332 is strongly reduced and hemidesmosomes are rudimentary or absent. There is no cure for JEB and >40% of the patients succumb to the disease by adolescence1,2. Available symptomatic treatments can only relieve the devastating clinical manifestations.
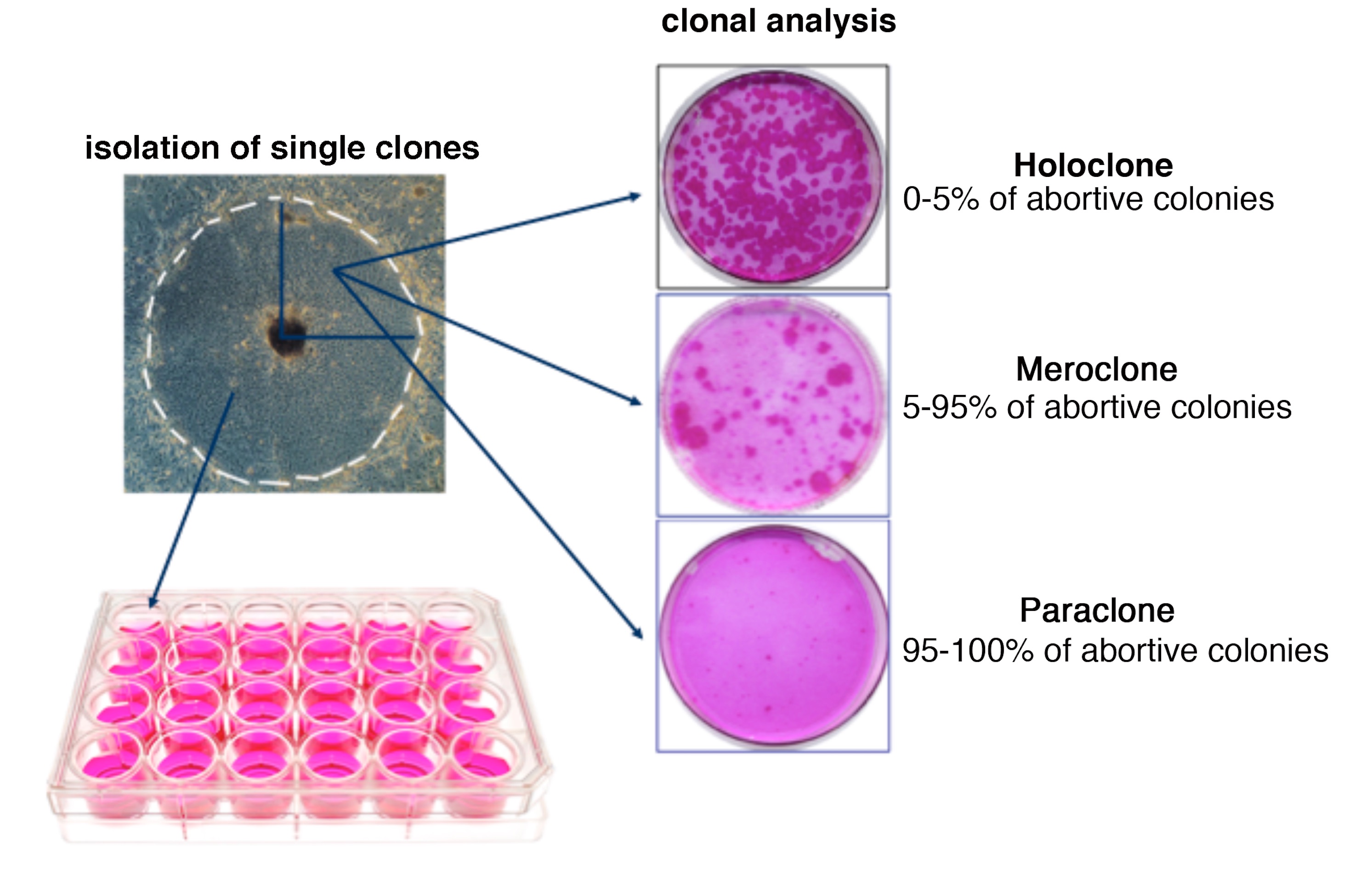
Monthly renewal and timely repair of human epidermis is sustained by epidermal stem cells, which generate colonies known as holoclones3,4. Holoclones produce meroclone- and paraclone-forming cells, which behave as transient amplifying (TA) progenitors3,4. Epithelial cultures harbouring holoclone-forming cells can permanently restore massive skin and ocular defects5–9. A phase I/II clinical trial (1 patient) and a single-case study provided compelling evidence that local transplantation of transgenic epidermal cultures can generate a functional epidermis, leading to permanent (the longest follow-up being of 12 years) correction of JEB skin lesions10–12. However, paucity of treated areas (a total of ~0.06 m2) did not significantly improve patients’ quality of life10–12.
A major criticism to this therapeutic approach has been its supposed unsuitability for the massive skin lesions marking generalized JEB. Here we show life-saving regeneration of virtually the entire epidermis (~0.85 m2) on a 7-year-old child suffering from a devastating form of JEB by means of autologous transgenic keratinocyte cultures. The regenerated epidermis remained robust, resistant to mechanical stress and did not develop blisters or erosions during 21 months follow-up. Such fully functional epidermis is entirely sustained by a limited number of transgenic epidermal stem cells, detected as holoclones, able to extensively self-renew in vitro and in vivo.
The patient
In June 2015, a 7-year-old child was admitted to the Burn Unit of the Children’s Hospital, Ruhr-University, Bochum, Germany. He carried a homozygous acceptor splice site mutation (C1977-1G > A, IVS 14-1G >A) within intron 14 of LAMB3. Since birth, the patient developed blisters all over his body, particularly on limbs, back and flanks. His condition severely deteriorated six weeks before admission, due to infection with Staphylococcus aureus and Pseudomonas aeruginosa. Shortly after admission, he suffered complete epidermal loss on ~60% of the total body surface area (TBSA). During the following weeks, all therapeutic approaches failed and the patient’s short-term prognosis was unfavourable (Methods). After the parents’ informed consent, the regional regulatory authorities and the ethical review board of the Ruhr-University authorised the compassionate use of combined ex vivo cell and gene therapy. The parents of the patient also consented on the publication of the photographs and medical information included in this paper.
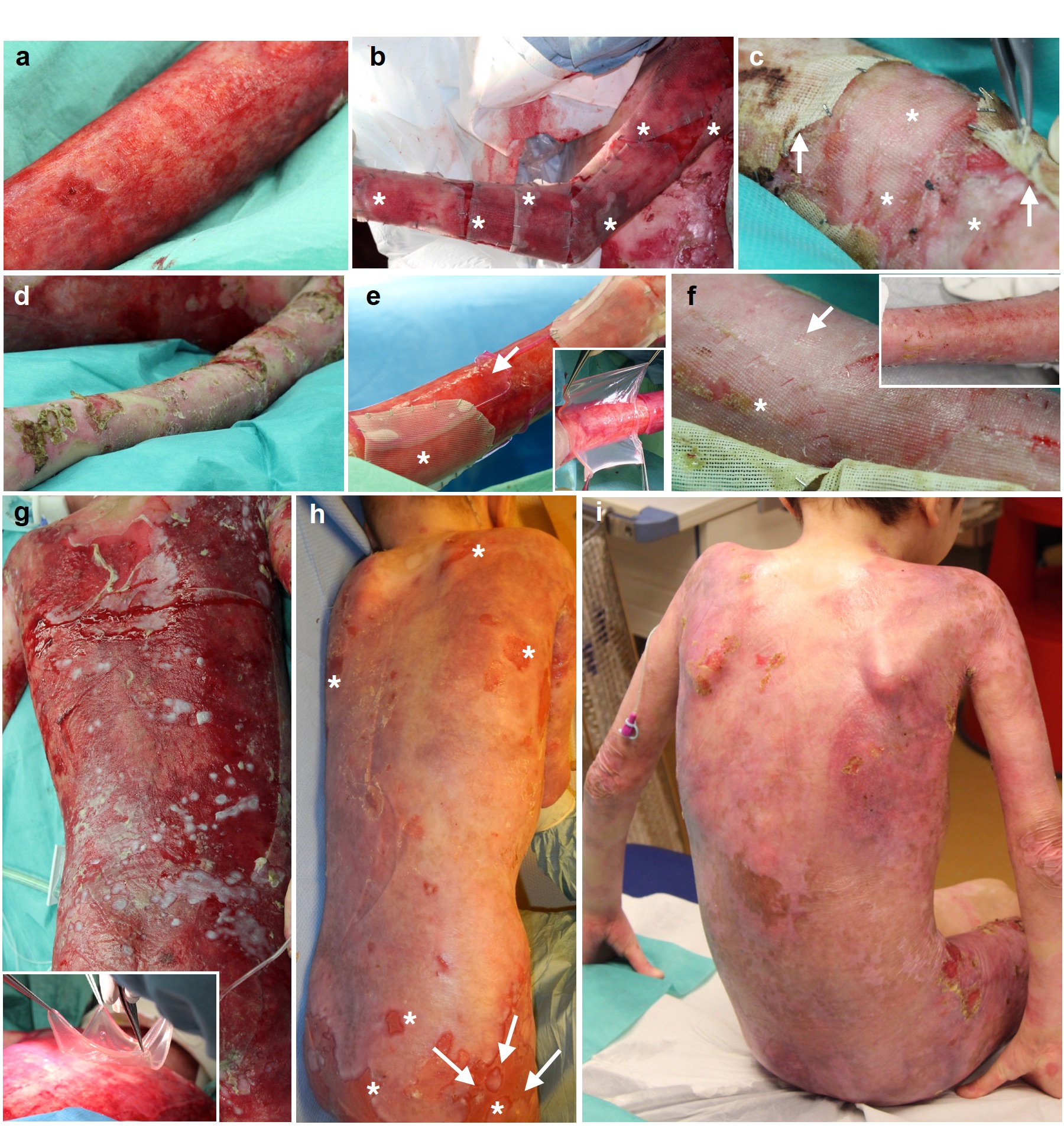
At the first surgery, the patient had complete epidermal loss on ~80% TBSA (Fig. 1a, b).
Figure 1. Regeneration of the transgenic epidermis.

a, Clinical picture of the patient showing massive epidermal loss. b, Schematic representation of the clinical picture. The denuded skin is indicated in red, while blistering areas are indicated in green. Flesh-colored areas indicate currently non-blistering skin. Transgenic grafts were applied on both red and green areas. c, Restoration of patient’s entire epidermis, with the exception of very few areas on the right thigh, buttocks, upper shoulders/neck and left axilla (asterisks, altogether ≤2% of TBSA). d, Normal skin functionality and elasticity. e, Absence of blister formation at sites where some of post-graft biopsies were taken (arrow).
Regeneration of a functional epidermis by transgenic epidermal cultures
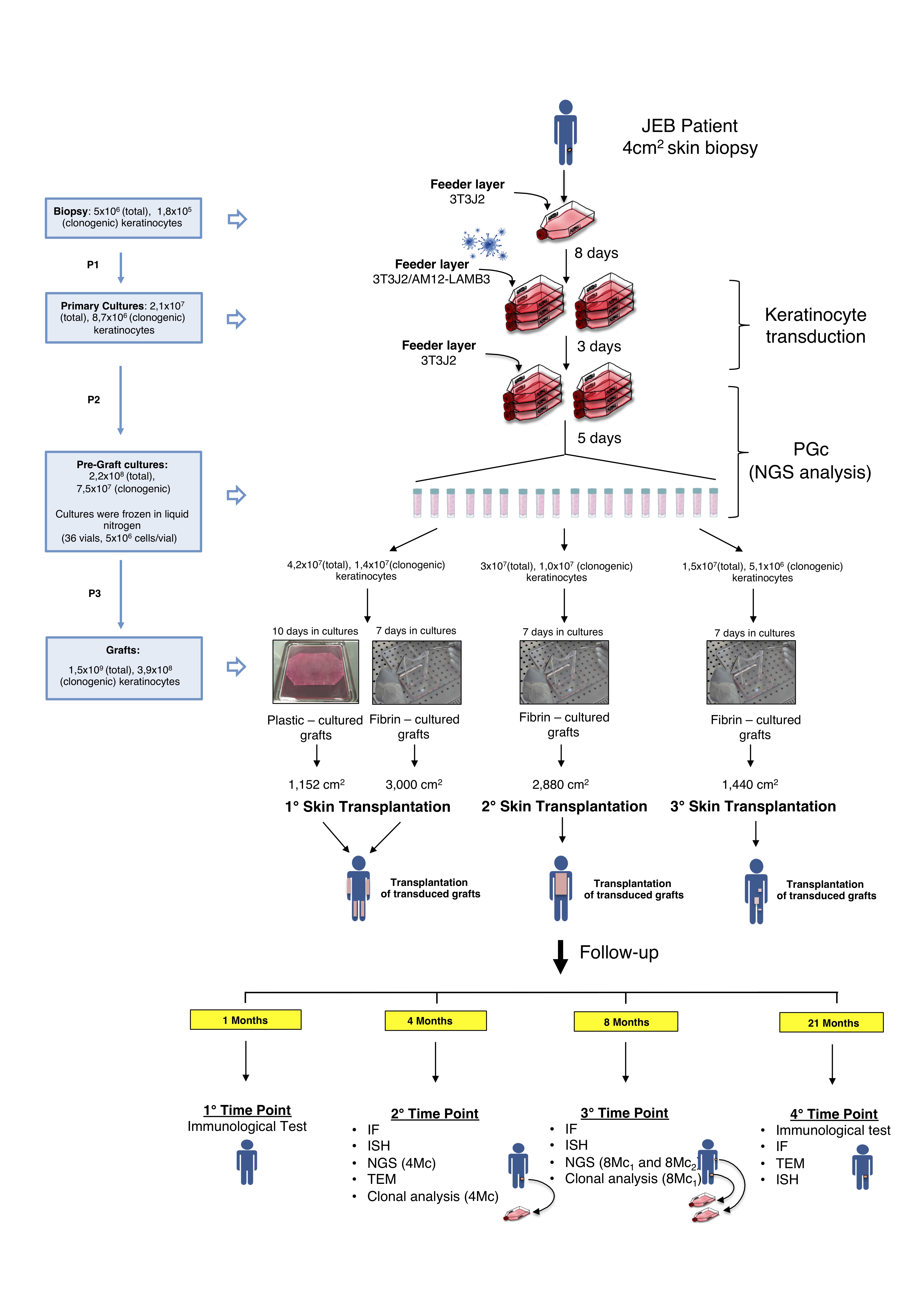
On September 2015, a 4-cm2 biopsy, taken from a currently non-blistering area of patient’s left inguinal region, was used to establish primary keratinocyte cultures, which were then transduced with a retroviral vector (RV) expressing the full-length LAMB3 cDNA under the control of the Moloney leukaemia virus (MLV) long terminal repeat13 (Methods, Extended Data Fig. 1 and Supplementary Information). Sequentially, 0.85 m2 transgenic epidermal grafts, enough to cover all patient’s denuded body surface, were applied on a properly prepared dermal wound bed (Extended Data Fig. 2a). All limbs, flanks and the entire back were grafted on October and November 2015. Some of the remaining denuded areas were grafted on January 2016.
Previously, transgenic epidermal sheets were cultivated on plastic, enzymatically detached from the vessel and mounted on a non-adhering gauze10–12. Keratinocyte cultivation on a fibrin substrate – currently used to treat massive skin and ocular burns6,8,9 - eliminates cumbersome procedures for graft preparation and transplantation and avoids epidermal shrinking, allowing the production of larger grafts using the same number of clonogenic cells needed to produce plastic-cultured grafts. Since degradation of fibrin after transplantation, which is critical to allow cell engraftment, was never assessed in a JEB wound bed, at the first surgery we compared plastic- and fibrin-cultured grafts (Methods, Extended Data Fig. 1).
The left arm received plastic-cultured grafts (Extended Data Fig. 2b, asterisks). Upon removal of the non-adhering gauze (10 days post-grafting, Extended Data Fig. 2c, arrows), epidermal engraftment was evident (asterisks). Epidermal regeneration, evaluated at 1 month, was stable and complete (Extended Data Fig. 2d). The left leg received both plastic- and fibrin-cultured grafts (Extended Data Fig. 2e, asterisk and arrow, respectively), both of which showed full engraftment at 10 days (Extended Data Fig. 2f, asterisk and arrow, respectively) and complete epidermal regeneration at 1 month (Extended Data Fig. 2f, inset). Similar data were obtained on the other limbs. Thus, the patient’s denuded back (Extended Data Fig. 2g) received only fibrin-cultured grafts (inset). As shown in Extended Data Fig. 2h, virtually complete epidermal regeneration was observed at 1 month, with the exception of some areas (asterisks), some of which contained islands of newly formed epidermis (arrows). Over the following weeks, the regenerated epidermis surrounding the open lesions and those epidermal islands spread and covered most of the denuded areas (Extended Data Fig. 2i). We then transplanted the remaining defects on flanks, thorax, right thigh, right hand and shoulders. Epidermal regeneration was attained in most of those areas.
Thus, ~80% of the patient’s TBSA was restored by the transgenic epidermis (Fig. 1c). During the 21 months follow-up (over 20 epidermal renewing cycles), the regenerated epidermis firmly adhered to the underlying dermis, even after induced mechanical stress (Fig. 1d and video in Supplementary Information), healed normally and did not form blisters, also in areas where follow-up biopsies were taken (Fig. 1e, arrow).
The patient was discharged in February 2016 and is currently leading a normal social life. His epidermis is currently stable, robust, does not blister, itch, or require ointment or medications.
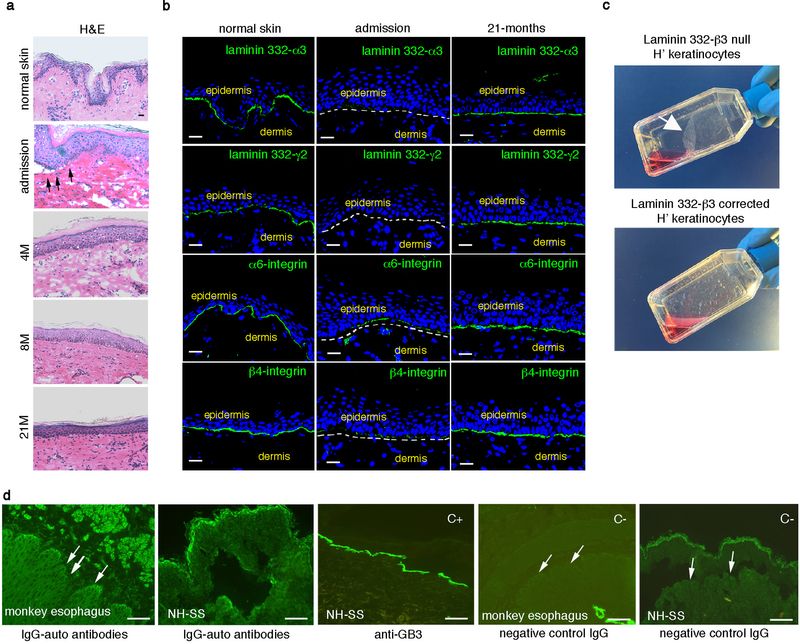
Ten punch biopsies were randomly taken, 4, 8 and 21 months after grafting. The epidermis had normal morphology and we could not detect blisters, erosions or epidermal detachment from the underlying dermis (Extended Data Fig. 3a). In situ hybridization using a vector specific t-LAMB3 probe showed that the regenerated epidermis consisted only of transgenic keratinocytes (Fig. 2a). At admission, laminin 332-β3 was barely detectable in patient’s skin (Fig. 2b). In contrast, control and transgenic epidermis expressed virtually identical amounts of laminin 332-β3, which was properly located at the epidermal-dermal junction (Fig. 2b). The basal lamina contained normal amounts of laminin 332-α3 and γ2 chains and α6β4 integrins, all of which were strongly decreased at admission (Extended Data Fig. 3b). Thus, transduced keratinocytes restored a proper adhesion machinery (Extended Data Fig. 3c). Indeed, the transgenic epidermis revealed normal thickness and continuity of the basement membrane (Fig. 2c, arrowheads) and normal morphology of hemidesmosomes (Fig. 2c, arrows). At 21 months follow-up, the patient’s serum did not contain autoantibodies directed against the basement-membrane zone (Extended Data Fig. 3d).
Figure 2. Restoration of a normal epidermal-dermal junction.

Skin sections were prepared from normal skin, patient’ affected (admission) and transgenic skin at 4, 8 and 21 months follow-up. a, In situ hybridization was performed using a transgene-specific probe (t-LAMB3) on 10-μm-thick sections. E-cadherin-specific probe (Cdh1) was used as a control. Scale bars, 40 μm. b, Immunofluorescence of laminin 332-β3 was performed with 6F12 moAbs on 7-μm-thick sections. DAPI (blue) marks nuclei. Dotted line marks the epidermal-dermal junction. Scale bars, 20 μm. c, Electron-microscopy was performed on 70-nm-thick skin sections. A regular basement membrane (arrows) and normal hemidesmosomes (arrowheads, higher magnification in the inset) are evident in patient’ transgenic skin. Scale bars, 1 μm.
In summary, transgenic epidermal cultures generated an entire functional epidermis in a JEB patient. This is consistent with the notion that keratinocyte cultures have been used for decades to successfully treat life-threatened burn victims on up to 98% of TBSA5,6,9,14. It can be argued that the patient’s clinical picture (massive epidermal loss, critical conditions, poor short-term prognosis) was unusual and our aggressive surgery (mandatory for this patient) unthinkable for the clinical course of most EB patients. But progressive replacement of diseased epidermis can be attained in multiple, less invasive surgical interventions on more limited body areas. EB has the advantage of a preserved dermis (not available in deep burns), which allows good functional and cosmetic outcomes. This approach would be optimal for newly diagnosed patients early in their childhood. A bank of transduced epidermal stem cells taken at birth could be used to treat skin lesions while they develop, thus preventing, rather than restoring, the devastating clinical manifestations rising through adulthood. Currently, combined ex vivo cell and gene therapy cannot be applied to lesions of the internal mucosae, which, however, are usually more manageable than those on skin, perhaps with the exception of oesophageal strictures.
Integration profile of transgenic epidermis
Pre-graft transgenic cultures (PGc) were generated by ~8.7x106 primary clonogenic cells and consisted of 2.2x108 keratinocytes (divided in 36 vials), ~45% of which were seeded to prepare 0.85 m2 transgenic epidermal grafts (Extended Data Fig. 1).
To investigate the genome-wide integration profile, 3 PGc samples were sequenced using two independent LTR-primers (i.e., 3pIN and 3pOUT, Supplementary Table 1) for library enrichment (n=12; see Methods). High-throughput sequencing recovered a total of 174.9M read pairs and the libraries obtained using the two LTR-primers showed similar number of reads and comparable insertion counts (Pearson R>0.92, p<0.005). After merging all integration sites from the two independent priming systems, we identified 27,303 integrations in PGc (Fig. 3a, bars) with an average coverage of 2.5 reads/insertion (Fig. 3a, lines and Supplementary Table 4). The same analysis was performed on primary cultures initiated from 3 biopsies (~0.5 cm2 each) taken at 4 (left leg) and 8 (left arm and left leg) months after grafting, referred to as 4Mc, 8Mc1, and 8Mc2, respectively (Methods).
Figure 3. Integration profile of transgenic epidermis.

a, Integrations were identified in libraries obtained using two LTR-primers (3pIN, light grey bars; 3pOUT, dark grey bars; Supplementary Table 1) and in the merged set (black bars). Lines (secondary axis) depict the average integration coverage, calculated after removal of PCR duplicates. b, Venn diagram of the number of shared integrations across samples. c, percentage of integrations mapped to: promoters, exons, introns, and intergenic regions (left); epigenetically defined active and weak promoters and enhancers, or genomic regions with no histone marks (right); (p-value>0.05; Pearson's Chi-squared test). d, Dot plot of the top 5 enriched GO Biological Process terms for each sample. Dot colour indicates statistical significance of the enrichment (q-value); dot size represents the fraction of genes annotated to each term.
Strikingly, we detected only 400, 206, and 413 integrations in 4Mc, 8Mc1, and 8Mc2, respectively (Fig. 3a, bars) with an average coverage of 27.3, 19.5, and 20.4 (Fig. 3a, lines).
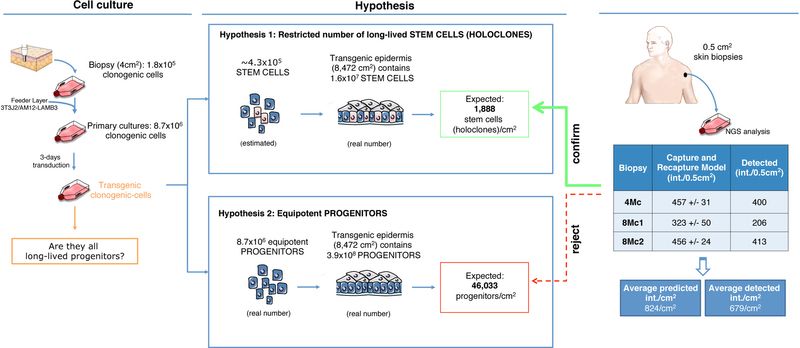
To exclude that the major difference in the number of integrations found in pre- and post-graft samples could be ascribable to PCR reactions causing unbalanced representation of event-specific amplicons, or to spatiality-effect of punch biopsies, we estimated the expected number of PGc, 4Mc, 8Mc1, and 8Mc2 integrations using the Chapman-Wilson capture-recapture model on the data obtained from the independent libraries (Supplementary Information)15. In PGc, the model estimated 65,030±2,120 integrations, i.e. approximately twice the actual number of detected insertions. The same model estimated 457±31, 323±50, and 457±24, independent integrations in 4Mc, 8Mc1, and 8Mc2, respectively (confidence level of 99%, α=0.01), which is highly consistent with the number of events actually detected. Of note, 58%, 43% and 37% of 4Mc, 8Mc1 and 8Mc2 integrations, respectively, were identified in PGc (Fig. 3b), which is consistent with the percentage (~50%) of insertions detected in PGc by NGS analysis.
Integrations were mapped to promoters (defined as 5 kb regions upstream the transcription start site of RefSeq genes), exons, introns, and intergenic regions. In all pre- and post-graft samples, ~10% of events were located within promoters. The majority of integrations were either intronic (~47%) or intergenic (~38%) and less than 5% were found in exons (Fig. 3c, left panel). We also annotated integrations in epigenetically defined transcriptional regulatory elements (Methods and Supplementary Information). As shown in Fig. 3c (right panel), ~27% of integrations were associated to active promoters or enhancers and no significant difference in the distribution of insertions was detected in pre- and post-graft samples (p-value>0.05; Pearson's Chi-squared test). Thus, the integration pattern was maintained in vivo and epidermal renewal did not determine any clonal selection.
Genes containing an integration were not functionally enriched in Gene Ontology categories related to cancer-associated biological processes16, with the exception of cell migration and small GTPase mediated signal transduction (Fig. 3d and Extended Data Table 1). These findings are however expected, since our culture conditions are optimized to foster keratinocyte proliferation and migration, to sustain clonogenic cells and to avoid premature clonal conversion and terminal differentiation, all of which are instrumental for the proper clinical performance of cultured epidermal grafts14. Thus, similarly to what has been reported in transgenic hematopoietic stem cells17,18, our high-throughput analyses revealed a cell-specific vector preference that is related to the host cell status in terms of chromatin state and transcriptional activity at the time of transduction19.
MLV-RV vectors raised concerns about insertional genotoxicity, which has been reported with hematopoietic stem cells, but in specific disease contexts17,20–22. Indeed, a γRV vector, similar to ours, obtained a marketing authorization for ex vivo gene therapy of adenosine deaminase severe combined immunodeficiency and has been approved for PhaseI/II clinical trials on RDEB (https://clinicaltrials.gov/ct2/show/NCT02984085)23. The patient’s integration profile confirmed absence of clonal selection both in vitro and in vivo. Likewise, we never observed immortalization events related to specific proviral integrations in many serially cultivated MLV-RV-transduced keratinocytes. Two JEB patients, receiving a total of ~1x107 clonogenic transgenic keratinocytes in selected body sites (3.5 and 12 years follow-up)10–12, and the patient, receiving ~3.9x108 transgenic clonogenic cells all over his body (Extended Data Fig. 1), did not manifest tumour development or other related adverse events. Therefore, based on in vivo data, the frequency of a detectable transformation event (if any) in MLV-RV-transduced keratinocytes would be less than 1 out of 1x107 during the first 12 years follow-up. Although the follow up of this patient is shorter and does not allow drawing definitive conclusions, the frequency of detectable insertional mutagenesis events to date is less than 1 out of 3.9x108. In evaluating the risk/benefit ratio, it should also be considered that severely affected JEB patients are likely to develop aggressive squamous cell carcinoma as a consequence of the progression of the disease.
The transgenic epidermis is sustained by self-renewing stem cells (holoclones)
The percentage of clonogenic cells, including holoclones, remained relatively constant during the massive cell expansion needed to produce the grafts (Extended Data Fig. 1 and Extended Data Table 3). The patient received ~3.9x108 clonogenic cells, ~1.6x107 of which were holoclone-forming cells, to cover ~0.85 m2 of his body (Extended Data Fig. 1 and 4 and Extended Data Table 3). Thus, ~4.6x104/cm2 clonogenic cells or ~1.8x103/cm2 stem cells were transplanted on the patient’s body surface (Extended Data Fig. 4).
If originally transduced clonogenic cells were all long-lived equipotent progenitors, (i) we would have recovered thousands of integrations per cm2 of regenerated epidermis; (ii) all clonogenic cells contained in 4Mc, 8Mc1 and 8Mc2 cultures would have independent integrations, irrespectively of the clonal type. Instead, if the transgenic epidermis was sustained only by a restricted number of long-lived stem cells (continuously generating pools of TA progenitors), (i) we would have recovered, at most, only few hundreds of integrations per cm2; (ii) mero- and paraclones contained in 4Mc, 8Mc1 and 8Mc2 cultures would have the same integrations found in the corresponding holoclones.
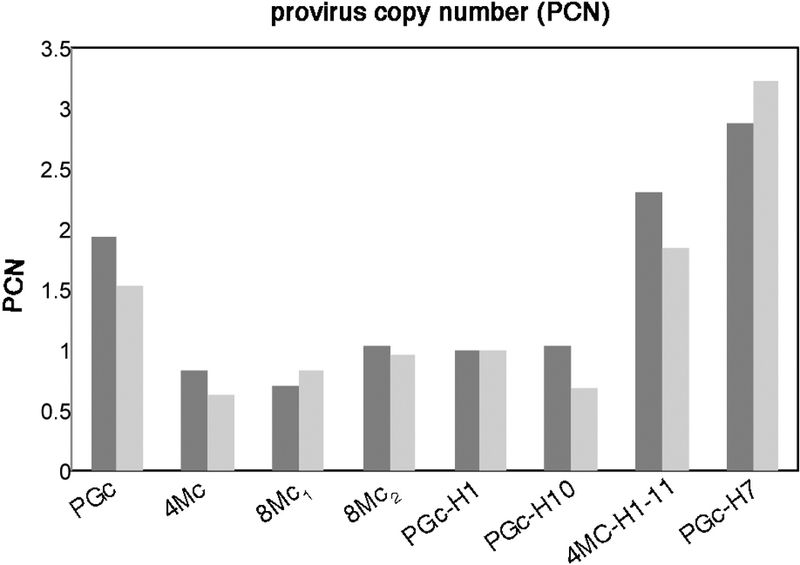
The number of integrations detected in post-graft cultures (Fig. 3a) is consistent with the number of stem cells that have been transplanted (Extended Data Fig. 4), hence it strongly supports the latter hypothesis, which was verified by proviral analyses at clonal level (Extended Data Fig. 5) on PGc, 4Mc and 8Mc1. A total of 687 clones (41 holoclones and 646 mero/paraclones) were analysed. PGc, 4Mc and 8Mc1 generated 20, 14 and 7 holoclones and 259, 264 and 123 mero/paraclones, respectively. Thus, PGc, 4Mc and 8Mc1 contained 7.2%, 5.0% and 5.4% holoclone-forming cells, respectively (Extended Data Table 3). Each clone was cultivated for further analysis. Libraries of vector-genome junctions, generated by linear-amplification-mediated (LAM) PCR followed by pyrosequencing, retrieved 31 independent integrations unambiguously mapped on the genome of holoclones (Extended Data Table 2). One holoclone (4Mc) was untransduced, 28, 11 and 1 holoclones contained 1, 2 and 3 integrations, respectively. Eleven holoclones in 4Mc shared the same integration pattern. The same happened for two couples of holoclones in 8Mc1. Holoclones’ copy numbers were confirmed by RTq-PCR (Extended Data Fig. 6). Strikingly, 75% and 80% of integrations found in 4Mc and 8Mc1 holoclones were retrieved in PGc, respectively (Fig. 4a), supporting the NGS-based survey as well as a representative sampling. The integration pattern observed in holoclones confirms absence of selection of specific integrations during epidermal renewal in vivo (Fig. 4b) and mirrors the pattern found in their parental cultures (Fig. 3c), including absence of genes associated to cell cycle control, cell death, or oncogenesis (Fig. 3d and Extended Data Table 1).
Figure 4. Integration profile of stem and TA cells.

a, Percentage of holoclone integrations recovered in the PGc bulk population. b, Holoclone integrations mapped to: promoters, exons, and introns, and intergenic regions (left); epigenetically defined active and weak promoters and enhancers, or genomic regions with no histone marks (right). c, The PGc pie chart (grey segment) shows that 91% of mero/paraclones did not contain the same integrations detected in the corresponding holoclones (each indicated by different blue segments). The 4Mc and 8Mc1 pie charts (grey segments) show that such percentage decreased to 37% and 17%, respectively.
Clonal tracing was then performed by PCR, using genomic coordinates of holoclone insertions. As expected, the vast majority of PGc meroclones and paraclones (91%) did not contain the same integrations detected in the corresponding holoclones (Fig. 4c, PGc). Such percentage decreased to 37% already at 4 months after grafting (Fig. 4c, 4Mc). Strikingly, virtually the entire clonogenic population of primary keratinocyte cultures established at 8 months contained the same integrations detected in the corresponding holoclones (Fig 4c, 8Mc1). Thus, the in vivo half-live of TA progenitors is of approximately 3-4 months. These data formally show that the regenerated epidermis is sustained only by long-lived stem cells (holoclones) and underpins the notion that meroclones and paraclones are short-lived progenitors continuously generated by the holoclones, both in vitro and in vivo. The high percentage of holoclone integrations retrieved in PGc, together with the number of shared events across cultures (Fig. 3b), suggests that the average coverage of the NGS analysis in PGc allowed to preferentially identify integrations in holoclones and in TA cells deriving from such holoclones already during the cultivation process.
In summary, as depicted in Extended Data Fig. 7, altogether these findings demonstrate that (i) PGc consisted of a mixture of independent transgenic holoclones, meroclones and paraclones, (ii) meroclones and paraclones (which can be isolated directly from a skin biopsy) are TA progenitors, do not self-renew and are progressively lost during cultivation and in vivo epidermal renewal, hence do not contribute to long-term maintenance of the epidermis; (iii) the transgenic epidermis is sustained only by long-lived stem cells detected as holoclones; (iv) founder stem cells contained in the original primary culture must have gone extensive self-renewal (in vitro and in vivo) to ultimately sustain the regenerated epidermis, as confirmed by the number of shared events across samples and across holoclones.
Discussion
The entire epidermis of a JEB patient can be replaced by autologous transgenic epidermal cultures harbouring an appropriate number of stem cells. Both stem and TA progenitors are instrumental for proper tissue regeneration in mammals24. However, the nature and the properties of mammalian epidermal stem cells and TA progenitors are a matter of debate25,26. Although epidermal cultures have been used for 30 years in the clinic14, a formal proof of the engraftment of cultured stem cells has been difficult to obtain. Similarly, the identification of holoclones as human epithelial stem cells and mero/paraclones as TA progenitors and their role in long-term human epithelial regeneration have been inferred from compelling, yet indirect evidence6,8,9,27. Using integrations as clonal genetic marks, we show that the vast majority of TA progenitors are progressively lost within a few months after grafting and the regenerated epidermis is indeed sustained only by a limited number of long-lived, self-renewing stem cells. Similar data have been produced with transgenic hematopoietic stem cells28. This notion argues against a model positing the existence of a population of equipotent epidermal progenitors that directly generate differentiated cells during the lifetime of the animal25 and fosters a model where specific stem cells persist during the lifetime of the human and contribute to both renewal and repair by giving rise to pools of progenitors that persist for various periods of time, replenish differentiated cells and make short-term contribution to wound healing26. Hence, the essential feature of any cultured epithelial grafts is the presence (and preservation) of an adequate number of holoclone-forming cells. The notion that the transgenic epidermis is sustained only by engrafted stem cells further decreases the potential risk of insertional oncogenesis.
In conclusion, transgenic epidermal stem cells can regenerate a fully functional epidermis virtually indistinguishable from a normal epidermis, so far in the absence of related adverse events. The different forms of EB affect approximately 500,000 people worldwide (http://www.debra.org). The successful outcome of this study paves the road to gene therapy of other types of EB and provides a blueprint that can be applied to other stem cell-mediated combined ex vivo cell and gene therapies.
Methods
Ethics statement
Five weeks after the patient’s admission, we considered a palliative treatment, as the clinical situation had deteriorated. The patient’s father asked for possible experimental treatments. We informed the parents on the possibility of the transplantation of genetically modified epidermal cultures. With the help of an interpreter, the parents were informed that the aforementioned procedure had been applied only on two patients with epidermolysis bullosa and on limited body sites. They were also informed that, given the patient’s critical conditions, the complexity of the entire surgical procedure needed for graft application could have been itself lethal. The potential risk of tumour development within the transplant was also discussed. As the parents still expressed their wish to use this experimental procedure, the local research ethics committee of the Medical Faculty of the Ruhr-University Bochum, contacted in July 2015, gave its approval to perform the procedure if responsible authorities approved the proposed treatment in our patient. We contacted the Paul-Ehrlich-Institut, which referred the request to the District Council of Arnsberg. The District Council of Arnsberg, North Rhine-Westphalia, Germany, which was responsible for the approval of committed treatments with new medical products, authorized the compassionate use of combined ex vivo cell and gene therapy in August 2015. The District Council of Duesseldorf, North Rhine-Westphalia, Germany, approved the genetic engineering work according to the Act on Genetic Engineering §9 Abs. 2 GenTG on the basis of the pre-existing approval for the Gene Technology Lab Security Level 2, which had been amended to the operating room of the BG University Hospital Bergmannsheil, Ruhr-University Bochum in August 2015.
The entire procedure used to prepare the transgenic epidermis has been previously scientifically reviewed and evaluated by the Italian Ministry of Health and approved by the ethical review board of the University of Modena and Reggio Emilia, both of which approved a phase I/II clinical trial with the very same transgenic cultures in June 2015. Similarly, the Austrian regulatory authorities scientifically reviewed and approved 2 additional clinical trials envisaging the use of very similar transgenic cultures, the only difference being the transgene used in the vector.
All procedures were performed in adherence to the last available (2008) version of the International Society for Stem Cell Research (ISSCR) “Guidelines for the Clinical Translation of Stem Cells”. Since all legal requirements currently required in Germany to obtain the approval for the treatment were fully met and the clinical condition of the patient was rapidly deteriorating, we opted to proceed with the life saving treatment, which was started in September 2015, after obtaining the parents’ informed consent. All documents were presented to the parents in German and their native language translated by an accredited translator. The patient’s parents also consented on the publication of photographs and medical information included in this publication. All photographs were presented to them before signing the consent forms.
Patient, clinical course, surgical, and post-operative procedures
Since birth, the patient repeatedly developed blisters, upon minor trauma, on the back, the limbs and the flanks, which occasionally caused chronic wounds persisting up to one year. Six weeks before the actual exacerbation, his condition deteriorated with the development of massive skin lesions. One day prior to admission, he developed fever followed by massive epidermal loss. He was admitted to a tertiary care hospital where topical wound care was performed using absorbable foam dressings (Mepilex, Mölnlycke Healthcare, Erkrath, Germany). As the patient appeared septic with elevated infection parameters, he initiated systemic antibiotic treatment with meropenem and vancomycin. Severe electrolyte imbalances required parenteral substitution of sodium, potassium, and magnesium. Swabs revealed Staphylococcus aureus and Pseudomonas aeruginosa. Due to the large wound area and further deterioration of his clinical condition, the patient was transferred to the paediatric burn centre of the Ruhr-University 4 days later. At admission, he suffered complete epidermal loss on ~60% of total body surface area (TBSA), affecting all limbs, the back and the flanks. The patient was febrile, cachectic, with a total body weight of 17 kg (below 3rd percentile), had signs of poor perfusion and C-reactive protein (CRP) was 150 mg/L. Antibiotic treatment was continued according to microbiologic assessment with flucloxacilline and ceftazidime. Retrospectively, the diagnosis of staphylococcal scalded skin syndrome was suspected due to flaky desquamations appearing 10 d after the symptoms began and Staphylococcus aureus was found on swabs. The iscorEB clinician score29 was rated at 47. We initiated aggressive nutritional therapy by nasogastric tube (1100-1300 kcal/d) and additional parenteral nutrition (700 kcal/d kcal/kg/d, glucose 4 g/kg/d, amino acids 3 g/kg/d, fat 1.5 g/kg/d) according to his nutritional demands calculated using the Galveston formula. A necessary intake of about 1800 kcal/d was determined. Vitamins and trace elements were substituted as needed since zinc, selenium, and other trace elements were below the detection threshold. Beta-adrenergic blockade with propranolol was also started, as with severe burns30. Due to bleeding during dressing changes and on-going loss of body fluids from the widespread skin erosions, the transfusion of 300 ml packed red blood cells was required every 7 to 12 days to keep the Hb value above 6-7 g/dl, and 20 g albumin were substituted once per week to keep albumin levels above 2.0 g/dl. Patient care was performed in accordance with the epidermolysis bullosa treatment guidelines31. The patient was bathed in povidone-iodine (PVP) solution or rinsed with polyhexanide-biguanide solution (PHMB) under general anaesthesia, first on a daily basis and subsequently every other day. We also employed several topical wound dressings and topic antimicrobials, including PHMB-gel and PVP ointment, without any significant impact on wound healing. However, wounds became cleaner and Staphylococcus aureus were no longer detectable for several weeks. The patient had persistent systemic inflammatory response syndrome (SIRS) with spiking fevers, wasting, and high values of acute-phase proteins (CRP, ferritin). He had chronic pain necessitating comprehensive drug management using fentanyl, dronabinol, gabapentin, amitryptiline and NSAIDs. Antibiotic treatment was continued according to swabs taken once weekly; swabs revealed intermittent wound infection with Pseudomonas aeruginosa and in the course Enterobacter cloacae, Enterococcus faecalis and again Staphylococcus aureus. Treatment was changed biweekly omitting glycopeptides, carbapenemes and other drugs of last resort using mainly ceftazidime, cefepime, ampicilline, flucloxacilline, and tobramycin. Due to his life-threatening condition, we performed an unsuccessful allotransplantation of split-thickness skin grafts taken from his father. Despite an initial engraftment, complete graft loss occurred 14 days post-transplantation. Treatment attempts with Suprathel (Polymedics Innovation GmbH, Denkendorf, Germany), amnion, and glycerol preserved donor skin (Glyaderm, Euro Tissue Bank, Beverwijk, Netherlands) were unsuccessful as well. Further treatment attempts were judged to be futile by several experts in this field. After 5 weeks at the intensive care unit, the patient no longer tolerated nutrition via nasogastric or duodenal tube and began to vomit after small amounts of food. Due to massive hepatosplenomegaly, a PEG or PEJ was not feasible. A Broviac catheter was implanted and total parenteral nutrition was begun (1500 kcal/d, glucose 14 g/kg/d, amino acids 4 g/kg/d, fat 2 g/kg/d). Following an attempt of increased fat administration via parenteral nutrition, the patient developed a pancreatitis that resolved after omitting fat from the parenteral nutrition for a few days. With this nutritional regimen the patient’s weight remained stable and blood glucose below 150 mg/dl was obtained without insulin administration. At this point, palliative care seemed the only remaining option. Because of the very poor short-term prognosis, we decided to start an experimental therapy approach using autologous epidermal stem cell-mediated combined ex-vivo cell and gene therapy (see Ethics Statement). Transgenic grafts were prepared, free of charge, under Good Manufacturing Practices (GMP) standards by Holostem Terapie Avanzate S.r.l. at the the Centre for Regenerative Medicine “Stefano Ferrari”, University of Modena and Reggio Emilia, Modena, Italy. On October 2015, we performed the first transplantation of transgenic cultures on the 4 limbs (and part of the flanks). At that time, the patient suffered complete epidermal loss on ~80% of his body and still needed transfusion of 300 ml packed red blood cells every 7 to 12 days and 20 g albumin once per week to keep the albumin level above 2.0 g/dl. He continued suffering from spiking fevers, wasting, and high values for acute-phase proteins (CRP, Ferritin). Wounds were colonized with Staphylococcus aureus and Escherichia coli. Perioperative antibiotic therapy was performed with flucloxacilline, ceftazidime and ciprofloxacine. Under general anaesthesia, a careful and thorough disinfection with octenidine dihydrochloride (Schuelke & Mayr, Norderstedt, Germany) and surgical debridement of all limbs and flanks was performed, both with copper sponges and surgical knife. The debrided areas demonstrated a good perfusion with intact dermis. After achieving haemostasis using epinephrine soaked gauze, all debrided areas were washed thoroughly with saline to prevent epinephrine contact with cultured grafts. Grafts were carefully transplanted on the denuded, debrided areas and covered with Adaptic, a non-adhering dressing (Systagenix Wound Management, Gargrave, UK) and sterile dressing. Post-operatively, as total immobilization was recommended after the transplantation, the patient was maintained under continuous isoflurane sedation for 12 days using the AnaConDa system (SedanaMedical, Uppsala, Sweden). A catheter related blood-stream infection was successfully treated with vancomycin and meropeneme. Despite the use of clonidine and propofol, the patient developed a severe delirium after the isoflurane sedation, which was solved by levomepromazine. Engraftment was evaluated at 8-14 days. Epidermal regeneration was evaluated at 1 month (see text). Following the first transplantation, regular weekly transfusion of red blood cells and infusion of albumin was no longer necessary. The general condition improved and enteral nutrition became feasible again with the patient tolerating up to 400 kcal/d via nasogastric tube complementing the parenteral nutrition (1500 kcal/d, glucose 14 g/kg/d, amino acids 4 g/kg/d, fat 2 g/kg/d)32. On November 2015, a second transplantation was performed on the dorsum, the buttocks (and small areas on the shoulders and the left hand). These wounds were colonized with Staphyloccus epidermidis and Enterococcus faecium at the time of transplantation. Antibiotic treatment was done with vancomycin and ceftazidime due to suspected infection of the Broviac catheter. However, due to the high risk and severe side effects of long-term sedation, the patient was not sedated after the second transplantation. All dressings at the back and the buttocks had to be removed due to infection with enterococcus faecium four days after transplantation. Topical antimicrobial therapy using polihexanide was started. On the dorsum, the graft healed in the following four weeks despite the early infection, and a stable skin without blister formation appeared (see text). Four weeks after the second transplantation, the CRP values remained below 100 mg/L and the patient was no longer febrile (Extended Data Fig. 8). Complete enteral nutrition became feasible again. The affected body surface area remained below 10% TBSA. On January 2016, we performed a third procedure in a similar fashion covering the remaining defects on flanks, thorax, right thigh, right hand, and shoulders. These wounds were colonized with Staphylococcus epidermidis. The transplanted cells engrafted well. The patient could be withdrawn from his analgesics. The Broviac catheter was removed and the patient was discharged 7 ½ months after admission. At this time, he still had minor defects on the right thigh and the buttocks (Fig. 1 and Extended Data Fig. 2). The iscorEB clinical score was 12. The transplanted skin was clinically stable and not forming blisters. The child returned back to regular elementary school on March 2016.
Cell lines
3T3J2 cell line
Mouse 3T3-J2 cells were a gift from Prof. Howard Green, Harvard Medical School (Boston, MA, USA). A clinical grade 3T3-J2 cell bank was established under GMP standards by a qualified contractor (EUFETS, GmbH, Idar-Oberstein, Germany), according to the ICH guidelines. GMP-certified 3T3-J2 cells have been authorized for clinical use by national and European regulatory authorities and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% irradiated calf serum, glutamine (4 mM) and penicillin-streptomycin (50 IU/ml).
MFG-LAMB3-Packaging cell line
A retroviral vector expressing the full-length 3.6-kb LAMB3 cDNA under the control of the MLV LTR was constructed by cloning a 3.6-kb of LAMB3 cDNA (Gene Bank Accession #Q13751) into MFG-backbone 13. A 5’ fragment of LAMB3 cDNA (563bp) from the ATG to StuI site was obtained by PCR using as template the LB3SN plasmid 33. The PCR product was cloned into NcoI and BamHI sites of MFG-vector. The second fragment of LAMB3 cDNA (3050bp) was obtained from LB3SN by enzyme digestion from StuI to XmnI and cloned into MGF-vector into StuI site. The entire cDNA of LAMB3 was fully sequenced. The Am12-MGFLAMB3 producer cell lines were generated by transinfection in the amphotropic Gp+envAm12 packaging cell line 34. Briefly, plasmid DNA was introduced into the GP+E86 ecotropic packaging cell line 34 by standard calcium phosphate transfection. Forty-eighth ours after transfection, supernatant was harvested and used to infect the amphotropic packaging cell line GP+envAml2 ATCC n° CRL 9641 13 for 16h in the presence of 8 ug/ml Polybrene. Infected Am12 cells were clonally selected in HXM medium supplemented with 10% FCS, and containing 0.8mg/ml G418 and 0.2mg/ml hygromycin B (Sigma). Single colonies were screened for human LAMB3 production by immunofluorescence using an antibody specific for LAMB3 6F12 monoclonal antibody (from Dr. Patricia Rousselle, CNRS, Lyon) and for viral titer. The resulting producer cell lines showed a viral titer of 2X106 colony-forming units (cfu). A master cell bank of a high-titer packaging clone (Am12-LAMB3 2/8) was made under GMP standards by a qualified contractor (Molmed S.p.A, Milan, Italy) according to the ICH guidelines and cultured in DMEM supplemented with 10% irradiated fetal bovine serum, glutamine (2 mM), and penicillin-streptomycin (50 IU/ml). All certifications, quality and safety tests (including detection on viruses and other micro-organisms both in vitro and in vivo) were performed under GMP standards for both cell lines.
Generation of genetically corrected epidermal sheets and graft preparation
Primary cultures were initiated from skin biopsy taken from a non-blistering area of inguinal region. Transgenic cultured epidermal grafts were prepared under GMP standards by Holostem Terapie Avanzate S.r.l. at the Centre for Regenerative Medicine “Stefano Ferrari”, University of Modena and Reggio Emilia, Modena, Italy. Briefly, a 4-cm2 skin biopsy was minced and trypsinized (0.05% trypsin and 0.01% EDTA) at 37°C for 3h. Cells were collected every 30 min, plated (2.7x104 cells/cm2) on lethally irradiated 3T3-J2 cells (2.66x104 cells/cm2) and cultured in 5% CO2 and humidified atmosphere in keratinocyte growth medium (KGM): DMEM and Ham’s F12 media (2:1 mixture) containing irradiated fetal bovine serum (10%), insulin (5 μg/ml), adenine (0.18 mM), hydrocortisone (0.4 μg/ml), cholera toxin (0.1 nM), triiodothyronine (2 nM), glutamine (4 mM), epidermal growth factor (10 ng/ml), and penicillin-streptomycin (50 IU/ml). Sub-confluent primary cultures were trypsinized (0.05% trypsin and 0.01% EDTA) at 37°C for 15-20 minutes and seeded (1.33x104 cells/cm2) onto a feeder-layer (8x104 cells/cm2) composed of lethally irradiated 3T3-J2 cells and producer GP+envAm12-LAMB3 cells12 (a 1:2 mixture) in KGM. After 3 days of cultivation, cells were collected and cultured in KGM onto a regular 3T3-J2 feeder-layer. Sub-confluent transduced cultures were pooled, re-suspended in KGM supplemented with 10% glycerol, aliquoted, and frozen in liquid nitrogen (36 vials, 5x106 cells/vial). At each step, efficiency of colony formation (CFE) by keratinocytes was determined by plating 1000 cells, fixing colonies with 3.7% formaldehyde 12 days later and staining them with 1% Rhodamine B.
For the preparation of plastic-cultured grafts, transduced keratinocytes were thawed and plated (1x104 cells/cm2) on 100 mm culture dishes containing lethally irradiated 3T3-J2 cells and grown to confluence in KGM with no penicillin-streptomycin. Grafts were then detached with Dispase II, 2.5 mg/ml (Roche Diagnostics S.p.a.) and mounted basal side up on sterile non-adhering gauze (Adaptic, Systagenix Wound Management, Gargrave, UK). For fibrin-cultured grafts, fibrin gels were prepared in 144 cm2 plates (Greiner, Stuttgart, Germany) as described10,12,35. Fibrin gels consisted of fibrinogen (23.1 mg/ml) and thrombin (3.1IU/ml) in NaCl (1%), CaCl2 (1mM) and Aprotinin (1786 KIU/ml). Transduced keratinocytes were thawed and plated (1x104 cells/cm2) on lethally irradiated 3T3-J2 cells onto the fibrin gels and grown as above. Grafts were washed twice in DMEM containing 4 mM glutamine, and placed in sterile, biocompatible, non-gas-permeable polyethylene boxes containing DMEM and 4 mM glutamine. Boxes were closed, thermo-sealed and packaged into a sealed, sterile transparent plastic bag for transportation to the hospital.
Immunofluorescence (IF), In situ hybridization (ISH), transmission electron microscopy (TEM) and Hematoxylin/Eosin staining
The following antibodies were used for IF: mouse 6F12 monoclonal antibody to laminin 332-β, laminin 332-α3 BM165 mAb (both from Dr. Patricia Rousselle, CNRS, Lyon), laminin 332-γ2 D4B5 mAb (Chemicon), α6 integrin 450-30A mAb and β4 integrin 450-9D mAb (Thermo Fisher Scientific).
For immunofluorescence, normal skin biopsies were obtained as anonymized surgical waste, typically from abdominoplasties or mammoplasty reduction and used as normal control. Ethical approval for obtaining the tissue, patient information sheets, and consent forms have been obtained and approved by our institutions (Comitato Etico Provinciale, Prot. N° 2894/C.E.). The patient’s skin biopsies were taken randomly, upon agreement patient information sheets and consent forms, at 4, 8 and 21 months. Skin biopsies were washed in PBS, embedded in Killik-OCT (Bio-Optica) and frozen. Immunofluorescence was performed on 7μm skin sections (fixed in PFA 3%, permeabilized with PBS/triton 0.2% for 15 min at r.t. and blocked 1h at r.t with BSA 2% in PBS/triton 0.2%) using the previous described antibodies in BSA 2% in PBS/triton 0.2% and added to skin sections for 30 min at 37°C. Sections were washed 3 times in PBS/triton 0.1% and incubated with Alexa Fluor 488 goat anti-mouse (Life Technologies), diluted 1:2,000 in BSA 2%, PBS/triton 0.2% for 30 min at 37°C. Cell nuclei were stained with DAPI. Glasses were then mounted with Dako Mounting medium and fluorescent signals were monitored under a Zeiss confocal microscope LSM510meta with a Zeiss EC Plan-Neofluar 40x/1.3 oil immersion objective.
To assess the percentage of transduced colonies, 10,000 cells from the sub-confluent transduced PGc pool were plated on a chamber slide and cultivated for 5 days as above. Chamber slides were fixed in methanol 100% for 10 min at -20°C and immunofluorescence analysis was performed as above. Laminin 332-β positive colonies were counted under a Zeiss Microscope AXIO ImagerA1 with EC-Plan Neofluar 20x/0.5 objective.
In situ hybridization (ISH) was performed on 10μm skin sections. DIG-RNA probe synthesis was performed according to the manufacturer’s instructions (Roche, DIG Labelling MIX). Primer pairs with Sp6/T7 promoter sequences (MWG Biotech) were used to obtain DNA templates for in vitro transcription. The following vector-specific primers were used: 5’-Sp6-AGTAACGCCATTTTGCAAGG-3’ (Tm 60°C) and 5’-T7-AACAGAAGCGAGAAGCGAAC-3’ (Tm 58°C) 11,12. OCT sections were fixed in PFA 4% and permeabilized with proteinase K 5μg/ml and post-fixed in PFA 4%. Sections were then incubated in hybridization solution (50% formamide, 4x SSC, Yeast RNA 500 μg/ml, 1x Denhard’s solution, 2 mM EDTA, 10% dextran sulfate in DEPC treated water) at 37°C for 1 h. DIG-probes were diluted in pre-heated hybridization solution at 80°C for 2 min and added to the slice for 20 h at 37°C. Sections were washed, blocked in Antibody buffer (1% blocking reagent from Roche in PBS tween 0.1%) containing 10% sheep serum for 1 h at RT. Anti-DIG antibody 1:200 was diluted in the same blocking solution and added to the slide for 4 h at room temperature. Signals were developed with BM-Purple solution ON at RT until signal reached the desired intensity. Slices were then mounted in 70% glycerol and visualized with Zeiss Cell Observer microscope with EC-Plan Neofluar 20x/0.5 objective.
For transmission electron microscopy, skin biopsies were fixed in 2.5% glutaraldehyde in Tyrode’s saline pH 7.2 (24 hr at 4C°), post fixed in 1% osmium tetroxide (Electron Microscopy Sciences) for 2 hr at room temperature, dehydrated in ethanol and propylene oxide, and embedded in Spurr resin (Polysciences). Ultrathin 70nm-thick sections were collected on copper grids, stained with uranyl acetate and lead citrate, and observed with a Jeol 1200 EXII (Jeol Ltd, Akishima, Japan) electron microscope.
For H&E staining, sections (7μm) were stained with H&E (Harris hematoxylin for 2 min, running tap water for 1 min, eosin Y for 2 min, 70% ethanol for 1 min, 95% ethanol for 1 min, 100% ethanol for 1 min, two rinses in 100% xylene for 1 min each) and observed with Zeiss Microscope AXIO ImagerA1 with EC-Plan Neofluar 20x/0.5 objective.
Clonal Analysis and DNA Analysis
Clonal analysis was performed as described34 and shown in Extended Data Fig. 5. Sub-confluent epidermal cultures were trypsinized, serially diluted and plated in 96 wells plates (0.5 cells/well). After 7 d of cultivation, single clones were identified under an inverted microscope and trypsinized. A quarter of the clone was cultured for 12 days onto a 100 mm (indicator) dish, which was then fixed and stained with Rhodamine B for the classification of clonal type3. The remaining part of the clone (3/4) was cultivated on 24-multiwell plates for genomic DNA extraction and further analysis (Extended Data Fig. 5).
Library preparation and sequencing
Illumina barcoded libraries were obtained from 3 independent pre-graft cultures (PGc, generated by 3 vials, each containing ~220,000 clonogenic keratinocytes) and 3 post-graft cultures (4Mc, 8Mc1, and 8Mc2). For each sample, 2 tubes with 500 ng of genomic DNA were sheared in 100 μl of water applying 3 sonication cycles of 15 sec/each in a Bioruptor (Diagenode) to obtain fragments of 300-500 bp. Fragmented DNA was recovered through purification with 0.8 volumes of Agencourt AMPure XP beads, two washing steps with 80% ethanol, and elution in Tris-HCl 10 mM. Repair of DNA ends and A-tailing of blunt ends were both performed using Agilent SureSelectXT reagents (Agilent Technologies), according to manual specifications, followed by purification with 1.2 volumes of AMPure XP beads. A custom universal adapter was generated by annealing <Phos-TAGTCCCTTAAGCGGAG - C3> oligo and <GTAATACGACTCACTATAGGGCNNNNNNCTCCGCTTAAGGGACTAT> oligo on a thermocycler from 95°C to 21°C, with decrease of 1°C/min in a 10 mM Tris-HCl, 50 mM NaCl buffer. Ligation of universal adapter to A-tailed DNA was carried out in a reaction volume of 30μl with 400 U of T4 DNA ligase (New England Biolabs) with respective T4 DNA ligase buffer 1X and 35 pmol of dsDNA universal adapter and incubated at 23°C for 1 h, at 20°C for 1 h, and finally heat inactivated at 65° C for 20 min. Each ligation product was purified with 1.2 volumes of AMPure XP beads as described above. Eluate of each reaction was split in 3 different tubes to perform independent PCR reaction in order to mitigate reaction-specific complexity reduction. Each tube was amplified by PCR with a combination of I7-index primers (701/702/703), to multiplex samples on the same Illumina sequencing lane, and of two I5 LTR-primers (501/502) to barcode specific enrichments of MLV-LTR sequences (Supplementary Table 1). PCR reaction was carried out in a final volume of 25 μL, with 20 pmoles of each primer and Phusion High-Fidelity master mix 1X (New England Biolabs). PCR products were purified with 0.8 AMPure XP beads and all amplification products from the same sample (2 fragmentations, 3 PCR reactions) were pooled and quantified on Bioanalyzer 2100 high sensitivity chip. Paired-end 125 bp sequencing was performed on Illumina HiSeq2500 (V4 chemistry). Illumina barcodes on the whole Illumina lanes were combined to maintain a minimum hamming-distance of at least 3 nucleotides. Extraction and de-multiplexing of reads was obtained using CASAVA software (v. 1.8.2) applying a maximum barcode mismatch of 1 nucleotide and considering the dual indexing of I7-I5 sequences. Reads were processed using the bioinformatics pipeline described in details in the Methods. Briefly, reads were first inspected with cutadapt36 to verify specific enrichments, then trimmed using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) and bbduk2 (http://jgi.doe.gov/data-and-tools/bbtools/) to remove adaptors and primers, and mapped to the human genome reference sequence GRCh37/hg19 using BWA MEM37 with default parameters and the -M flag. Finally, the start coordinate of the alignment was used as the putative integration site.
Genomic and functional annotation of integration events
Annotation of integration sites to gene features was performed using the ChIPseeker R package36. Insertion sites were mapped to promoters (defined as 5 kb regions upstream of the transcription start site), exons, and introns of RefSeq genes, and intergenic regions. Functional enrichment in GO Biological Processes of genes harboring an integration site was performed using the clusterProfiler R package36, setting a q-value threshold of 0.05 for statistical significance. Annotation of integration sites to epigenetically defined transcriptional regulatory elements was performed with the BEDTools suite 38 using publicly available ChIP-seq data of histone modifications (H3K4me3, H3K4me1, and H3K27ac) in human keratinocyte progenitors (GSE64328)36.
Linear amplification-mediated (LAM) PCR, NGS on holoclones, PCR on mero/paraclones and integration site analysis
100 ng of DNA of transduced keratinocytes was used as template for LAM-PCR. LAM-PCR product was initiated with a 50-cycle linear PCR and digested with 2 enzymes simultaneously without splitting the DNA amount using 1μl MseI (5U/μl) and 1μl PstI (5U/μl) (Thermo Fisher, Waltham, US) and ligation of a MseI restriction site–complementary linker cassette. LAM-PCR was digested with 2 enzymes simultaneously without splitting the DNA amount. The second enzyme PstI was introduced to eliminate the undesired 5´LTR-LAMB3 sequences. The first exponential biotinylated PCR product was captured via magnetic beads and reamplified by a nested second PCR. LAM-PCR primers for MLV-LAMB3 used are in table 2. For the initial LAM-PCR, the 5′-biotinylated oligonucleotide complementary to the 3´-LTR sequence (5´-GGTACCCGTGTATCCAATAA-3´) was used for the linear amplification step. The 2 sequential exponential amplification steps were performed with nested oligonucleotides complementary to the 3´- LTR sequence (5´- GACTTGTGGTCTCGCTGTTCCTTGG-3´); (5´-GGTCTCCTCTGAGTGATTGACTACC-3´), each coupled with the oligonucleotides complementary to the linker cassette (Supplementary Table 2). LAM-PCR amplicons were either separated on 2% standard agarose gels (Biozym, Hessisch Oldendorf, Germany) and the excised bands cloned into the StrataClone PCR Cloning Kit (Agilent Technologies, Santa Clara), PCR-purified using High Pure PCR Product Purification Kit (Roche, Basel, Switzerland), shotgun cloned, and sequenced by Sanger, or used as unpurified PCR product as template for NGS library preparation. The fragments were end-repaired, adaptor-ligated, nick-repaired and purified by using the Ion Plus Fragment Library Kit (Life Technologies, Carlsbad, US). The template preparation and the sequencing run on the machine were also performed according to the protocols of Life Technologies. A mean vertical coverage was planned to reach at least 2000 reads.
Screening of the integration sites of the meroclones and paraclones was done by PCR using a combination of the FW primer MLV 3´LTR control F (5’-GGACCTGAAATGACCCTGTG-3’) of the LTR and a specific reverse primer (Supplementary Table 3) in the proximity of the integration site. Genomic DNA from the holoclones was used as positive controls.
Provirus copy number (PCN)
TaqMan PCR analysis was performed with TaqMan Universal PCR Master Mix and vector-specific LAMB3 and GAPDH probes (LAMB3: Hs00165078_m1; GAPDH: Hs03929097_g1, Applied Biosystems). The amplicon for LAMB3 was located between adjacent exons to recognize only provirus LAMB3. Reactions were performed with ABI Prism 7900 Sequence Detection System (Applied Biosystems), using 10 ng of genomic DNA. The relative quantity that relates the PCR signal of the target provirus was normalized to the level of GAPDH (internal control gene) in the same genomic DNA by using the 2-ΔΔCT quantification.
Bioinformatics analysis of sequencing data
To process the sequencing reads we assembled a custom bioinformatics pipeline composed of standard tools for NGS data analysis. In particular, we first used cutadapt (v1.14; https://cutadapt.readthedocs.io/en/stable/)36 to verify the presence, in read pairs, of specific sequences indicative of a successful enrichment. Specifically, in the read harboring the I5 LTR-primer sequence (read 1), we searched for the primer sequence and, at its 3’-end, for the remainder LTR sequence. Instead, in the read harboring the I7 indexing primer (read 2), we searched for the presence of the common adapter sequence preceding the 6 indexing bases. Pairs containing both sequences were retained for analysis after trimming the I5 primer and the remainder LTR sequence in read 1 and the common adapter sequence in read 2. Then, we used FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) to remove from read 2 the first 6 indexing bases, utilized as de-duplicator component during de-multiplexing. Since half of the amplification products are expected to be non-informative in the detection of the insertion site, given the identity of the two LTRs of the MLV genome, we applied bbduk2 (http://jgi.doe.gov/data-and-tools/bbtools/) to identify and remove read pairs representing inward-facing LTR primer enrichment events. In bbduk2 we set the kmer length to 27 (k=27) and the edit distance and the maxbadkmers parameters both to 1. Reads were aligned on the human genome reference sequence GRCh37/hg19 using BWA MEM 37 with default parameters and the -M flag (to include multiple-mapping signature in the BAM file). Read pairs sharing the same mapping coordinates and the same de-duplicator component were labeled as PCR duplicates and removed. Aligned read pairs were further filtered to retain only those mapping at a distance comprised between 150 and 600 bp (corresponding to the expected library insert size), allowing a maximum of 1 bp soft-clip (unaligned) on all ends, with the exception of the 5’ end of read 2 where we allowed 20 bp soft clip since it contains the 18 bp untrimmed common adapter sequence. Finally, we retained read 1 sequences with a minimum mapping quality of 40 and extracted and counted the alignment coordinates of their first base, representing the putative insertion site. Insertion sites within 10 bp from one another were treated as a single insertion, their counts summed using BEDTools (v2.15; http://bedtools.readthedocs.io/en/latest/content/bedtools-suite.html) 38 and the summed count assigned to left coordinate. When intersecting insertion sites across samples, we considered overlapping those insertion events closer than 30bp.
Calculation of the expected number of integrations
The expected number of integrations (i.e., the expected population size) in PGc, 4Mc, 8Mc1, and 8Mc2 samples was calculated in R applying a capture-recapture model based on the Chapman’s estimate and its confidence intervals 15,39:
where is the estimated number of integrations, n1 is the number of integrations found in the 3pIN library, n2 those found in the 3pOUT library, n11 the number of overlapping integrations, n12 and n21 the insertion respectively exclusive of 3pIN and 3pOUT, respectively, and Z1−α/2 = 2.56 for α=0.01.
Genomic and functional annotation of insertion events
To annotate the integration sites to gene features, we used the ChIPseeker R package (v1.10.3, https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html) 40. The integration sites were mapped to promoters, defined as 5 kb regions upstream of transcription start sites (TSS), exons, and introns of RefSeq genes, and to intergenic regions.
We performed functional annotation of genes harboring an integration sites using the clusterProfiler R package (v3.2.14; https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html) 41, setting a q-value threshold of 0.05 to define enriched Gene Ontology (GO) Biological Processes.
To annotate the integration sites to epigenetically defined transcriptional regulatory elements (promoters and enhancers), we used the BEDTools suite (v2.15; http://bedtools.readthedocs.io/en/latest/content/bedtools-suite.html) 38. We define promoters and enhancers using publicly available ChIP-seq data of histone modifications (H3K4me3, H3K4me1 and H3K27ac) produced in human keratinocyte progenitors 42. Briefly, bed files containing the coordinates of genomic regions enriched for each histone modification (peaks) were downloaded from the Gene Expression Omnibus database (GSM1568245 for H3K4me3, GSM1568244 for H3K4me1 and GSM1568247 for H3K27ac). H3K4me3 peaks close to the TSS (<5 kb) of RefSeq genes were defined as promoters, while H3K4me1 peaks far from TSS (>5 kb) were defined as enhancers. Promoters and enhancers were classified as “active” if they overlap with H3K27ac peaks, otherwise are classified as “weak”. Finally, integration sites were mapped to active and weak promoters and enhancers.
Differences in the annotation of integration sites to gene features and regulatory elements were tested using the chisq.test function (Pearson's Chi-squared test) of the stat R package.
Bioinformatics analysis of NGS data from holoclones
Analysis of the data was implemented with single read sequences of the BAM file. Output results with ≥ 5% of query cover, ≥ 95% identity, and a size of ≥ 48bp were taken into account for confirming as integration site with control PCR. Sequences were aligned to the human genome (Genome Reference Consortium GRCh37) using the NCBI BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Identification of the nearest gene was performed with dedicated PERL scripts. Visualization of the RTCGD CIS integrations as a feature on the UCSC BLAT output was achieved by connecting to UCSC through the RTCGD web interface (http://rtcgd.abcc.ncifcrf.gov); map position of each of the retroviral integrations was automatically loaded as custom tracks on the UCSC BLAT search engine.
Statistical analyses and data visualization
Statistical analyses were implemented in R (v3.3.1, http://www.r-project.org/). Figure 3d was generated using the ggplot2 R package (v2.2.1, https://cran.r-project.org/web/packages/ggplot2/index.html).
Data availability
All high-throughput sequencing data of the integration profiles have been deposited in the Sequence Read Archive (SRA) under accession number SRP110373. All data used to generate main and supplementary figures are provided as source data files.
Extended Data
Extended Data Table 1.
a. Enrichment of cancer-related biological process in genes harboring an insertion. Statistically significant enrichments at a 95% confidence level (q-value≤0.05 in a Fisher’s exact test) are in bold. GO categories were selected to represent the cancer hallmarks described in Hanahan D, Weinberg RA. Cell. 2011 Mar 4;144(5):646-74.
b. Genomic and functional annotations of integrations in holoclones.
| Cancer-related biological process | GO ID | Description | q-value (FDR) | |||
|---|---|---|---|---|---|---|
| PGc | 4Mc | 8Mc1 | 8Mc2 | |||
| Cell death and apoptosis | GO:0070265 | necrotic cell death | 0.28 | 0.58 | 0.56 | 0.65 |
| GO:0010939 | regulation of necrotic cell death | 0.31 | 0.53 | 0.53 | 0.64 | |
| GO:0097300 | programmed necrotic cell death | 0.25 | 0.54 | 0.53 | 0.66 | |
| GO:2001233 | regulation of apoptotic signaling pathway | --- | 0.52 | 0.67 | 0.72 | |
| DNA repair | GO:0006282 | regulation of DNA repair | 0.06 | 0.67 | --- | --- |
| GO:0006298 | mismatch repair | 0.53 | 0.54 | --- | 0.66 | |
| GO:0006302 | double-strand break repair | 0.64 | 0.57 | 0.72 | 0.91 | |
| GO:0006289 | nucleotide-excision repair | 0.82 | 0.75 | --- | 0.83 | |
| GO:0036297 | interstrand cross-link repair | 0.84 | 0.58 | --- | 0.72 | |
| Angiogenesis | GO:0001525 | angiogenesis | 9.54E-05 | 0.52 | 0.59 | 0.74 |
| GO:0045765 | regulation of angiogenesis | 0.53 | 0.73 | 0.73 | 0.72 | |
| Migration | GO:0090130 | tissue migration | 7.82E-06 | 0.50 | 0.53 | 0.04 |
| GO:0090132 | epithelium migration | 3.64E-06 | 0.50 | 0.53 | 0.04 | |
| GO:0010631 | epithelial cell migration | 3.26E-06 | 0.49 | 0.53 | 0.04 | |
| GO:0010632 | regulation of epithelial cell migration | 2.43E-06 | 0.44 | 0.53 | 0.05 | |
| GO:0051546 | keratinocyte migration | 0.22 | --- | 0.53 | 0.65 | |
| GO:0001667 | ameboidal-type cell migration | 3.19E-08 | 0.52 | 0.53 | 0.06 | |
| Inflammation | GO:0002526 | acute inflammatory response | 0.85 | 0.58 | 0.63 | --- |
| GO:0002544 | chronic inflammatory response | 0.82 | 0.45 | --- | --- | |
| GO:0050727 | regulation of inflammatory response | 0.80 | 0.69 | 0.61 | 0.80 | |
| Telomerase activity | GO:0000723 | telomere maintenance | 0.48 | 0.77 | 0.63 | 0.72 |
| GO:0007004 | telomere maintenance via telomerase | 0.38 | --- | --- | 0.73 | |
| GO:0032204 | regulation of telomere maintenance | 0.69 | --- | --- | 0.65 | |
| GO:0051972 | regulation of telomerase activity | 0.66 | --- | --- | --- | |
| Cell cycle | GO:0000075 | cell cycle checkpoint | 0.14 | 0.60 | 0.74 | --- |
| GO:1901976 | regulation of cell cycle checkpoint | 0.18 | --- | --- | --- | |
| GO:1901987 | regulation of cell cycle phase transition | 0.02 | 0.48 | 0.83 | --- | |
| GO:0045786 | negative regulation of cell cycle | 0.01 | 0.57 | 0.60 | --- | |
| Proliferation | GO:0050673 | epithelial cell proliferation | 4.06E-03 | 0.52 | 0.65 | 0.75 |
| GO:0050678 | regulation of epithelial cell proliferation | 0.01 | 0.52 | 0.60 | 0.72 | |
| GO:0043616 | keratinocyte proliferation | 1.24E-03 | 0.56 | 0.55 | --- | |
| GO:0010837 | regulation of keratinocyte proliferation | 0.01 | 0.54 | 0.53 | --- | |
| GO:0072089 | stem cell proliferation | 0.25 | 0.73 | 0.60 | --- | |
| Glycolysis | GO:0006096 | glycolytic process | 0.15 | 0.65 | --- | 0.75 |
| GO:0006110 | regulation of glycolytic process | 0.22 | 0.54 | --- | 0.66 | |
| Sample | chr | start | end | ID Holoclone | Annotation to genes | Gene symbol | Annotation to regulatory elements | Recoin I |
|---|---|---|---|---|---|---|---|---|
| PGc | chr5 | 131410002 | 131410003 | PGc_H1 | Intron | CSF2 | no mark | no |
| chr2 | 144859325 | 144859326 | PGc_H2 | Intron | GTDC1 | no mark | no | |
| chr4 | 101941589 | 101941590 | PGc_H3 | Intergenic | - | weak enhancer | no | |
| chr4 | 39355299 | 39355300 | PGc_H4 | Intron | RFC1 | weak enhancer | no | |
| chr19 | 17908000 | 17908001 | PGc_H5 | Intron | B3GNT3 | no mark | no | |
| chr19 | 42615156 | 42615157 | PGc_H6 | Intron | POU2F2 | no mark | no | |
| chr5 | 150977858 | 150977859 | PGc_H7* | Intergenic | - | active enhancer | yes | |
| chr17 | 80832738 | 80832739 | PGc_H7* | Intron | TBCD | active enhancer | yes | |
| chr16 | 56726522 | 56726523 | PGc_H7* | Intergenic | - | no mark | yes | |
| chr2 | 8999619 | 8999620 | PGc_H8 | Intron | MBOAT2 | no mark | no | |
| chr3 | 47024025 | 47024026 | PGc_H9 | Promoter | CCDC12 | active promoter | yes | |
| chrY | 18367597 | 18367598 | PGc_H10 | Intergenic | - | no mark | no | |
| chr6 | 160458524 | 160458525 | PGc_H11 | Intron | IGF2R | no mark | no | |
| chr14 | 91711334 | 91711335 | PGc_H12 | Promoter | GPR68 | active promoter | yes | |
| LOC10192813 | ||||||||
| chr11 | 13946563 | 13946564 | PGc_H13 | Promoter | 2 | no mark | yes | |
| chr14 | 33789922 | 33789923 | PGc_H14 | Intron | NPAS3 | no mark | no | |
| chr13 | 20693331 | 20693332 | PGc_H15 | Intergenic | - | weak enhancer | no | |
| chr6 | 136930722 | 136930723 | PGc_H16 | Intron | MAP3K5 | weak enhancer | yes | |
| chr18 | 65398639 | 65398640 | PGc_H17 | Intron | LOC643542 | no mark | no | |
| chr4 | 11625725 | 11625726 | PGc_H18 | Intergenic | - | active enhancer | no | |
| chr20 | 22743911 | 22743912 | PGc_H19 | Intergenic | - | no mark | yes | |
| chr8 | 48293010 | 48293011 | PGc_H20 | Intron | SPIDR | active enhancer | no | |
| 4Mc | chrl | 183130951 | 183130952 | 4Mc H1-11** | Intergenic | - | no mark | yes |
| chr9 | 103188807 | 103188808 | 4Mc_H1-11** | Promoter | MSANTD3 | active promoter | yes | |
| chr14 | 105213201 | 105213202 | 4Mc_H12 | Intron | ADSSL1 | no mark | no | |
| chr15 | 39577423 | 39577424 | 4Mc_H13 | Intergenic | - | no mark | yes | |
| 8MC1 | chr8 | 67025314 | 67025315 | 8Mc1_H1-2 | Intergenic | - | active enhancer | yes |
| chr9 | 125129763 | 125129764 | 8Mc1_H3 | Promoter | PTGS1 | no mark | yes | |
| chr17 | 76158277 | 76158278 | 8Mc1_H4-5 | Intron | C17orf99 | no mark | no | |
| chrX | 114601642 | 114601643 | 8Mc1_H6 | Intergenic | - | no mark | yes | |
| chr5 | 135342207 | 135342208 | 8Mc1_H7 | Intergenic | - | no mark | yes | |
Extended Data Table 2.
Clonal analysis was performed on pre-graft cultures (PGc), a graft ready for transplantation (Graft) and on primary cultures established at 4 (4Mc) and 8 (8Mc1) months after grafting. H, M and P indicate holoclones, meroclones and paraclones, respectively. Frequency indicates the percentage of holoclones detected in the population of clonogenic keratinocytes. Graft was not used for LAM-PCR or NGS analyses but for holoclone quantification as part of quality control.
| ID sample | ID replicate | Total number of clones | H | M/P | H Frequency (%) |
|---|---|---|---|---|---|
| PGc | PG-1 | 88 | 10 | 78 | 11.36 |
| PG-2 | 63 | 4 | 59 | 6.35 | |
| PG-3 | 62 | 3 | 59 | 4.84 | |
| PG-4 | 66 | 3 | 63 | 4.55 | |
| Total | 279 | 20 | 259 | 7.17 | |
| Graft | Graft | 95 | 4 | 91 | 4.21 |
| 4Mc | 4Mc-1 | 121 | 8 | 113 | 6.61 |
| 4Mc-2 | 105 | 5 | 100 | 4.76 | |
| 4Mc-3 | 52 | 1 | 51 | 1.92 | |
| Total | 278 | 14 | 264 | 5.04 | |
| 8Mc1 | 8Mc1-1 | 56 | 4 | 52 | 7.14 |
| 8Mc1-2 | 74 | 3 | 71 | 4.05 | |
| Total | 130 | 7 | 123 | 5.38 | |
Supplementary Material
Supplementary Information is available in the on line version of the paper.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This article is dedicated to the memory of Professor Howard Green, whose passion and commitment to excellence in science and education inspired the researchers of the Centre for Regenerative Medicine.
Holostem Terapie Avanzate s.r.l. met all costs of GMP production and procedures of transgenic epidermal grafts.
This work was partially supported by Italian Ministry of Education, University and Research (MIUR), n. CTN01_00177_888744; Regione Emilia-Romagna, Asse 1 POR-FESR 2007-13; Fondazione Cassa di Risparmio di Modena; DEBRA Südtirol - Alto Adige; DEBRA Austria; European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Program (Grant Agreement No. 670126-DENOVOSTEM) and ERC under the European Union's Seventh Framework Programme (Grant Agreement No. 294780-NOVABREED); Epigenetics Flagship project CNR-MIUR grants. We thank Ole Goertz, MD for his contribution to the surgical procedures, the Department of Anaesthesiology, in particular Peter Zahn, MD and Tim Maecken, MD and the entire OR staff, in particular Sarah Taszarski and Victoria Stroh, for their dedicated perioperative care, the nurses of ward PÄD1 for continuous and devoted assistance to the child. We thank Anna Neumayer and Jeremias Frank for technical assistance in defining clone integrations, Birgit Mussnig for performing indirect immunofluorescence, Maria Carmela Latella for determining the average number of integrations in pre and post-graft cultures and M. Forcato for critical feedback on the bioinformatics analyses.
Footnotes
Author contributions T.H., T.R., N.T., J.B., G.P., who contributed equally to this work, defined strategic procedures, performed transplantation of the transgenic grafts, surgical and medical procedures and clinical follow-up; L.D.R. performed immunofluorescence data and imaging analysis, analysed the data and assembled all input data, prepared the figures and edited the manuscript, D.S., I.J., M.M. performed integration profile of transgenic epidermis; R.C., J.R. A.K., and D.K. performed experiments of clonal tracing in epidermal cells; O.R. and S.Bi. conducted all bioinformatics analyses, A.S.S and E.E. performed in situ hybridization experiments, S.C. and S.Bo. performed all cultures procedures and preparation of genetically modified epidermal graft; F. J., T.L., M.L, M.F., M, K carried out the follow-up of patient, D.Q. performed electron microscopy analysis; M.D.L. coordinated the study, defined strategic procedures, administered the experiments and wrote the manuscript.
Competing financial interests G.P. and M.D.L. are co-founders and member of the Board of Directors of Holostem Terapie Avanzate (HTA), s.r.l, Modena, Italy; Chiesi Farmaceutici S.p.A. (a co-founder of HTA), holds an Orphan Drug Medicinal Product designation (EU/3/15/1465) for the transgenic cultures used in this paper.
References
- 1.Fine JD, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70:1103–1126. doi: 10.1016/j.jaad.2014.01.903. [DOI] [PubMed] [Google Scholar]
- 2.Fine JD, Johnson LB, Weiner M, Suchindran C. Cause-specific risks of childhood death in inherited epidermolysis bullosa. J Pediatr. 2008;152:276–280. doi: 10.1016/j.jpeds.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 3.Barrandon Y, Green H. Three clonal types of keratinocyte with different capacities for multiplication. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:2302–2306. doi: 10.1073/pnas.84.8.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pellegrini G, et al. Location and clonal analysis of stem cells and their differentiated progeny in the human ocular surface. J Cell Biol. 1999;145:769–782. doi: 10.1083/jcb.145.4.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gallico GG, 3rd, O'Connor NE, Compton CC, Kehinde O, Green H. Permanent coverage of large burn wounds with autologous cultured human epithelium. The New England journal of medicine. 1984;311:448–451. doi: 10.1056/NEJM198408163110706. [DOI] [PubMed] [Google Scholar]
- 6.Pellegrini G, et al. The control of epidermal stem cells (holoclones) in the treatment of massive full-thickness burns with autologous keratinocytes cultured on fibrin. Transplantation. 1999;68:868–879. doi: 10.1097/00007890-199909270-00021. [DOI] [PubMed] [Google Scholar]
- 7.Pellegrini G, et al. Long-term restoration of damaged corneal surfaces with autologous cultivated corneal epithelium. Lancet. 1997;349:990–993. doi: 10.1016/S0140-6736(96)11188-0. [DOI] [PubMed] [Google Scholar]
- 8.Rama P, et al. Limbal stem-cell therapy and long-term corneal regeneration. The New England journal of medicine. 2010;363:147–155. doi: 10.1056/NEJMoa0905955. [DOI] [PubMed] [Google Scholar]
- 9.Ronfard V, Rives JM, Neveux Y, Carsin H, Barrandon Y. Long-term regeneration of human epidermis on third degree burns transplanted with autologous cultured epithelium grown on a fibrin matrix. Transplantation. 2000;70:1588–1598. doi: 10.1097/00007890-200012150-00009. [DOI] [PubMed] [Google Scholar]
- 10.Bauer JW, et al. Closure of a Large Chronic Wound through Transplantation of Gene-Corrected Epidermal Stem Cells. Journal of Investigative Dermatology. 137:778–781. doi: 10.1016/j.jid.2016.10.038. [DOI] [PubMed] [Google Scholar]
- 11.De Rosa L, et al. Long-term stability and safety of transgenic cultured epidermal stem cells in gene therapy of junctional epidermolysis bullosa. Stem Cell Reports. 2014;2:1–8. doi: 10.1016/j.stemcr.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mavilio F, et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. 2006;12:1397–1402. doi: 10.1038/nm1504. [DOI] [PubMed] [Google Scholar]
- 13.Markowitz D, Goff S, Bank A. Construction and use of a safe and efficient amphotropic packaging cell line. Virology. 1988;167:400–406. [PubMed] [Google Scholar]
- 14.De Luca M, Pellegrini G, Green H. Regeneration of squamous epithelia from stem cells of cultured grafts. Regenerative medicine. 2006;1:45–57. doi: 10.2217/17460751.1.1.45. [DOI] [PubMed] [Google Scholar]
- 15.Chapman DG, Robbins H. Minimum Variance Estimation Without Regularity Assumptions. The Annals of Mathematical Statistics. 1951;22:581–586. [Google Scholar]
- 16.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Aiuti A, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. The New England journal of medicine. 2009;360:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- 18.Biasco L, et al. Integration profile of retroviral vector in gene therapy treated patients is cell-specific according to gene expression and chromatin conformation of target cell. EMBO Mol Med. 2011;3:89–101. doi: 10.1002/emmm.201000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cavazza A, et al. Self-inactivating MLV vectors have a reduced genotoxic profile in human epidermal keratinocytes. Gene Ther. 2013;20:949–957. doi: 10.1038/gt.2013.18. [DOI] [PubMed] [Google Scholar]
- 20.Hacein-Bey-Abina S, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hacein-Bey-Abina S, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. The New England journal of medicine. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 22.Howe SJ, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siprashvili Z, et al. Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With Recessive Dystrophic Epidermolysis Bullosa. JAMA. 2016;316:1808–1817. doi: 10.1001/jama.2016.15588. [DOI] [PubMed] [Google Scholar]
- 24.Hsu YC, Li L, Fuchs E. Transit-amplifying cells orchestrate stem cell activity and tissue regeneration. Cell. 2014;157:935–949. doi: 10.1016/j.cell.2014.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clayton E, et al. A single type of progenitor cell maintains normal epidermis. Nature. 2007;446:185–189. doi: 10.1038/nature05574. [DOI] [PubMed] [Google Scholar]
- 26.Mascre G, et al. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature. 2012;489:257–262. doi: 10.1038/nature11393. [DOI] [PubMed] [Google Scholar]
- 27.Pellegrini G, et al. Biological parameters determining the clinical outcome of autologous cultures of limbal stem cells. Regenerative medicine. 2013;8:553–567. doi: 10.2217/rme.13.43. [DOI] [PubMed] [Google Scholar]
- 28.Biasco L, et al. In Vivo Tracking of Human Hematopoiesis Reveals Patterns of Clonal Dynamics during Early and Steady-State Reconstitution Phases. Cell Stem Cell. 2016;19:107–119. doi: 10.1016/j.stem.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwieger-Briel A, et al. Instrument for scoring clinical outcome of research for epidermolysis bullosa: a consensus-generated clinical research tool. Pediatr Dermatol. 2015;32:41–52. doi: 10.1111/pde.12317. [DOI] [PubMed] [Google Scholar]
- 30.Herndon DN, et al. Long-term propranolol use in severely burned pediatric patients: a randomized controlled study. Ann Surg. 2012;256:402–411. doi: 10.1097/SLA.0b013e318265427e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldschneider KR, et al. Pain care for patients with epidermolysis bullosa: best care practice guidelines. BMC Med. 2014;12:178. doi: 10.1186/s12916-014-0178-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez NA, Jeschke MG, Williams FN, Kamolz LP, Herndon DN. Nutrition in burns: Galveston contributions. JPEN J Parenter Enteral Nutr. 2011;35:704–714. doi: 10.1177/0148607111417446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dellambra E, et al. Corrective transduction of human epidermal stem cells in laminin-5-dependent junctional epidermolysis bullosa. Hum Gene Ther. 1998;9:1359–1370. doi: 10.1089/hum.1998.9.9-1359. [DOI] [PubMed] [Google Scholar]
- 34.Mathor MB, et al. Clonal analysis of stably transduced human epidermal stem cells in culture. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:10371–10376. doi: 10.1073/pnas.93.19.10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guerra L, et al. Treatment of "stable" vitiligo by Timedsurgery and transplantation of cultured epidermal autografts. Arch Dermatol. 2000;136:1380–1389. doi: 10.1001/archderm.136.11.1380. [DOI] [PubMed] [Google Scholar]
- 36.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011;17:10–12. doi: 10.14806/ej.17.1.200. 2011. [DOI] [Google Scholar]
- 37.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Eprint Arxiv. 2013 [Google Scholar]
- 38.Quinlan AR. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics. 2014;47(11 12):11–34. doi: 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chapman DG University of California, B. Some properties of the hypergeometric distribution with applications to zoological sample censuses. University of California Press; 1951. [Google Scholar]
- 40.Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 41.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cavazza A, et al. Dynamic Transcriptional and Epigenetic Regulation of Human Epidermal Keratinocyte Differentiation. Stem Cell Reports. 2016;6:618–632. doi: 10.1016/j.stemcr.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All high-throughput sequencing data of the integration profiles have been deposited in the Sequence Read Archive (SRA) under accession number SRP110373. All data used to generate main and supplementary figures are provided as source data files.