

Abstract
Pancreatic cancer is a malignancy with an extremely poor prognosis. Chronic pancreatitis is a well-known risk factor for pancreatic cancer. Inflammation is thought to influence carcinogenesis through DNA damage and activation of intracellular signaling pathways. Many transcription factors and signaling pathways co-operate to determine and maintain cell identity at each phase of pancreatic organogenesis and cell differentiation. Recent studies have shown that carcinogenesis is promoted through the suppression of transcription factors related to differentiation. Pancreatitis also demonstrates transcriptional changes, suggesting that multifactorial epigenetic changes lead to impaired differentiation. Taken together, these factors may constitute an important framework for pancreatic carcinogenesis. In this review, we discuss the role of inflammation and de-differentiation in the development of pancreatic cancer, as well as the future of novel therapeutic applications.
Keywords: Pancreatitis, Inflammation, Organogenesis, Differentiation, Transcription factor, Pancreatic cancer
Core tip: Inflammation is involved in carcinogenesis by causing DNA damage. Recent studies show that carcinogenesis is promoted by reprogramming factors and by suppressing transcription factors related to acinar cell differentiation. Pancreatitis also shows such transcriptional changes, suggesting that epigenetic changes by several causes leading to the impaired differentiation may constitute an important framework for pancreatic carcinogenesis. New diagnostic, preventive and/or treatment strategies based on the findings described in this review are expected to be clinically applied in the near future.
INTRODUCTION
The worldwide incidence of pancreatic cancer is approximately 330000 cases in 2012, with trends indicating that rates are higher in men and in developed countries. The number of deaths due to pancreatic cancer are also estimated to be approximately 330000 people a year; it ranks 11th in cancer-related deaths[1,2]. The mean survival time is 19 mo and the 5-year survival rate is 5% or less; these figures indicate that pancreatic cancer has one of the worst prognoses across all forms of malignancy[2,3]. The early stages of pancreatic cancer are almost always asymptomatic. As a result, by the time symptoms become apparent, the disease is already at a very advanced stage. Because the 5-year survival rates of stage I and IV pancreatic cancers are 43% and 7.7% respectively, early diagnosis and treatment are especially crucial to improve the overall prognosis of the disease.
One option, to aid in the earlier diagnosis of pancreatic cancer, is to elucidate more thoroughly the mechanism of carcinogenesis and identify high-risk groups to follow carefully. Well-known risk factors include smoking, obesity, diabetes, and chronic pancreatitis[4]. In particular, the risk of pancreatic cancer in patients with chronic pancreatitis is 13.3 times greater than that of healthy controls, suggesting that inflammation is deeply involved in the pathogenesis of pancreatic cancer[5,6].
It is well known that the KRAS mutation and mutational inactivation of the CDKN2A, TP53, and SMAD4 tumor suppressors play important roles in the development of pancreatic cancer[4,7-9]. Furthermore, recent studies have shown that carcinogenesis is promoted by reprogramming factors and by suppression of transcription factors related to differentiation[10,11]. Interestingly, pancreatitis also shows the above transcriptional changes, suggesting that multifactorial epigenetic changes that result in impaired differentiation have an important role in pancreatic carcinogenesis.
In this review, we will discuss the mechanisms of pancreatic carcinogenesis from the perspective of pancreatic inflammation and cell differentiation.
INFLAMMATION AND PANCREATIC CARCINOGENESIS
In 1863, Rudolph Virchow first reported inflammatory cells in cancer tissues and hypothesized that inflammation promoted carcinogenesis[12]. In 1915, Yamagiwa induced skin cancer on the ears of rabbits by repeatedly painting them with coal tar, and experimentally revealed a case of carcinogenesis due to inflammation[13]. Furthermore, several cancers are known to be epidemiologically related to inflammatory diseases. For example, Helicobacter pylori-related gastritis patients have a 2.6-fold increased risk of gastric cancer[14]. Viral hepatitis and inflammatory bowel disease are risk factors for liver cancer and colon cancer, respectively. Previous epidemiological studies have demonstrated that non-steroidal anti-inflammatory drugs such as aspirin, lowers the overall risk of colon cancer[15,16]. Taken together, these results suggest that inflammation is frequently associated with carcinogenesis.
Chronic pancreatitis is a risk factor for pancreatic cancer[6]. Patients with hereditary pancreatitis, a rare cause of chronic pancreatitis and a strong risk for pancreatic cancer (49% of the patients develop pancreatic cancer by age 75 years), suffer from recurrent pancreatitis with pancreatic exocrine insufficiency and diabetes mellitus from a young age[17]. Mutations of the cationic trypsinogen (PRSS1) and serine protease inhibitor Kazal type 1 (SPINK1) genes cause hereditary pancreatitis[18,19]. Because the risk of developing pancreatic cancer does not change with the presence or absence of PRSS1 or SPINK1 gene mutations, it is unlikely that the gene itself functions as an oncogene or tumor-suppressor gene[19,20]. The increased carcinogenic risk in hereditary pancreatitis patients is presumed to be carcinogenesis due to prolonged inflammation.
Notably, Bailey et al[21] conducted unsupervised clustering of pancreatic cancer RNA sequencing data, and they classified pancreatic cancers into four subtypes: Squamous, pancreatic progenitor, immunogenic, and aberrantly differentiated endocrine exocrine. Each subtype differently expresses unique transcription factors and downstream targets, which are important in lineage specification and differentiation during pancreas development. Among them, the immunogenic subtype is associated with a significant immune infiltrate[21], which may be associated with pancreatitis and carcinogenesis.
The relationship between pancreatic cancer and inflammation has also been explored in experiments using genetically engineered mice. When Kras mutations were introduced during the embryonic stage in mice, pancreatic intraepithelial neoplasia (PanIN) formation was promoted while pancreatic cancer developed at a lower frequency[22]. The introduction of Kras mutations alone induced pancreatic cancer development over 1 year; however, when Trp53 and Cdkn2a defects were introduced, it only took 7 and 18 wk, respectively, to develop pancreatic cancer[23]. Furthermore, when pancreatitis was induced by administering caerulein in Kras mutant mice, carcinogenesis occurred at 12 wk[24]. These results demonstrated that some secondary abnormalities in Kras-mutated mice are necessary for rapid progression to invasive cancer, and that inflammation promotes carcinogenesis in conjunction with Kras mutations.
DNA damage caused by inflammation may contribute to carcinogenesis (Figure 1)[25]. Inflammatory cytokines produce reactive oxygen species, which randomly oxidize DNA to cause genetic mutation[26]. NO, induced by inflammation, also inhibits DNA repair enzymes to promote mutations[27]. In fact, the duration of chronic pancreatitis correlates positively with the incidence of KRAS mutations, suggesting that DNA damage accumulates due to the persistence of inflammation, promoting further carcinogenesis[28].
Figure 1.
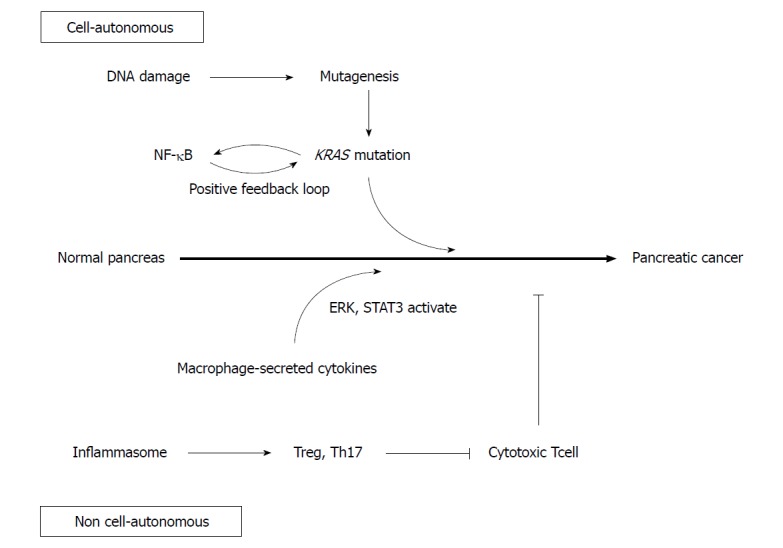
Inflammation induces carcinogenesis both cell-autonomously and non-cell-autonomously. DNA damage caused by inflammation contributes to mutagenesis. Nuclear factor κB and KRAS activate each other and sustained KRAS activity promotes carcinogenesis. Macrophage-secreted cytokines activate the ERK and STAT3 signaling pathways in epithelial cells. Inflammasomes inactivate cytotoxic T cells via the activation of Th17 and regulatory T cells. NF-κB: Nuclear factor κB.
Although random genetic mutations caused by inflammation may contribute to carcinogenesis, work done by Guerra et al[29] suggests an alternative role of inflammation in the development of carcinogenesis. Guerra et al[29] used a Cre Tet-off system to control the expression of mutant Kras in mice. When mutant KRas was expressed during the embryonic stage, PanIN was formed at 1-3 mo. However, when mutant KRas was expressed in adult mice 2 mo after birth, PanIN was not formed. Furthermore, even when Cdkn2a or Trp53 gene deficiencies were simultaneously introduced into adult mutant Kras mice, PanIN did not develop[29]. These results suggest that the carcinogenic potential through genetic mutation differs between the embryonic and adult stages in mice. Moreover, when pancreatitis was induced by administering caerulein to adult mutant Kras mice, PanIN developed and rapidly progressed to pancreatic cancer[30]. However, PanIN did not develop after deletion of Cdkn2a or Trp53 in adult mice accompanied with caerulein pancreatitis. From these results, mutant Kras and inflammation are necessary components of pancreatic carcinogenesis in adult mice, with inflammation contributing to carcinogenesis by means other than the introduction of specific gene mutations as a result of DNA damage. Recent studies revealing the association of various signaling pathways and microenvironments with inflammation and pancreatic carcinogenesis may support this concept.
CELL-AUTONOMOUS INTRACELLULAR SIGNALING PATHWAYS IN PANCREATIC INFLAMMATION AND CARCINOGENESIS
Nuclear factor κB (NF-κB) is involved not only in inflammation but also in cell differentiation and proliferation, both of which are activated in pancreatic cancer[31,32]. Mutant KRas is known to activate interleukin-1alpha (IL-1α) via AP-1. IL-1α polyubiquitinates tumor necrosis factor receptor-associated factor 6 and activates IKK2/β, which activates NF-κB. NF-κB subsequently upregulates IL-1α and p62 transcription, which in turn re-activates NF-κB in a positive feedback loop[33]. Because activated NF-κB activates KRas, another positive feedback loop is generated, resulting in sustained KRas activity which may promote pancreatic cancer development[34].
Additionally, Toll like receptor 4 (TLR4) and TLR7 are upregulated within the pancreatic cancer microenvironment[35,36]. TLRs are receptors that recognize pathogen-associated molecular patterns and diverse byproducts of inflammation and cellular injury. Activated TLRs induce the activation of NF-κB pathway within acinar cells, which may further promote the development of pancreatic cancer.
NON CELL-AUTONOMOUS INTRACELLULAR SIGNALING PATHWAYS IN PANCREATIC INFLAMMATION AND CARCINOGENESIS
The IL-6 / STAT3 pathway is also involved in pancreatic cancer and inflammation[37]. While caerulein-induced pancreatitis transiently activates STAT3, prolonged activity and PanIN development were both observed in Kras mutant mice[38]. In these mice, pancreatic Kras-mutant epithelial cells recruited macrophages, which secreted IL-6, result in the STAT3 activation in epithelial cells and formation of PanIN. Conversely, inactivation of IL-6 trans-signaling or inhibition of STAT3 resulted in decreased PanIN formation[39].
Various studies have revealed that macrophages play an important role in pancreatitis, and are likely to be related to pancreatic carcinogenesis[40]. As mentioned above, macrophages secrete IL-6 and activate the STAT3 signaling pathway to promote pancreatic carcinogenesis. In addition, macrophages are observed around acinar ductal metaplasia (ADM) lesions, which are precancerous lesions formed in response to pancreatitis. They secrete inflammatory cytokines such as TNFα, and the chemokine regulated upon activation of normal T cell expressed and presumably secreted (RANTES). They also promote ADM formation through activation of NF-κB and matrix metalloproteinase-9[41,42]. Macrophages that migrate around ADM and PanIN are polarized dominantly from M1 to M2 by stimulation of IL-13. M2 macrophages secrete CCL2 and IL-1ra, which activate the ERK signaling pathway and promote the growth of PanIN[43].
Th17 is associated with many inflammatory conditions, such as inflammatory bowel diseases. In the pancreas, the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome is also activated in pancreatitis[44]. Macrophages expressing NLRP3 inactivate cytotoxic CD8+ T cells through the activation of Th17 and regulatory T cells, which also contribute to the promotion of pancreatic cancer development[45].
PANCREATIC ORGANOGENESIS AND DIFFERENTIATION
The Guerra et al[29] study demonstrated that KRAS gene mutations induce PanIN formation in the embryonic, but not the adult stage. From these results, cell differentiation status at the embryonic or adult stage may control organ carcinogenesis. Research on the inflammation and differentiation of pancreatic cells has been increasing in recent years, and elucidation of pancreatic embryology on a cellular level would provide great understanding to pancreatic cancer development.
Pancreatic development begins with the evagination of dorsal mesenchyme of foregut endoderm on embryonic day 26 (E26) in humans and E9.5 in mice[46-48] (Figure 2). The ventral pancreatic bud emerges at 6 d in humans and at 12 h in mice after the appearance of the dorsal pancreatic bud. Branching begins immediately after evagination. Stalk elongation and gut rotation occur on the ventral and dorsal side, while fusion of the ventral and dorsal pancreas occurs during E12 to E13 in mice and E37 to E42 in humans. During E13-14 in mice, there is a dramatic increase in endocrine cells, particularly β-cells, known as “secondary transition”. Similarly, acinar cells develop and acinar enzyme gene expression increases. After E15 in mice, the destiny of pancreatic cells is determined.
Figure 2.

Pancreatic organogenesis and cell differentiation. The pancreatic bud arises from the endoderm foregut. During early branching morphogenesis, the branch tip is composed of multipotent progenitor cells that change into acinar cells. The trunk region is composed of bipotential progenitor cells that can differentiate into either duct or endocrine cells. As differentiation continues, the expression of PTF1A, NR5A2, and MIST1 is restricted in acinar cells.
Pancreatic tissue consists of acinar, duct, and endocrine cells. Lineage tracing using CreERT mice revealed that multipotent progenitor cells differentiate into respective cell populations[49]. Multipotent progenitor cells co-express homeobox protein PDX1, Sry-box protein SOX9, and basic helix-loop-helix (bHLH) protein PTF1A. As differentiation continues, the expression of PDX1, SOX9 and PTF1A are restricted in endocrine, duct, and acinar cells respectively. During early branching morphogenesis, the branch tip is composed of PDX1, PTF1A, and Cpa1 positive multipotent progenitor cells that can differentiate into all three type of cells; however, cells in the tip area lose their multipotency and change into pro-acinar cells after E14[50]. The trunk region is composed of bipotent progenitor cells that can differentiate into either duct or endocrine cells[51]. Some of these cells express neurogenin 3 (NGN3) and will differentiate further into endocrine cells[52].
Various transcription factors and signaling pathways are involved in acinar cell development. NR5A2 is a member of the nuclear hormone receptor family, and is responsible for pancreatic exocrine secretion in the mature pancreas[53]. NR5A2 regulates the various stages of development and is required for OCT4 expression in the epiblast[54]. It is also required for gastrulation and acinar cell maturation during secondary transition[55,56]. Since there is decreased expression of pancreas-related transcription factors during secondary transition, NR5A2 is thought to regulate pancreatic differentiation in cooperation with other transcription factors at this stage[56].
MIST1 is a bHLH transcription factor, highly expressed in acinar cells, as well as the stomach, prostate, and seminal vesicles[57]. Mice with Mist1 gene knockout developed highly disorganized acinar cells with impaired exocytosis[58]. Furthermore, ADM formation and susceptibility to caerulein pancreatitis were increased in these mice[59]. MIST1 is thought to be required for the maintenance of acinar cell identity.
The Wnt/β catenin signaling pathway is necessary for differentiation of acinar cells. Pancreatic hypoplasia was observed in β-catenin knockout mice[60]. Furthermore, acinar cell proliferation was promoted by deficiencies in the Apc gene, which has endogenous β-catenin inhibitory activity. Because this abnormal proliferation stops when c-myc is deleted, c-myc is considered to be an important downstream component of the Wnt/β catenin pathway[61].
The Hippo signaling pathway has been associated with pancreatic development. Deletion of the core Hippo kinase genes Mst1 and Mst2 induced pancreatic hypoplasia via YAP, the downstream mediator. Interestingly, in Mst1 and Mst2 double knockout mice, expression levels of MIST1, PTF1A, and NR5A2 were equivalent to those seen in wild type mice, with normal pancreatic sizes at birth. However, after 1 mo, the acinar cells changed to duct-like cells while the overall size of the pancreas was approximately half that of wild type mice. This suggests that the Hippo signaling pathway is necessary to maintain acinar cell identity and pancreas size after birth in mice[62].
The Notch signaling pathway is also indirectly related to acinar cell differentiation via lateral inhibition. NGN3 is a transcription factor that promotes differentiation to endocrine cells. Cells expressing NGN3 upregulate the expression of DLL1, which is a Notch ligand. DLL1 binds to the Notch receptor of surrounding cells and activates the Notch signaling pathway, thereby upregulating HES1 expression. HES1 inhibits NGN3 and suppresses endocrine cell proliferation. HES1 also maintains the expression level of PTF1A in multipotent progenitor cells and is thought to contribute to multipotent progenitor cell proliferation[63].
PANCREATIC CELL DE-DIFFERENTIATION, INFLAMMATION, AND CARCINOGENESIS
As described above, pancreatic cell differentiation and their identities are maintained by the cooperation of various transcription factors and signaling pathways. However, recent research has revealed that differentiated pancreatic cells show plasticity under specific circumstances. Acinar cells transdifferentiate or dedifferentiate into duct cells and endocrine cells after pancreatic duct ligation. During this change, cells express SOX9 and HNF1β multipotency factors[64,65]. The conversion from an acinar cell to embryonic progenitor phenotype that exhibits ductal markers, is called ADM. ADM is thought to be a reversible process and is frequently observed in pancreatic inflammation and injury. However, it becomes irreversible when combined with a Kras mutation. This alteration results in a lesion that is considered a precancerous stage of pancreatic cancer[66,67].
Epigenetic factors play crucial roles in differentiation and carcinogenesis. A recent study showed that Brg1, a catalytic ATPase subunit of the SWI/SNF chromatin remodeling complex, is inactivated in approximately 10% of pancreatic cancer[68]. Brg-1 binds to the SOX9 promoter and regulates the expression of SOX9. Acinar cell-specific deletion of Brg-1 attenuates ADM/PanIN formation in Kras mutant mice[69].
NR5A2 suppression and forced expression of SOX9 or PDX1 can induce ADM[11,70-72]. These results suggest that transcriptional changes that cause the loss of acinar cell identity promote ADM formation. Interestingly, although the pancreatic tissues of Nr5a2+/- mice are histologically normal, transcriptome analyses of Nr5a2+/- mice show inflammasome upregulation. In humans, similar transcriptomic changes occur in the pancreas with low levels of NR5A2 expression. Furthermore, NR5A2 is relocated from the promoters of differentiation-specific genes to the promoters of inflammation-related genes. AP-1 is upregulated in these mice and the deletion of Jun results in the downregulation of AP-1 and NR5A2 binding to AP-1 and inflammatory gene promoters[73].
In another study, temporal activation of reprogramming factors (Oct3/4, Sox2, Klf4, c-Myc) in the pancreas of Kras mutant mice promoted ADM formation and pancreatic cancer[10] (Figure 3). In previous transcriptome analyses, when the reprogramming factors are activated, acinar cell-related genes Ptf1a and Mist1 were downregulated. In addition, when pancreatitis was induced via caerulein administration in Kras mutant mice, similar transcriptional patterns were observed. Conversely, forced expression of Ptf1a or Mist1 in Kras mutant mice with caerulein-induced pancreatitis suppressed PanIN formation. These results demonstrate the crucial role of epigenetic regulation in the initiation of pancreatic carcinogenesis.
Figure 3.
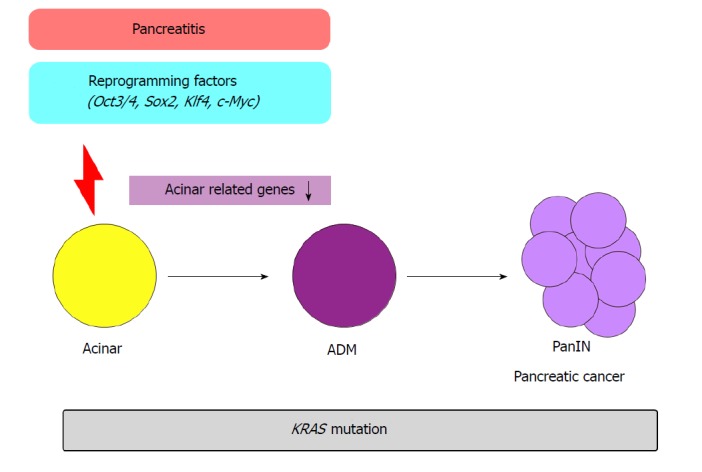
Pancreatic cell de-differentiation, inflammation, and carcinogenesis. Carcinogenesis is promoted by reprogramming factors (Oct3/4, Sox2, Klf4, and c-Myc). When the reprogramming factors are activated, acinar cell-related genes are suppressed. Pancreatitis also shows such transcriptional changes.
FUTURE PERSPECTIVES
A growing body of research in pancreatic carcinogenesis demonstrates that the loss of acinar cell identity caused by the suppression of transcriptional networks by reprogramming factors plays a crucial role in ADM formation. In addition, Kras mutation and epigenetic regulation play important roles in pancreatic carcinogenesis. Furthermore, inflammation induces an intracellular transcriptional state similar to the de-differentiated state of pancreatic cells, implying that inflammation, cell differentiation, and carcinogenesis are very closely related.
Some questions remain to be resolved. Inflammation may induce not only de-differentiation but also stem cell damage and impaired differentiation, and subsequently cause carcinogenesis. Further research is needed to determine the origin of pancreatic cancer. There is a strong association between chronic pancreatitis and pancreatic cancer. However, only 1.34% of pancreatic cancers are thought to be caused by chronic pancreatitis[74]. Furthermore, pancreatic cancer concomitant with intraductal papillary mucinous neoplasm, a premalignant lesion of pancreatic cancer, is not associated with pancreatitis or pancreatic atrophy[75]. However, these epidemiological and pathological data do not completely deny the connection between carcinogenesis and inflammation. One possible explanation is that pro-inflammatory states may exist in the absence of histologically observed pancreatitis[73]. Further studies are required to clarify the inflammation-like changes in “inflammation-absent” pre-neoplastic pancreatic lesions. This may subsequently allow the identification of high-risk patients.
Many novel therapeutic strategies for pancreatic cancer are aimed at reprogramming pancreatic cancer cells to behave like normal pancreatic cells[76]. For example, PD 325901 inhibits MEK1/2 and induces PanIN re-differentiation into acinar cells[77]. Another study has shown that the overexpression of bHLH transcription factors E47 and PTF1A resulted in increased acinar cell gene expression, suppressing cancer proliferation[78,79]. It is highly expected that in the near future, new diagnostic and/or treatment strategies based on the findings described in this review will be clinically applied, improving the prognosis of patients with pancreatic cancer.
Footnotes
Conflict-of-interest statement: No potential conflicts of interest.
Manuscript source: Invited manuscript
Peer-review started: September 3, 2018
First decision: October 11, 2018
Article in press: November 15, 2018
Specialty type: Medicine, research and experimental
Country of origin: Japan
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P- Reviewer: Pierzchalski P, Jonckheere N S- Editor: Wang JL L- Editor: A E- Editor: Tan WW
Contributor Information
Takahiro Seimiya, Department of Gastroenterology, Graduate School of Medicine, The University of Tokyo, Tokyo 113-8655, Japan.
Motoyuki Otsuka, Department of Gastroenterology, Graduate School of Medicine, The University of Tokyo, Tokyo 113-8655, Japan. otsukamo-tky@umin.ac.jp.
Takuma Iwata, Department of Gastroenterology, Graduate School of Medicine, The University of Tokyo, Tokyo 113-8655, Japan.
Eri Tanaka, Department of Gastroenterology, Graduate School of Medicine, The University of Tokyo, Tokyo 113-8655, Japan.
Tatsunori Suzuki, Department of Gastroenterology, Graduate School of Medicine, The University of Tokyo, Tokyo 113-8655, Japan.
Kazuma Sekiba, Department of Gastroenterology, Graduate School of Medicine, The University of Tokyo, Tokyo 113-8655, Japan.
Mari Yamagami, Department of Gastroenterology, Graduate School of Medicine, The University of Tokyo, Tokyo 113-8655, Japan.
Rei Ishibashi, Department of Gastroenterology, Graduate School of Medicine, The University of Tokyo, Tokyo 113-8655, Japan.
Kazuhiko Koike, Department of Gastroenterology, Graduate School of Medicine, The University of Tokyo, Tokyo 113-8655, Japan.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol. 2016;22:9694–9705. doi: 10.3748/wjg.v22.i44.9694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwon W, He J, Higuchi R, Son D, Lee SY, Kim J, Kim H, Kim SW, Wolfgang CL, Cameron JL, et al. Multinational validation of the American Joint Committee on Cancer 8th edition pancreatic cancer staging system in a pancreas head cancer cohort. J Hepatobiliary Pancreat Sci. 2018;25:418–427. doi: 10.1002/jhbp.577. [DOI] [PubMed] [Google Scholar]
- 4.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039–1049. doi: 10.1056/NEJMra1404198. [DOI] [PubMed] [Google Scholar]
- 5.Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24:349–358. doi: 10.1016/j.bpg.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 6.Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144:1252–1261. doi: 10.1053/j.gastro.2013.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology. 2013;144:1220–1229. doi: 10.1053/j.gastro.2013.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, Hruban RH, Maitra A, Kinzler K, Vogelstein B, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142:730–733.e9. doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maitra A, Adsay NV, Argani P, Iacobuzio-Donahue C, De Marzo A, Cameron JL, Yeo CJ, Hruban RH. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol. 2003;16:902–912. doi: 10.1097/01.MP.0000086072.56290.FB. [DOI] [PubMed] [Google Scholar]
- 10.Shibata H, Komura S, Yamada Y, Sankoda N, Tanaka A, Ukai T, Kabata M, Sakurai S, Kuze B, Woltjen K, et al. In vivo reprogramming drives Kras-induced cancer development. Nat Commun. 2018;9:2081. doi: 10.1038/s41467-018-04449-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Figura G, Morris JP 4th, Wright CV, Hebrok M. Nr5a2 maintains acinar cell differentiation and constrains oncogenic Kras-mediated pancreatic neoplastic initiation. Gut. 2014;63:656–664. doi: 10.1136/gutjnl-2012-304287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 13.Fujiki H. Gist of Dr. Katsusaburo Yamagiwa’s papers entitled “Experimental study on the pathogenesis of epithelial tumors” (I to VI reports) Cancer Sci. 2014;105:143–149. doi: 10.1111/cas.12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet. 1993;341:1359–1362. [PubMed] [Google Scholar]
- 15.Thun MJ, Namboodiri MM, Calle EE, Flanders WD, Heath CW Jr. Aspirin use and risk of fatal cancer. Cancer Res. 1993;53:1322–1327. [PubMed] [Google Scholar]
- 16.Langman MJ, Cheng KK, Gilman EA, Lancashire RJ. Effect of anti-inflammatory drugs on overall risk of common cancer: case-control study in general practice research database. BMJ. 2000;320:1642–1646. doi: 10.1136/bmj.320.7250.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rebours V, Boutron-Ruault MC, Schnee M, Férec C, Maire F, Hammel P, Ruszniewski P, Lévy P. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: a national exhaustive series. Am J Gastroenterol. 2008;103:111–119. doi: 10.1111/j.1572-0241.2007.01597.x. [DOI] [PubMed] [Google Scholar]
- 18.Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, Martin SP, Gates LK Jr, Amann ST, Toskes PP, Liddle R, McGrath K, Uomo G, Post JC, Ehrlich GD. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14:141–145. doi: 10.1038/ng1096-141. [DOI] [PubMed] [Google Scholar]
- 19.Cazacu IM, Farkas N, Garami A, Balaskó M, Mosdósi B, Alizadeh H, Gyöngyi Z, Rakonczay Z Jr, Vigh É, Habon T, Czopf L, Lazarescu MA, Erőss B, Sahin-Tóth M, Hegyi P. Pancreatitis-Associated Genes and Pancreatic Cancer Risk: A Systematic Review and Meta-analysis. Pancreas. 2018;47:1078–1086. doi: 10.1097/MPA.0000000000001145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howes N, Lerch MM, Greenhalf W, Stocken DD, Ellis I, Simon P, Truninger K, Ammann R, Cavallini G, Charnley RM, Uomo G, Delhaye M, Spicak J, Drumm B, Jansen J, Mountford R, Whitcomb DC, Neoptolemos JP; European Registry of Hereditary Pancreatitis and Pancreatic Cancer (EUROPAC) Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol. 2004;2:252–261. doi: 10.1016/s1542-3565(04)00013-8. [DOI] [PubMed] [Google Scholar]
- 21.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 22.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 23.Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carrière C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun. 2009;382:561–565. doi: 10.1016/j.bbrc.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawanishi S, Ohnishi S, Ma N, Hiraku Y, Murata M. Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis. Int J Mol Sci. 2017;18:E1808. doi: 10.3390/ijms18081808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaiswal M, LaRusso NF, Burgart LJ, Gores GJ. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000;60:184–190. [PubMed] [Google Scholar]
- 27.Jaiswal M, LaRusso NF, Gores GJ. Nitric oxide in gastrointestinal epithelial cell carcinogenesis: linking inflammation to oncogenesis. Am J Physiol Gastrointest Liver Physiol. 2001;281:G626–G634. doi: 10.1152/ajpgi.2001.281.3.G626. [DOI] [PubMed] [Google Scholar]
- 28.Löhr M, Klöppel G, Maisonneuve P, Lowenfels AB, Lüttges J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. 2005;7:17–23. doi: 10.1593/neo.04445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernández-Porras I, Cañamero M, Rodriguez-Justo M, Serrano M, Barbacid M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19:728–739. doi: 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 31.Prabhu L, Mundade R, Korc M, Loehrer PJ, Lu T. Critical role of NF-κB in pancreatic cancer. Oncotarget. 2014;5:10969–10975. doi: 10.18632/oncotarget.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5:119–127. [PubMed] [Google Scholar]
- 33.Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, Li J, Peng B, Fleming JB, Wang H, et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105–120. doi: 10.1016/j.ccr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang H, Daniluk J, Liu Y, Chu J, Li Z, Ji B, Logsdon CD. Oncogenic K-Ras requires activation for enhanced activity. Oncogene. 2014;33:532–535. doi: 10.1038/onc.2012.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ochi A, Graffeo CS, Zambirinis CP, Rehman A, Hackman M, Fallon N, Barilla RM, Henning JR, Jamal M, Rao R, et al. Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J Clin Invest. 2012;122:4118–4129. doi: 10.1172/JCI63606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ochi A, Nguyen AH, Bedrosian AS, Mushlin HM, Zarbakhsh S, Barilla R, Zambirinis CP, Fallon NC, Rehman A, Pylayeva-Gupta Y, et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J Exp Med. 2012;209:1671–1687. doi: 10.1084/jem.20111706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steele CW, Kaur Gill NA, Jamieson NB, Carter CR. Targeting inflammation in pancreatic cancer: Clinical translation. World J Gastrointest Oncol. 2016;8:380–388. doi: 10.4251/wjgo.v8.i4.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fukuda A, Wang SC, Morris JP 4th, Folias AE, Liou A, Kim GE, Akira S, Boucher KM, Firpo MA, Mulvihill SJ, Hebrok M. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 2011;19:441–455. doi: 10.1016/j.ccr.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Klöppel G, Yoshimura A, Reindl W, Sipos B, Akira S, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 40.Liou GY. Inflammatory Cytokine Signaling during Development of Pancreatic and Prostate Cancers. J Immunol Res. 2017;2017:7979637. doi: 10.1155/2017/7979637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wörmann SM, Diakopoulos KN, Lesina M, Algül H. The immune network in pancreatic cancer development and progression. Oncogene. 2014;33:2956–2967. doi: 10.1038/onc.2013.257. [DOI] [PubMed] [Google Scholar]
- 42.Liou GY, Döppler H, Necela B, Krishna M, Crawford HC, Raimondo M, Storz P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-κB and MMPs. J Cell Biol. 2013;202:563–577. doi: 10.1083/jcb.201301001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liou GY, Bastea L, Fleming A, Döppler H, Edenfield BH, Dawson DW, Zhang L, Bardeesy N, Storz P. The Presence of Interleukin-13 at Pancreatic ADM/PanIN Lesions Alters Macrophage Populations and Mediates Pancreatic Tumorigenesis. Cell Rep. 2017;19:1322–1333. doi: 10.1016/j.celrep.2017.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoque R, Sohail M, Malik A, Sarwar S, Luo Y, Shah A, Barrat F, Flavell R, Gorelick F, Husain S, et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology. 2011;141:358–369. doi: 10.1053/j.gastro.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daley D, Mani VR, Mohan N, Akkad N, Pandian GSDB, Savadkar S, Lee KB, Torres-Hernandez A, Aykut B, Diskin B, et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J Exp Med. 2017;214:1711–1724. doi: 10.1084/jem.20161707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gittes GK. Developmental biology of the pancreas: a comprehensive review. Dev Biol. 2009;326:4–35. doi: 10.1016/j.ydbio.2008.10.024. [DOI] [PubMed] [Google Scholar]
- 47.Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn. 2011;240:530–565. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- 48.Shih HP, Wang A, Sander M. Pancreas organogenesis: from lineage determination to morphogenesis. Annu Rev Cell Dev Biol. 2013;29:81–105. doi: 10.1146/annurev-cellbio-101512-122405. [DOI] [PubMed] [Google Scholar]
- 49.Murtaugh LC, Keefe MD. Regeneration and repair of the exocrine pancreas. Annu Rev Physiol. 2015;77:229–249. doi: 10.1146/annurev-physiol-021014-071727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Q, Law AC, Rajagopal J, Anderson WJ, Gray PA, Melton DA. A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell. 2007;13:103–114. doi: 10.1016/j.devcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 51.Solar M, Cardalda C, Houbracken I, Martín M, Maestro MA, De Medts N, Xu X, Grau V, Heimberg H, Bouwens L, et al. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev Cell. 2009;17:849–860. doi: 10.1016/j.devcel.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 52.Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000;97:1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holmstrom SR, Deering T, Swift GH, Poelwijk FJ, Mangelsdorf DJ, Kliewer SA, MacDonald RJ. LRH-1 and PTF1-L coregulate an exocrine pancreas-specific transcriptional network for digestive function. Genes Dev. 2011;25:1674–1679. doi: 10.1101/gad.16860911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gu P, Goodwin B, Chung AC, Xu X, Wheeler DA, Price RR, Galardi C, Peng L, Latour AM, Koller BH, et al. Orphan nuclear receptor LRH-1 is required to maintain Oct4 expression at the epiblast stage of embryonic development. Mol Cell Biol. 2005;25:3492–3505. doi: 10.1128/MCB.25.9.3492-3505.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Labelle-Dumais C, Jacob-Wagner M, Paré JF, Bélanger L, Dufort D. Nuclear receptor NR5A2 is required for proper primitive streak morphogenesis. Dev Dyn. 2006;235:3359–3369. doi: 10.1002/dvdy.20996. [DOI] [PubMed] [Google Scholar]
- 56.Hale MA, Swift GH, Hoang CQ, Deering TG, Masui T, Lee YK, Xue J, MacDonald RJ. The nuclear hormone receptor family member NR5A2 controls aspects of multipotent progenitor cell formation and acinar differentiation during pancreatic organogenesis. Development. 2014;141:3123–3133. doi: 10.1242/dev.109405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pin CL, Bonvissuto AC, Konieczny SF. Mist1 expression is a common link among serous exocrine cells exhibiting regulated exocytosis. Anat Rec. 2000;259:157–167. doi: 10.1002/(SICI)1097-0185(20000601)259:2<157::AID-AR6>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 58.Pin CL, Rukstalis JM, Johnson C, Konieczny SF. The bHLH transcription factor Mist1 is required to maintain exocrine pancreas cell organization and acinar cell identity. J Cell Biol. 2001;155:519–530. doi: 10.1083/jcb.200105060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kowalik AS, Johnson CL, Chadi SA, Weston JY, Fazio EN, Pin CL. Mice lacking the transcription factor Mist1 exhibit an altered stress response and increased sensitivity to caerulein-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1123–G1132. doi: 10.1152/ajpgi.00512.2006. [DOI] [PubMed] [Google Scholar]
- 60.Wells JM, Esni F, Boivin GP, Aronow BJ, Stuart W, Combs C, Sklenka A, Leach SD, Lowy AM. Wnt/beta-catenin signaling is required for development of the exocrine pancreas. BMC Dev Biol. 2007;7:4. doi: 10.1186/1471-213X-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strom A, Bonal C, Ashery-Padan R, Hashimoto N, Campos ML, Trumpp A, Noda T, Kido Y, Real FX, Thorel F, et al. Unique mechanisms of growth regulation and tumor suppression upon Apc inactivation in the pancreas. Development. 2007;134:2719–2725. doi: 10.1242/dev.02875. [DOI] [PubMed] [Google Scholar]
- 62.Gao T, Zhou D, Yang C, Singh T, Penzo-Méndez A, Maddipati R, Tzatsos A, Bardeesy N, Avruch J, Stanger BZ. Hippo signaling regulates differentiation and maintenance in the exocrine pancreas. Gastroenterology. 2013;144:1543–1553, 1553.e1. doi: 10.1053/j.gastro.2013.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ahnfelt-Rønne J, Jørgensen MC, Klinck R, Jensen JN, Füchtbauer EM, Deering T, MacDonald RJ, Wright CV, Madsen OD, Serup P. Ptf1a-mediated control of Dll1 reveals an alternative to the lateral inhibition mechanism. Development. 2012;139:33–45. doi: 10.1242/dev.071761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pan FC, Bankaitis ED, Boyer D, Xu X, Van de Casteele M, Magnuson MA, Heimberg H, Wright CV. Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development. 2013;140:751–764. doi: 10.1242/dev.090159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rooman I, Real FX. Pancreatic ductal adenocarcinoma and acinar cells: a matter of differentiation and development? Gut. 2012;61:449–458. doi: 10.1136/gut.2010.235804. [DOI] [PubMed] [Google Scholar]
- 66.Storz P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol. 2017;14:296–304. doi: 10.1038/nrgastro.2017.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, Feldmann G, Stoffers DA, Konieczny SF, Leach SD, et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A. 2008;105:18913–18918. doi: 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marquez-Vilendrer SB, Thompson K, Lu L, Reisman D. Mechanism of BRG1 silencing in primary cancers. Oncotarget. 2016;7:56153–56169. doi: 10.18632/oncotarget.10593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tsuda M, Fukuda A, Roy N, Hiramatsu Y, Leonhardt L, Kakiuchi N, Hoyer K, Ogawa S, Goto N, Ikuta K, et al. The BRG1/SOX9 axis is critical for acinar cell-derived pancreatic tumorigenesis. J Clin Invest. 2018;128:3475–3489. doi: 10.1172/JCI94287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Prévot PP, Simion A, Grimont A, Colletti M, Khalaileh A, Van den Steen G, Sempoux C, Xu X, Roelants V, Hald J, et al. Role of the ductal transcription factors HNF6 and Sox9 in pancreatic acinar-to-ductal metaplasia. Gut. 2012;61:1723–1732. doi: 10.1136/gutjnl-2011-300266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miyatsuka T, Kaneto H, Shiraiwa T, Matsuoka TA, Yamamoto K, Kato K, Nakamura Y, Akira S, Takeda K, Kajimoto Y, et al. Persistent expression of PDX-1 in the pancreas causes acinar-to-ductal metaplasia through Stat3 activation. Genes Dev. 2006;20:1435–1440. doi: 10.1101/gad.1412806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Flandez M, Cendrowski J, Cañamero M, Salas A, del Pozo N, Schoonjans K, Real FX. Nr5a2 heterozygosity sensitises to, and cooperates with, inflammation in KRas(G12V)-driven pancreatic tumourigenesis. Gut. 2014;63:647–655. doi: 10.1136/gutjnl-2012-304381. [DOI] [PubMed] [Google Scholar]
- 73.Cobo I, Martinelli P, Flández M, Bakiri L, Zhang M, Carrillo-de-Santa-Pau E, Jia J, Sánchez-Arévalo Lobo VJ, Megías D, Felipe I, et al. Transcriptional regulation by NR5A2 links differentiation and inflammation in the pancreas. Nature. 2018;554:533–537. doi: 10.1038/nature25751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Duell EJ, Lucenteforte E, Olson SH, Bracci PM, Li D, Risch HA, Silverman DT, Ji BT, Gallinger S, Holly EA, et al. Pancreatitis and pancreatic cancer risk: a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4) Ann Oncol. 2012;23:2964–2970. doi: 10.1093/annonc/mds140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yagi Y, Masuda A, Zen Y, Shiomi H, Toyama H, Sofue K, Takenaka M, Kobayashi T, Nakagawa T, Yamanaka K, et al. Pancreatic inflammation and atrophy are not associated with pancreatic cancer concomitant with intraductal papillary mucinous neoplasm. Pancreatology. 2018;18:54–60. doi: 10.1016/j.pan.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 76.Wong CH, Li YJ, Chen YC. Therapeutic potential of targeting acinar cell reprogramming in pancreatic cancer. World J Gastroenterol. 2016;22:7046–7057. doi: 10.3748/wjg.v22.i31.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Collins MA, Yan W, Sebolt-Leopold JS, Pasca di Magliano M. MAPK signaling is required for dedifferentiation of acinar cells and development of pancreatic intraepithelial neoplasia in mice. Gastroenterology. 2014;146:822–834.e7. doi: 10.1053/j.gastro.2013.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim S, Lahmy R, Riha C, Yang C, Jakubison BL, van Niekerk J, Staub C, Wu Y, Gates K, Dong DS, et al. The basic helix-loop-helix transcription factor E47 reprograms human pancreatic cancer cells to a quiescent acinar state with reduced tumorigenic potential. Pancreas. 2015;44:718–727. doi: 10.1097/MPA.0000000000000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jakubison BL, Schweickert PG, Moser SE, Yang Y, Gao H, Scully K, Itkin-Ansari P, Liu Y, Konieczny SF. Induced PTF1a expression in pancreatic ductal adenocarcinoma cells activates acinar gene networks, reduces tumorigenic properties, and sensitizes cells to gemcitabine treatment. Mol Oncol. 2018;12:1104–1124. doi: 10.1002/1878-0261.12314. [DOI] [PMC free article] [PubMed] [Google Scholar]