

Abstract
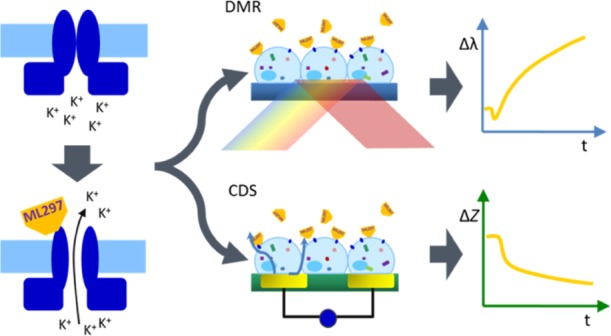
Dynamic mass redistribution (DMR) and cellular dielectric spectroscopy (CDS) are label-free biosensor technologies that capture real-time integrated cellular responses upon exposure to extra- and intracellular stimuli. They register signaling routes that are accompanied by cell shape changes and/or molecular movement of cells proximal to the biosensor to which they are attached. Here, we report the unexpected observation that robust DMR and CDS signatures are also elicited upon direct stimulation of G protein-activated inwardly rectifying potassium (GIRK) channels, which are involved in the regulation of excitability in the heart and brain. Using ML297, a small-molecule GIRK activator, along with channel blockers and cytoskeletal network inhibitors, we found that GIRK activation exerts its effects on cell shape by a mechanism which depends on actin but not the microtubule network. Because label-free real-time biosensing (i) quantitatively determines concentration dependency of GIRK activators, (ii) accurately assesses the impact of GIRK channel blockers, (iii) is high throughput-compatible, and (iv) visualizes previously unknown cellular consequences downstream of direct GIRK activation, we do not only provide a novel experimental strategy for identification of GIRK ligands but also an entirely new angle to probe GIRK (ligand) biology. We envision that DMR and CDS may add to the repertoire of technologies for systematic exploitation of ion channel function and, in turn, to the identification of novel GIRK ligands in order to treat cardiovascular and neurological disorders.
Introduction
As G protein-gated inwardly rectifying potassium (GIRK, Kir3) channels are implicated in an increasing number of pathologies, they are gaining focus as targets for pharmacological intervention.1,2 They exist as hetero- or homotetrameric structures comprised of one or more of four subunits (GIRK1-4), depending on tissue distribution.3,4 GIRKs are activated by Gβγ subunits of stimulated Gi protein-coupled receptors, thereby inducing neurons and cardiac pacemaker cells to hyperpolarize and, as such, regulate cellular excitability within heart and brain. GIRK channels have also been linked to pathologies related to perturbations of rhythmic action potential firing, such as epilepsy, cardiac arrhythmias, and Alzheimer’s disease among others.1,3−6 In order to combat these diseases, unrelenting search for new pharmacological treatment options has been underway. The recent development of the small molecule GIRK activator ML297 is one such example that has shown positive effects in preclinical models of epilepsy,7 anxiety,8 and Alzheimer’s disease.9 Yet, identification of new GIRK channel agonists with subtype selectivity and tissue specificity is still an ongoing endeavor in search for molecules with therapeutic potential.
Currently available methods for the investigation of ion channels and their ligands include automated electrophysiology and ion-specific fluorescence dyes.10,11 The former requires electrical access to the cell interior by either sealing a microelectrode onto the cell surface with gigaohm resistance (classical patch clamp) or dislodging the cells from their substrate in order to perform automated measurements (automated patch clamp).10 The latter relies on dyes which are loaded onto and trapped within cells, where they react sensitively to influx of specific ions or changes in potential.11 Both techniques, with patch clamping still considered as gold standard, are powerful, yet technically challenging, time consuming, and only offer an insight into a channel’s conducting function and upstream regulatory elements.
Optical-based dynamic mass redistribution (DMR) and cellular dielectric spectroscopy (CDS) are label-free biosensor platforms. They are well established for the detection of integrated responses in real-time when living cells are exposed to pharmacologically active stimuli.12−28 Rather than relying on specific endpoints, such as changes in electrical potentials or accumulation of ions, both biosensors deliver more complex time-resolved activity profiles of entire cells, without the need for physical access to cells or artificial labels (Figure 1a,b). In addition to their original purpose of visualizing activity of signaling-competent proteins within living cells, we here show that DMR and CDS also serve to monitor the cellular consequences that occur upon direct GIRK channel activation. We present the molecular underpinnings associated with GIRK-mediated cell shape changes and raise the possibility that this mode of activation may be mechanistically distinct from the endogenous Gαi-βγ activation pathway. Thereby, our results do not only present a novel method for detection of previously unrecognized GIRK-mediated downstream effects but also have important implications for GIRK ligand drug discovery.
Figure 1.

Label-free readouts visualize ML297-induced responses in a GIRK1/2-specific manner. (a) DMR assay: cells are located on top of a resonant waveguide grating biosensor and exposed to polychromatic light at wavelengths between 827 and 832.5 nm. The composition of the optical grating and properties of the cells result in penetration of light ∼150 nm into the cells37−39 (area of detectable DMR). In this area, a specific wavelength of light is reflected, whereas the rest is absorbed. If addition of pharmacological stimuli leads to changes in cellular morphology, the optical density within the “detection zone” is altered leading to a change in the reflected wavelength [λ]. Compounds that cause a decrease in mass proximal to the biosensor (yellow trace) shift the reflected light to shorter wavelengths, whereas an increase of mass (purple) results in a shift to longer wavelengths.13 (b) CDS assay: a monolayer of cells is cultivated on top of an electrical biosensor. Alternating voltages at set frequencies are applied through electrodes, which generate transcellular and extracellular currents (dark blue arrows). Addition of biologically active compounds influences the morphological structure of the monolayer. This change might include spreading and migration of the cells to cover more of the electrode’s surface area, or modifications in intercellular connections, which in turn leads to an alteration of the well’s impedance [Z].25−27,40,41 In the following, only the change in extracellular impedance, measured at frequencies between 3320 and 24 700 Hz (factory default setting), is shown. (c,f) Exemplary DMR and impedance traces of ML297-stimulated HEK cells either lacking or recombinantly expressing GIRK1/2. (d,g) Concentration-effect-curves calculated from the area under the curve (AUC) of DMR and impedance between 0 and 3600 s and normalized to the AUC of 100 μM Cch (n = 3). For pEC50 values, see Table S1. (e,h) Control signals of ATP and Cch in the absence or presence of GIRK1/2.
Results and Discussion
GIRK Activation Elicits Specific Label-Free Responses
Label-free methods are neither widely used nor considered appropriate to investigate ion channel behavior because the movement of ions per se through the plasmalemma is not believed to register as mass movement. On the other hand, evidence is available to suggest that selected ion channels can directly influence cellular signaling mechanisms through processes that are not directly linked to their ion flux function.29,30 We were intrigued to elicit robust label-free signatures upon stimulation of GIRK1/2, the neuronal GIRK channel prototype, with ML297, a small molecule GIRK activator selective for GIRK1-containing channels.8 Thus far, the GIRK subclass of inwardly rectifying potassium channels has been linked to “conducting functions” related to ion flow but not to “nonconducting functions” related to cellular signaling mechanisms.1,4,7,8,31 Therefore, we set out to investigate the molecular basis underlying GIRK channel activation-induced label-free whole cell responses. In HEK cells recombinantly expressing the functional GIRK1/2 subunit combination, we found that treatment with ML297 evoked concentration-dependent DMR response profiles that were characterized by an initial transient negative phase followed by a rapid ascending phase (Figure 1c,d). No changes in DMR were observed when native HEK cells were exposed to ML297, confirming the GIRK-specific origin of mass movement. Control stimulus carbachol (Cch) that acts via endogenous muscarinic M3 receptors was equally effective in native and GIRK1/2 expressing cells, corroborating that ML297 responses were no artifacts of altered cellular responsiveness of GIRK-containing cells (Figure 1e). Similar results were obtained with CDS, another label-free technique, which translates intracellular signaling events into bioimpedance signals.15,16,18,25,26 Therein, activation of GIRK1/2 with ML297 led to characteristic noninverting and concentration-dependent negative signals with similar steady kinetics over 3600 s in a strictly GIRK-dependent manner (Figure 1f,g). Control stimulus ATP was equally effective in both cell lines again confirming that ML297 recordings were no consequence of generally enhanced responsiveness in GIRK-expressing cells (Figure 1h). Notably, both methods revealed similar pEC50 values (Table S1), 5.45 and 5.50 for DMR and CDS, respectively, which is an order of magnitude lower than those observed in patch clamp8 and thallium flux32 readouts. Together, these data show that direct activation of GIRK channels is associated with a cellular event whose persistent slow time scope differs considerably from what would be expected from a channel that allows ions to flow rapidly through the plasma membrane.
Label-Free GIRK1/2 Response Is Sensitive to Inhibitors
To further corroborate that the label-free records specifically originated from GIRK1/2 channel activation, and to investigate whether mass movement is functionally linked to ion flux, we pretreated GIRK1/2 expressing cells with three distinct channel blockers: (i) the small molecule SCH23390 (SCH), initially described as dopamine receptor agonist but known to also block GIRK currents,33 (ii) Tertiapin Q (TPNQ), the stable form of the bee toxin peptide, which inhibits channel function via interaction with the base of the external pore,34,35 and (iii) extracellular barium (Ba2+), which effectively blocks the channel pore.36 Incubation of cells with increasing concentrations of SCH (Figure 2a,b) and TPNQ (Figure 2c,d) dampened the ML297 responses in a concentration-dependent manner without shifting pEC50 values (Table S2), compatible with noncompetitive antagonism. Ba2+ only partially blocked the ML297-derived signal even at maximal concentration (Figure 2e,f), reminiscent of findings previously reported in electrophysiological recordings.7 Inhibitor effects were specifically mediated via GIRK1/2 channels because cell-intrinsic viability controls prostaglandin E1 (PGE1) and ATP were unaltered in inhibitor pretreated cells (Figures 2g–i and S1). Equivalent findings were obtained when GIRK1/2 cells were pretreated with SCH and TPNQ using bioimpedance measurements (Figure 3a–d). These results show that both label-free platforms display specific ML297-mediated cell responses that are impaired (Ba2+) or absent (SCH and TPNQ) in the presence of GIRK blockers in striking resemblance to results obtained with electrophysiological or ion-specific fluorescence dye assays.8 We conclude that label-free DMR and CDS portray the cellular consequences of GIRK channel opening and thereby reveal an apparent molecular link between ion flow and cell morphology changes, the molecular basis of DMR and CDS detection.18,20,25,26 Indeed, images extracted from time-lapse videos of living HEK GIRK1/2 cells revealed profound flattening of cells in a strictly ML297-specific manner consistent with the movement of mass towards the biosensor that was detected by DMR (Figure 4a,b). This motion was not observed when cells were pretreated with SCH (Figure 4c) and was absent in cells lacking the GIRK1/2 channel (Figure S2, for full time-lapse videos see the Supporting Information).
Figure 2.

GIRK1/2-dependent DMR response to ML297 can be diminished using known GIRK channel inhibitors. (a,c,e) Exemplary DMR traces of ML297-stimulated HEK cells stably expressing GIRK1/2 channels after preincubation with indicated concentrations of ionic barium, SCH, or TPNQ. (b,d,f) Concentration-effect relationships calculated from AUC of DMR recordings (n = 3) (a,c,e), between 0–4800 (a), 300–3900 (c), and 0–8000 s (e). For pEC50 values, see Table S2. (g–i) AUC quantification of DMR traces of PGE1 (100 μM) and ATP (100 μM) (n = 2), calculated between 0–4800 (g), 300–3900 (h), and 0–8000 s (i).
Figure 3.

GIRK channel inhibitors suppress the impedance response to ML297. (a) Impedance profiles of ML297-activated GIRK1/2 in HEK cells pretreated with buffer, SCH, and TPNQ. (b) Concentration–effect curves of impedance AUC over 3600 s derived from traces in (a) (n = 3), pEC50 value of buffer-treated cells was determined as 5.29 ± 0.10. (c) Representative impedance traces in response to Cch (100 μM) and ATP (100 μM) in the presence of GIRK inhibitors. (d) Quantification of impedance records derived from (c) (n = 3).
Figure 4.

Time-lapse imaging of living HEK GIRK1/2 cells reveals ML297-mediated cell morphology changes. Representative bright-field images of HEK cells stably expressing GIRK1/2 at 0, 1, 2, and 3 h after treatment with buffer (a), 10 μM ML297 (b), or pretreated with 10 μM SCH prior to ML297 (10 μM) application (c). Changes in cell morphology from oval to flat shape are marked by arrows. One representative out of three biological replicates is shown. Scale bar, 50 μm, applied to all.
Label-Free Readouts of GIRK1/4 Heterotetramers Display Inhibitor-Sensitive Responses to ML297
Patch clamp and thallium flux assays report ML297 activity only if functional channel complexes bear the GIRK1 subunit.8,31 To interrogate whether the same molecular requirements apply to label-free whole cell biosensing, we took advantage of previously described functional and nonfunctional ML297-sensitive and nonsensitive GIRK subunit combinations.8,31 We obtained ML297-mediated and SCH-sensitive DMR recordings in HEK cells stably expressing GIRK1/4, the prototypical isoform of the cardiovascular system42 (Figure 5a). Cells expressing GIRK1 which contains the ML297 binding site but does not form functional homomultimeric channels (Figure 5b), or GIRK2, which forms functional channels but lacks the relevant ML297 binding site3,4,8 (Figure 5c) failed to produce any change in mass distribution readouts. Comparable results were obtained in CDS recordings where ML297-dependent SCH-sensitive traces were evoked only in GIRK1/4 but not in GIRK1 or GIRK2 expressing cells (Figure 5d–f). The observation that both readouts only detect ML297-mediated GIRK activation upon its binding to functional GIRK1 bearing channels confirms this molecular prerequisite to be shared for both label-free and electrophysiological measurements.
Figure 5.

Label-free readouts of GIRK1/4 heterotetramers display inhibitor-sensitive responses to ML297. Representative DMR (a–c) and impedance (d–f) recordings of HEK cells stably transfected with the indicated combination of GIRK1, GIRK2, and GIRK4 subunits incubated with buffer or SCH as indicated prior to addition of ML297 and control compounds (n = 3).
ML297-mediated and SCH-sensitive whole cell responses were also evoked in HL-1 cells, an immortalized murine cardiac cell line that endogenously expresses GIRK1/4 channels.43,44 More specifically, we observed concentration-dependent DMR profiles in response to ML297 reminiscent of those observed in the HEK background (Figure 6a). ML297 (Figure 6b) but not control stimuli (Figure 6c,d) were blunted by SCH pretreatment, suggesting GIRK-specific inhibition of cellular responses. We also noted enhanced fluctuations within DMR profiles in all SCH-pretreated samples (Figure 6b,d) which likely reflect the increase in spontaneous beating because of endogenous GIRK1/4 inhibition by SCH. From these experiments, we concluded that GIRK channel-mediated cell morphology changes are not restricted to the HEK system but likely are of more general relevance for cells that express GIRK under native conditions.
Figure 6.
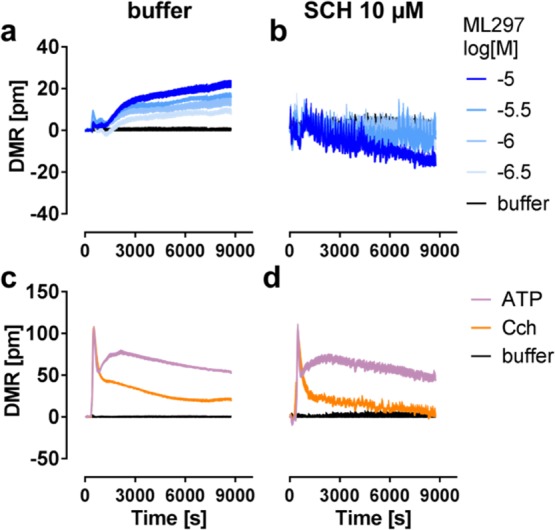
HL-1 cells endogenously expressing GIRK1/4 channels respond to ML297. Representative DMR responses of HL-1 cells to ML297 (a,b) or 100 μM of control stimuli ATP and Cch (c,d) in the absence (a,c) or presence (b,d) of SCH.
Screening of Structural and Functional Analogs of ML297
If DMR and CDS biosensors faithfully recapitulate the cellular effects triggered by direct GIRK activation, we reasoned that label-free activity rankings of GIRK activators with different potencies should match the profile obtained in thallium flux or patch clamp assays. To this end, we characterized a small library of structural ML297 analogs, all of which were previously characterized in thallium assays and/or whole-cell patch clamp electrophysiology in GIRK1/2-HEK cells7,32 (Table S3). Both DMR and CDS recordings revealed specific responses to 551 which were sensitive to inhibitor treatment and absent in native HEK cells (Figures 7 and S3). Although DMR also detected weak but significant activation by 553 (Figure 7a–c), this compound was inactive in CDS measurements across all tested concentrations (Figure 7d–f). Efficacy of direct GIRK activators varied somewhat between the two platforms (compare Figure 7c with 7f), yet pEC50 calculation portrayed 551 as more potent than ML297 and 553 for both methods, and, moreover, classified all remaining analogs as inactive. Thus, overall potency rankings were retained in label-free assays but concentration–effect curves for all active GIRK ligands were again shifted to lower concentrations as compared with thallium flux assays.
Figure 7.

Screening of structural analogs of ML297 for agonistic activity on GIRK1/2 channels. (a,d) Exemplary DMR (a) and impedance (d) traces of HEK GIRK1/2 cells after addition of 10 μM of a test compound structurally derived from ML297, either in the absence or presence of channel inhibitor SCH. In the following, the test compounds are referred to by the last three digits of their Vanderbilt University code. (b,e) AUCs from (a,d), respectively, and normalized to the indicated concentration of ML297. (c,f) are concentration–response curves derived for hit compounds using the DMR (c) or impedance (f) platform (n = 3). pEC50 values for DMR and impedance were determined as 5.07 ± 0.13; 5.22 ± 0.13 for ML297, 5.50 ± 0.16; 5.82 ± 0.16 for 551, and 5.06 ± 0.11; (too low to determine) for 553.
From these results, we concluded (i) that DMR and CDS are competent to differentiate between true and false positives akin to methods routinely applied for GIRK ligand characterization,8,11,31 and (ii) corroborate existence of a molecular link between direct GIRK activation and cell morphology changes.
Gi-βγ Does Not Visibly Contribute to Direct ML297-Induced GIRK Activation in DMR Detection
A mechanism alternative to direct GIRK activation is used endogenously by excitable cells and relies on Gβγ subunits that are released upon stimulation of Gi-coupled G protein-coupled receptors (GPCRs). Because it is known from whole-cell and single-channel patch clamp experiments that GPCRs—via Gβγ—and direct GIRK stimuli activate GIRK channels in distinct manners,8 we wondered whether DMR would recapitulate these mechanistic differences. One such difference consists in the necessity of βγ itself, which is required for GPCR—but not ML297-mediated GIRK activation.8 We used cell lines coexpressing the GIRK1/2 subunits along with the Gi-sensitive OXE-receptor (OXE-R) and Gi/o inhibitor pertussis toxin (PTX) to block liberation of Gβγ from Gi-βγ heterotrimers. Indeed, ML297-mediated DMR responses and thallium flux in PTX pretreated cells were likewise unaffected or slightly diminished (Figure 8a,b). On the contrary, Gαi-βγ-dependent thallium flux induced by 5-oxo-ETE stimulation of OXE-R was completely blunted by PTX, and additionally by the specific OXE-R blocker Gore27, confirming its receptor origin (Figure 8c). From these data, we concluded that ML297-mediated DMR traces in GIRK expressing cells do not rely on Gαi-βγ akin to ML297-mediated GIRK currents in thallium flux assays,8 again suggesting mechanistic commonalities between direct ion flux assay techniques and the more distal DMR detection. Because GIRK channels are pharmacological targets to reduce activity of excitable cells in cardiological and neurological disorders, discovery of ionotropic downstream consequences following their direct activation should be of considerable relevance to GIRK channel drug development.
Figure 8.

Gi-βγ does not visibly contribute to direct ML297-induced GIRK activation. (a–c) Representative traces and averages of independent data sets of (a) DMR recordings and (b,c) thallium flux showing HEK GIRK1/2 OXE-R cells stimulated with 10 μM ML297 (a,b) or 1 μM 5-oxo-ETE (c) in the presence and absence of PTX (a), or PTX and Gore27 (b,c), presented as AUC between 0–4800 (a) or 0–30 s (b,c), respectively (n = 3). Two-tailed student t-tests compared the inhibitor-treated with the respective untreated values.
ML297-Induced DMR in GIRK1/2 Cells Requires an Intact Actin Cytoskeleton
Changes registered with DMR or CDS biosensors are a consequence of altered morphology and/or cell motion that occurs when cells spread or “contract” on the surface of these biosensors.20,25,45 To investigate the cellular origin that accounts for the observed mass movements in response to ML297, we preincubated cells with actin or tubulin polymerization inhibitors prior to DMR detection. Because inhibition of actin- or tubulin-dependent cytoskeletal restructuring may lead to cell detachment and register as loss of mass, this experiment was recorded in suspension instead of the adherent mode and with sufficient time to allow for baseline equilibration before ML297 addition (Figure S4a–c).
Actin inhibitors cytochalasin B46 and latrunculin A,47,48 but not the microtubule inhibitor nocodazole49 attenuated ML297-mediated DMR responses in a concentration-dependent manner (Figure 9a–c). Importantly, inhibitor-treated cells remained viable during DMR detection (Figure S4d) and responsive to external stimuli that do not require an intact actin cytoskeleton such as Ca2+ release from intracellular stores50 (Figure S4e). From these results, we concluded that the ML297 signal portrayed by DMR originated from changes in actin network rearrangement downstream of ionotropic channel activity rather than channel conductance in its own right. Thereby, holistic DMR and CDS detection uncovered a previously unknown link between direct GIRK activation and reorganization of the actin cytoskeleton. We speculate that optical and impedance-based biosensing may become more widely applied in the field of ion channel research for those channels that elicit cellular effects beyond the simple conduction of ions.
Figure 9.

Inhibition of actin network rearrangement abolishes ML297-driven DMR changes. (a–c, left panels) Monitored wavelength shift (in pm) of HEK GIRK1/2 in response to 100 μM ML297 after 1 h incubation with indicated concentrations of (a) Cytochalasin B, (b) latrunculin A, and (c) nocodazole, shown as mean + SEM. (a–c, right panels) The summaries portray the AUC as mean + SEM normalized to 100 μM Cch (n = 3). Two-tailed student t-tests compared the inhibitor treated with the respective untreated values.
Conclusions
The major finding of the present study is centered on the discovery that optical and impedance-based biosensing of living cells sheds light on molecular properties of GIRK channels and their ligands that were previously not seen. Routinely, ion channel targeting drugs are characterized using methods that record direct ion flow with patch clamp being the gold standard because of its unrivalled precision and accuracy.10 Our study advances the field of ion channel drug discovery in an important way: we identified label-free DMR and CDS as additional technology platforms competent to portray GIRK channel activity beyond the mere conducting function. Apparently, methods that record only ion flux per se provide an incomplete picture of the spectrum of pharmacological activities of GIRK modulators. Indeed, several channels are known to engage in channel–protein interactions with cytoplasmic signaling molecules beyond the simple transport of ions through the plasma membrane.19,30,51 These would be in need of additional methodologies to portray their biological actions in a more comprehensive manner. Slack (KNa1.1, KCNT1, SLO2.2), a sodium-activated K+ channel and member of the inward rectifying K+ channel family, is one such example.30 On the other hand, K+ channels such as BK (KCNMA1, SLO1) and slick (KNa1.2, KCNT2, SLO2.1) do not produce DMR upon direct stimulation, indicating that label-free whole cell responses are no general consequence of increased K+ conductance.23 Because BK and slick—upon direct activation—do not produce DMR, and slack triggers mass movement independent of ion flux,30 GIRK channels stand out among voltage-sensitive K+ channels in that they convert direct activation into cell morphology changes, the basis of DMR and CDS detection.
One caveat deserves particular mention here: the majority of recordings were obtained in HEK cells which express GIRK subunits recombinantly. The striking correlation between DMR profiles (this study) and GIRK electrophysiology8,31 in this very cellular background, however, strongly suggests that a similar mechanism may operate in primary cardiac or neuronal cells. Indeed, HEK cells haven proven an ideal non-neuronal host for functional expression and electrophysiological study of GIRK channels in the past.8,31 Therefore our data do not only extend the repertoire of pharmacological assays for characterization of GIRK channel-modifying drugs by label-free noninvasive whole-cell biosensing but also identify GIRKs as a particular subgroup within inwardly rectifying K+ channels that connect ion flux to cytoskeleton-dependent cellular mechanisms. G protein-independent, direct GIRK activation is still poorly understood. The effects observed for ML297 in DMR and CDS may therefore be an important step to deepen our understanding how other extrinsic and intrinsic factors like ethanol or cholesterol, which also bypass G protein-gating,52−55 contribute to regulation of excitable cells. The ability to capture previously unseen cellular mechanisms surrounding GIRK activation in phenotypic live cell assays, therefore, offers an unprecedented approach toward both identification of new GIRK ligands and mechanistic understanding of their biological effects. Given that DMR and CDS are high throughput-compatible, we anticipate that both hold much promise in the preclinical discovery pipeline for development of ion channel targeting drugs that regulate ion flux and, additionally, are in need of technologies to portray their nonconducting functions.
Materials and Methods
Chemicals and Reagents
Synthesis of structural ML297 analogs was described previously.7,32 The analogs are referred to by the last three digits of their Vanderbilt University Code. For the full length code, see Figure 7a,d. The OXE-R antagonist Gore27 was prepared following the synthetic route provided in ref (56) and detailed in the Supporting Information. The Thallos AM reagent was purchased from TEFLabs (Austin, TX, USA), and PTX was from Life Technologies (Darmstadt, GER). In all experiments, Hank’s balanced salt solution from Thermo Fisher (Darmstadt, GER) supplemented with 20 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (pH 7.2) was used as assay buffer.
Stable Cell Lines
Generation of HEK293 (HEK) cell lines stably expressing GIRK1/2 or GIRK2 were described previously.7 HEK cells stably expressing GIRK1/2 channels and the Gi-sensitive 5-oxo-ETE receptor OXE-R were prepared from HEK GIRK1/2 cells using selection with Geneticin/G418 followed by single clone isolation. A stable monoclonal HEK GIRK1/4 expressing cell line was generated from HEK GIRK1 cells upon transfection with a pcDNA3.1 plasmid coding for GIRK4 using selection with Geneticin/G418. The cardiac muscle cell line HL-1 was purchased from Merck Millipore (Darmstadt, GER).
Cell Culture
All cell lines were cultivated with 5% CO2 at 37 °C in a humidified atmosphere. All media were supplemented with 10% (v/v) FCS, penicillin (100 U/mL), and streptomycin (100 mg/mL). Native HEK cells were cultivated in Dulbecco’s modified Eagle’s medium, and HEK cells stably transfected with GIRK subunits were cultivated in MEM Alpha Medium (1×) + GlutaMAX-I. For cell lines expressing the GIRK1 subunit, Blasticidin (final concentration of 5 μg/mL) was added to the media, Puromycin (3 μg/mL) for GIRK2, and Geneticin/G418 (400 μg/mL) for GIRK4 and OXE-R, respectively. HL-1 cells were cultivated in Claycomb medium supplemented with FCS 10% (v/v), penicillin (100 U/mL), norepinephrin (0.1 mM), and l-glutamine (2 mM).
Label-Free Real-Time Biosensor Assays with Living Cells
DMR was recorded with the Corning Epic biosensor as described previously.13 The Epic biosensor detects changes in the local index of refraction upon stimulus-induced mass redistribution within a cell monolayer that is grown in 384-well Epic microplates, which contain a resonant wave guide grating biosensor at the bottom of each well. Cells were seeded at a density of 15 000 cells/well 48 h before the experiment, with a media change every 24 h (HL-1) or at 18 000 cells per well and cultivated overnight (HEK). DMR assays in the presence of cytoskeletal inhibitor compounds (Figure 9) were recorded in the suspension mode, using 20 000 cells per well that were spun down gently at 800g to adhere to the sensor surface.
Bioimpedance (CellKey) assays record the change in extracellular impedance of a monolayer of cells located on top of an electrical biosensor. The experiments were performed according to the previously published protocol,57 with the following modifications: 15 000 cells per well were seeded on PDL-coated 384-well biosensor plates for 18–24 h to grow to confluence. For all assays, cells were preincubated with assay buffer for 4 h prior to CDS measurements.
Time-Lapse Microscopy
HEK cells lacking or stably expressing GIRK1/2 were seeded in PDL-coated 12-well plates using 200 000 cells per well and cultivated in FCS-free medium overnight. Before imaging, cells were equilibrated in assay buffer with or without SCH for 1 h prior to ML297 or DMSO treatment. Time-lapse imaging was performed using an inverted brightfield phase-contrast microscope (DM IRE 2, Leica) [Leica DFC350 FX camera, N Plan L 20×/0.40 CORR LMC objective (Leica)] equipped with a heated chamber (37 °C) and CO2 controller (5%) for 3 h after treatment. Pictures were taken every 3 min. Image processing was performed with ImageJ software.
Thallium Flux Assay
Activation of GIRK channels was measured using a Thallos AM dye-based thallium flux assay kit8 and recorded with the FlexStation 3 MultiMode Bench Top reader. The results show channel opening as increase in fluorescence over time. For the exact procedure, see the Supporting Information.
Calcium Flux Assay
Intracellular Ca2+ mobilization was measured using the calcium 5 assay kit (molecular devices) as previously described.58
Cell Viability Assay
Viability of HEK cell lines was assessed using the CellTiter-Blue assay from Promega following the manufacturer’s instructions. See the Supporting Information for more details.
Data Analysis and Statistics
All real-time recordings show one representative biological replicate as mean + SEM of three technical replicates generated in parallel recordings. For quantification of DMR and CDS traces, at least three independent biological replicates were used, each consisting of three technical replicates. Concentration–effect curves were fitted using GraphPad Prism 7.0 (GraphPad Software Inc., San Diego, CA, USA) showing means ± SEM and are based on the number of biological replicates indicated in the figure legends. P values were determined, and P < 0.05 was considered significant (*), P < 0.01 very significant (**), and P < 0.001 extremely significant (***).
Acknowledgments
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—214362475/GRK1873/2.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b02254.
Complete procedure for synthesis of Gore27, thallium flux assay, CellTiter-Blue assay, pEC50 values of ML297, pEC50 values for GIRK inhibitors, activity of ML297 analogs, GIRK channel inhibitors, time-lapse imaging of native HEK, ML analogs, and actin and microtubule inhibitors (PDF)
Time-lapse video files (ZIP)
Author Present Address
⊥ Bayer AG, 51368 Leverkusen, Germany.
Author Present Address
# Research & Development, Cardiovascular II, Aprather Weg 18a, 42113 Wuppertal, Germany.
Author Present Address
¶ Institute of Condensed Matter and Nanosciences, UCL, Louvain la Neuve, Belgium.
Author Contributions
K.M.K. and E.M.P. contributed equally. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): C.D.W. receives royalties from the sale of the thallium-sensitive dye, Thallos, through a licensing agreement between Vanderbilt University and TEFlabs. All remaining authors declare no competing financial interests.
Supplementary Material
References
- Luján R.; de Velasco E. M. F.; Aguado C.; Wickman K. New insights into the therapeutic potential of Girk channels. Trends Neurosci. 2014, 37, 20–29. 10.1016/j.tins.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield J.; Blednov Y. A.; Harris R. A. Behavioral and Genetic Evidence for GIRK Channels in the CNS: Role in Physiology, Pathophysiology, and Drug Addiction. Int. Rev. Neurobiol. 2015, 123, 279–313. 10.1016/bs.irn.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C.; Slesinger P. A. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 2010, 11, 301–315. 10.1038/nrn2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H.; Inanobe A.; Furutani K.; Murakami S.; Findlay I.; Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- Posokhova E.; Wydeven N.; Allen K. L.; Wickman K.; Martemyanov K. A. RGS6/Gβ5 Complex Accelerates I KACh Gating Kinetics in Atrial Myocytes and Modulates Parasympathetic Regulation of Heart Rate. Circ. Res. 2010, 107, 1350–1354. 10.1161/circresaha.110.224212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masotti A.; Uva P.; Davis-Keppen L.; Basel-Vanagaite L.; Cohen L.; Pisaneschi E.; Celluzzi A.; Bencivenga P.; Fang M.; Tian M.; et al. Keppen-Lubinsky Syndrome Is Caused by Mutations in the Inwardly Rectifying K+ Channel Encoded by KCNJ6. Am. J. Hum. Genet. 2015, 96, 295–300. 10.1016/j.ajhg.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann K.; Romaine I.; Days E.; Pascual C.; Malik A.; Yang L.; Zou B.; Du Y.; Sliwoski G.; Morrison R. D.; et al. ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem. Neurosci. 2013, 4, 1278–1286. 10.1021/cn400062a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wydeven N.; de Velasco E. M. F.; Du Y.; Benneyworth M. A.; Hearing M. C.; Fischer R. A.; Thomas M. J.; Weaver C. D.; Wickman K. Mechanisms underlying the activation of G-protein-gated inwardly rectifying K+ (GIRK) channels by the novel anxiolytic drug, ML297. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 10755–10760. 10.1073/pnas.1405190111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Rodríguez I.; Temprano-Carazo S.; Nájera A.; Djebari S.; Yajeya J.; Gruart A.; Delgado-García J. M.; Jiménez-Díaz L.; Navarro-López J. D. Activation of G-protein-gated inwardly rectifying potassium (Kir3/GirK) channels rescues hippocampal functions in a mouse model of early amyloid-β pathology. Sci. Rep. 2017, 7, 14658. 10.1038/s41598-017-15306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop J.; Bowlby M.; Peri R.; Vasilyev D.; Arias R. High-throughput electrophysiology: an emerging paradigm for ion-channel screening and physiology. Nat. Rev. Drug Discovery 2008, 7, 358–368. 10.1038/nrd2552. [DOI] [PubMed] [Google Scholar]
- Weaver C. D. Thallium Flux Assay for Measuring the Activity of Monovalent Cation Channels and Transporters. Methods Mol. Biol. 2018, 1684, 105–114. 10.1007/978-1-4939-7362-0_9. [DOI] [PubMed] [Google Scholar]
- Schröder R.; Janssen N.; Schmidt J.; Kebig A.; Merten N.; Hennen S.; Müller A.; Blättermann S.; Mohr-Andrä M.; Zahn S.; et al. Deconvolution of complex G protein–coupled receptor signaling in live cells using dynamic mass redistribution measurements. Nat. Biotechnol. 2010, 28, 943–949. 10.1038/nbt.1671. [DOI] [PubMed] [Google Scholar]
- Schröder R.; Schmidt J.; Blättermann S.; Peters L.; Janssen N.; Grundmann M.; Seemann W.; Kaufel D.; Merten N.; Drewke C.; et al. Applying label-free dynamic mass redistribution technology to frame signaling of G protein–coupled receptors noninvasively in living cells. Nat. Protoc. 2011, 6, 1748–1760. 10.1038/nprot.2011.386. [DOI] [PubMed] [Google Scholar]
- Carter R. L.; Grisanti L. A.; Yu J. E.; Repas A. A.; Woodall M.; Ibetti J.; Koch W. J.; Jacobson M. A.; Tilley D. G. Dynamic mass redistribution analysis of endogenous β-adrenergic receptor signaling in neonatal rat cardiac fibroblasts. Pharmacol. Res. Perspect. 2014, 2, e00024 10.1002/prp2.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundmann M.; Tikhonova I. G.; Hudson B. D.; Smith N. J.; Mohr K.; Ulven T.; Milligan G.; Kenakin T.; Kostenis E. A Molecular Mechanism for Sequential Activation of a G Protein-Coupled Receptor. Cell Chem. Biol. 2016, 23, 392–403. 10.1016/j.chembiol.2016.02.014. [DOI] [PubMed] [Google Scholar]
- Simon K.; Merten N.; Schröder R.; Hennen S.; Preis P.; Schmitt N.-K.; Peters L.; Schrage R.; Vermeiren C.; Gillard M.; et al. The Orphan Receptor GPR17 Is Unresponsive to Uracil Nucleotides and Cysteinyl Leukotrienes. Mol. Pharmacol. 2017, 91, 518–532. 10.1124/mol.116.107904. [DOI] [PubMed] [Google Scholar]
- Merten N.; Fischer J.; Simon K.; Zhang L.; Schröder R.; Peters L.; Letombe A.-G.; Hennen S.; Schrage R.; Bödefeld T.; et al. Repurposing HAMI3379 to Block GPR17 and Promote Rodent and Human Oligodendrocyte Differentiation. Cell Chem. Biol. 2018, 25, 775–786.e5. 10.1016/j.chembiol.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke N.; Xi B.; Ye P.; Xu W.; Zheng M.; Mao L.; Wu M.-J.; Zhu J.; Wu J.; Zhang W.; et al. Screening and identification of small molecule compounds perturbing mitosis using time-dependent cellular response profiles. Anal. Chem. 2010, 82, 6495–6503. 10.1021/ac1007877. [DOI] [PubMed] [Google Scholar]
- Sun H.; Wei Y.; Deng H.; Xiong Q.; Li M.; Lahiri J.; Fang Y. Label-free cell phenotypic profiling decodes the composition and signaling of an endogenous ATP-sensitive potassium channel. Sci. Rep. 2014, 4, 4934. 10.1038/srep04934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y.; Ferrie A. M.; Fontaine N. H.; Mauro J.; Balakrishnan J. Resonant Waveguide Grating Biosensor for Living Cell Sensing. Biophys. J. 2006, 91, 1925–1940. 10.1529/biophysj.105.077818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen H. B.; Gloriam D. E.; Pedersen D. S.; Cowland J. B.; Borregaard N.; Bräuner-Osborne H. Applying label-free dynamic mass redistribution assay for studying endogenous FPR1 receptor signalling in human neutrophils. J. Pharmacol. Toxicol. Methods 2017, 88, 72–78. 10.1016/j.vascn.2017.07.003. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Ganesh T.; Du Y.; Thepchatri P.; Rojas A.; Lewis I.; Kurtkaya S.; Li L.; Qui M.; Serrano G.; et al. Neuroprotection by selective allosteric potentiators of the EP2 prostaglandin receptor. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 2307–2312. 10.1073/pnas.0909310107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb S.; Michaelis S.; Plank N.; Bernhardt G.; Buschauer A.; Wegener J. Label-free analysis of GPCR-stimulation: The critical impact of cell adhesion. Pharmacol. Res. 2016, 108, 65–74. 10.1016/j.phrs.2016.04.026. [DOI] [PubMed] [Google Scholar]
- Fang Y.; Ferrie A. M.; Fontaine N. H.; Yuen P. K. Characteristics of dynamic mass redistribution of epidermal growth factor receptor signaling in living cells measured with label-free optical biosensors. Anal. Chem. 2005, 77, 5720–5725. 10.1021/ac050887n. [DOI] [PubMed] [Google Scholar]
- Giaever I.; Keese C. R. Micromotion of mammalian cells measured electrically. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 7896–7900. 10.1073/pnas.88.17.7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever I.; Keese C. R. A morphological biosensor for mammalian cells. Nature 1993, 366, 591–592. 10.1038/366591a0. [DOI] [PubMed] [Google Scholar]
- Stamer W. D.; Piwnica D.; Jolas T.; Carling R. W.; Cornell C. L.; Fliri H.; Martos J.; Pettit S. N.; Wang J. W.; Woodward D. F. Cellular Basis for Bimatoprost Effects on Human Conventional Outflow. Invest. Ophthalmol. Visual Sci. 2010, 51, 5176–5181. 10.1167/iovs.09-4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdonk E.; Johnson K.; McGuinness R.; Leung G.; Chen Y.-W.; Tang H. R.; Michelotti J. M.; Liu V. F. Cellular dielectric spectroscopy: a label-free comprehensive platform for functional evaluation of endogenous receptors. Assay Drug Dev. Technol. 2006, 4, 609–619. 10.1089/adt.2006.4.609. [DOI] [PubMed] [Google Scholar]
- Kaczmarek L. K. Non-conducting functions of voltage-gated ion channels. Nat. Rev. Neurosci. 2006, 7, 761–771. 10.1038/nrn1988. [DOI] [PubMed] [Google Scholar]
- Fleming M. R.; Brown M. R.; Kronengold J.; Zhang Y.; Jenkins D. P.; Barcia G.; Nabbout R.; Bausch A. E.; Ruth P.; Lukowski R.; et al. Stimulation of Slack K+ Channels Alters Mass at the Plasma Membrane by Triggering Dissociation of a Phosphatase-Regulatory Complex. Cell Rep. 2016, 16, 2281–2288. 10.1016/j.celrep.2016.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber D. M.; Schönberger M.; Burgstaller J.; Levitz J.; Weaver C. D.; Isacoff E. Y.; Baier H.; Trauner D. Optical control of neuronal activity using a light-operated GIRK channel opener (LOGO). Chem. Sci. 2015, 7, 2347–2352. 10.1039/c5sc04084a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W.; Wu W.; Romaine I. M.; Kaufmann K.; Du Y.; Sulikowski G. A.; Weaver C. D.; Lindsley C. W. Discovery of “molecular switches” within a GIRK activator scaffold that afford selective GIRK inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 4562–4566. 10.1016/j.bmcl.2013.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzhikandathil E. V.; Oxford G. S. Classic D1 dopamine receptor antagonist R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH23390) directly inhibits G protein-coupled inwardly rectifying potassium channels. Mol. Pharmacol. 2002, 62, 119–126. 10.1124/mol.62.1.119. [DOI] [PubMed] [Google Scholar]
- Li D.; Chen R.; Chung S.-H. Molecular dynamics of the honey bee toxin tertiapin binding to Kir3.2. Biophys. Chem. 2016, 219, 43–48. 10.1016/j.bpc.2016.09.010. [DOI] [PubMed] [Google Scholar]
- Jin W.; Klem A. M.; Lewis J. H.; Lu Z. Mechanisms of Inward-Rectifier K+Channel Inhibition by Tertiapin-Q. Biochemistry 1999, 38, 14294–14301. 10.1021/bi991206j. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; MacKinnon R. The Barium Site in a Potassium Channel by X-Ray Crystallography: Figure 1. J. Gen. Physiol. 2000, 115, 269–272. 10.1085/jgp.115.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y.; Ferrie A. M.; Li G.; Verrier F.; Tran E.; Xi J.; Soni M.; Fontaine N.; Verkleeren R.. Dynamic Mass Redistribution: A Novel Physiological Signal of Cells for Cell Systems Biology and Pharmacology, 2007.
- Fang Y. Non-Invasive Optical Biosensor for Probing Cell Signaling. Sensors 2007, 7, 2316–2329. 10.3390/s7102316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y.; Ferrie A. M.; Fontaine N. H.; Lahiri J.; Yuen P. K.. Label Free Biosensors and Cells. U.S. Patent 20,090,142,790 A1, 2009.
- Ciambrone G. J.; Liu V. F.; Lin D. C.; McGuinness R. P.; Leung G. K.; Pitchford S. Cellular dielectric spectroscopy: a powerful new approach to label-free cellular analysis. J. Biomol. Screening 2004, 9, 467–480. 10.1177/1087057104267788. [DOI] [PubMed] [Google Scholar]
- Leung G.; Tang H.; McGuinnes R.; Verdonk E.; Michelotti J.; Liu V. Cellular Dielectric Spectroscopy: A Label-Free Technology for Drug Discovery. J. Biomol. Screening 2005, 10, 258–269. 10.1016/j.jala.2005.06.002. [DOI] [Google Scholar]
- Kloukina V.; Herzer S.; Karlsson N.; Perez M.; Daraio T.; Meister B. G-protein-gated inwardly rectifying K+ channel 4 (GIRK4) immunoreactivity in chemically defined neurons of the hypothalamic arcuate nucleus that control body weight. J. Chem. Neuroanat. 2012, 44, 14–23. 10.1016/j.jchemneu.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Claycomb W. C.; Lanson N. A.; Stallworth B. S.; Egeland D. B.; Delcarpio J. B.; Bahinski A.; Izzo N. J. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 2979–2984. 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobles M.; Sebastian S.; Tinker A. HL-1 cells express an inwardly rectifying K+ current activated via muscarinic receptors comparable to that in mouse atrial myocytes. Pflugers Archiv 2010, 460, 99–108. 10.1007/s00424-010-0799-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgovan N.; Peter B.; Bősze S.; Ramsden J. J.; Szabó B.; Horvath R. Dependence of cancer cell adhesion kinetics on integrin ligand surface density measured by a high-throughput label-free resonant waveguide grating biosensor. Sci. Rep. 2014, 4, 4034. 10.1038/srep04034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakatsuki T.; Schwab B.; Thompson N. C.; Elson E. L. Effects of cytochalasin D and latrunculin B on mechanical properties of cells. J. Cell Sci. 2001, 114, 1025–1036. [DOI] [PubMed] [Google Scholar]
- Coué M.; Brenner S. L.; Spector I.; Korn E. D. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 1987, 213, 316–318. 10.1016/0014-5793(87)81513-2. [DOI] [PubMed] [Google Scholar]
- Morton W. M.; Ayscough K. R.; McLaughlin P. J. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat. Cell Biol. 2000, 2, 376–378. 10.1038/35014075. [DOI] [PubMed] [Google Scholar]
- Samson F.; Donoso J. A.; Heller-Bettinger I.; Watson D.; Himes R. H. Nocodazole action on tubulin assembly, axonal ultrastructure and fast axoplasmic transport. J. Pharmacol. Exp. Ther. 1979, 208, 411–417. [PubMed] [Google Scholar]
- Peng G. E.; Wilson S. R.; Weiner O. D. A pharmacological cocktail for arresting actin dynamics in living cells. Mol. Biol. Cell 2011, 22, 3986–3994. 10.1091/mbc.e11-04-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer M.; Jahnke H.-G.; Krinke D.; Zitzmann F. D.; Hill K.; Schaefer M.; Robitzki A. A. Quantitative characterization of capsaicin-induced TRPV1 ion channel activation in HEK293 cells by impedance spectroscopy. Anal. Bioanal. Chem. 2016, 408, 8529–8538. 10.1007/s00216-016-9978-x. [DOI] [PubMed] [Google Scholar]
- Glaaser I. W.; Slesinger P. A. Dual activation of neuronal G protein-gated inwardly rectifying potassium (GIRK) channels by cholesterol and alcohol. Sci. Rep. 2017, 7, 4592. 10.1038/s41598-017-04681-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T.; Ikeda K.; Kojima H.; Niki H.; Yano R.; Yoshioka T.; Kumanishi T. Ethanol opens G-protein-activated inwardly rectifying K+channels. Nat. Neurosci. 1999, 2, 1091–1097. 10.1038/16019. [DOI] [PubMed] [Google Scholar]
- Lewohl J. M.; Wilson W. R.; Mayfield R. D.; Brozowski S. J.; Morrisett R. A.; Harris R. A. G-protein-coupled inwardly rectifying potassium channels are targets of alcohol action. Nat. Neurosci. 1999, 2, 1084–1090. 10.1038/16012. [DOI] [PubMed] [Google Scholar]
- Aryal P.; Dvir H.; Choe S.; Slesinger P. A. A discrete alcohol pocket involved in GIRK channel activation. Nat. Neurosci. 2009, 12, 988–995. 10.1038/nn.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore V.; Gravel S.; Cossette C.; Patel P.; Chourey S.; Ye Q.; Rokach J.; Powell W. S. Inhibition of 5-oxo-6,8,11,14-eicosatetraenoic acid-induced activation of neutrophils and eosinophils by novel indole OXE receptor antagonists. J. Med. Chem. 2014, 57, 364–377. 10.1021/jm401292m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennen S.; Wang H.; Peters L.; Merten N.; Simon K.; Spinrath A.; Blattermann S.; Akkari R.; Schrage R.; Schroder R.; et al. Decoding signaling and function of the orphan G protein-coupled receptor GPR17 with a small-molecule agonist. Sci. Signaling 2013, 6, ra93. 10.1126/scisignal.2004350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage R.; Schmitz A.-L.; Gaffal E.; Annala S.; Kehraus S.; Wenzel D.; Büllesbach K. M.; Bald T.; Inoue A.; Shinjo Y.; et al. The experimental power of FR900359 to study Gq-regulated biological processes. Nat. Commun. 2015, 6, 10156. 10.1038/ncomms10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.