

Abstract
We report a family of Saudi Arabian ancestry with two children presenting with global developmental delay, dystonia, disturbed sleep, and heat intolerance. By genome sequencing, we identified a nonsense variant in the first exon of PI4K2A that was homozygous in both affected individuals and was absent from, or heterozygous in, seven unaffected siblings. PI4K2A is highly expressed in the brain and a mouse model displays a neurological phenotype, implicating PI4K2A as a new disease gene for a neurological disorder.
Introduction
The molecular diagnostic rate for individuals with rare neurological or neurodevelopmental disorders is typically 20–40%, depending on the cohort examined and the methodologies used.1, 2 These disorders are highly heterogeneous with many underlying genes, suggesting that there are further genes yet to be discovered. Such disorders are well suited for genome‐wide analysis by either exome or genome sequencing since the high levels of genetic and phenotypic heterogeneity make hypothesis‐driven targeted gene testing challenging. Genome‐wide analysis in patients from consanguineous families with previously undiagnosed neurological conditions can facilitate the discovery of new genetic etiologies, particularly for rare autosomal recessive conditions.
Here, we report a family with an undiagnosed developmental and neurological disorder found by genome sequencing to carry a homozygous variant in PI4K2A, a gene not previously implicated in human disease.
Clinical Report
Subjects II‐8 and II‐9 (Fig. 1) are brothers born to first degree cousins of Saudi Arabian origin, presenting with global developmental delay, dystonia, heat intolerance, and sleep disruption. They are the last‐born children in a family of nine with the other seven siblings all unaffected. The parents are also asymptomatic.
Figure 1.
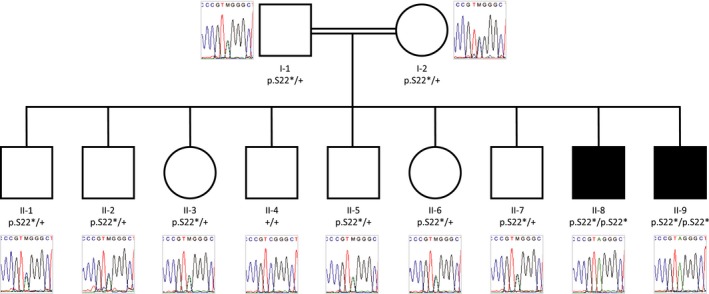
Pedigree of the family showing genotypes. “p.S22*” indicates the PI4K2A variant in isoform NM_018425 and “+” indicates no variant. Black shapes indicate affected individuals and unaffected family members are shown by white shapes.
The two affected boys were born following uncomplicated pregnancies and deliveries. They manifest subtle features of dysmorphism, they have hypotelorism, small eyes, micrognathia, and prominent ears. At 5 to 6 months of age they had failure to thrive, visual inattentiveness, loss of acquired skills and poor oral coordination and feeding. The remainder of infancy was accentuated by prolonged episodes of crying, irritability, and akathisia, which includes facial dyskinesias persisting until their present ages of 9 and 14 years. They have episodic arm dystonia with opisthotonic posturing that can last for hours on weekly basis (improved with Clonazepam). They are nonverbal and nonambulatory, they are unable to stand but are able to crawl and sit without support. Both developed myoclonus and generalized seizures at age 9, the latter controlled to date with levetiracetam. Both boys’ growth parameters remain at the third percentile. They are heat intolerant and have disturbed sleep in the form of prolonged sleep latency and short duration of up to 4 h per 24 h period; they sleep during the day and are awake through the night. They are hypotonic axially with increased appendicular tone and brisk reflexes. Normal strength was observed on individual muscle group testing. Pes cavus is evident and peripheral neuropathy is likely given bilaterally delayed responses to touch, pain, and vibration. Systemic examination was normal. Brain MRI for individual II‐8 at infancy is shown in Figure S1. EEG in both subjects showed slow background and bursts of central theta frequency waves with no frank epileptiform discharges. Cerebrospinal fluid neurotransmitter profile was normal.
Methods
All genomic analyses were performed on DNA extracted from whole blood. Exome sequencing (WES) was performed for subjects II‐8 and II‐9 (Fig. 1), using the Ion AmpliSeq Exome Kit (Life Technologies) with sequencing on the Ion Proton™semiconductor system following the manufacturer's instructions. Alignment and variant calling were performed using Torrent Suite (v4.0) on the Ion Proton Server using Ion Proton standard low‐stringency settings. Genome sequencing (WGS) and data analysis for II‐8 and II‐9 were performed as previously described.3 Briefly, the Illumina TruSeq Nano DNA Library Prep Kit was used for library preparation, omitting the PCR amplification step, followed by paired‐end sequencing on Illumina HiSeq X system as specified by the manufacturer. Base calling and analysis were performed using Illumina HiSeq Analysis Software v2‐2.5.55.1311. Reads were mapped to hg19 using Isaac alignment (SAAC00776.15.01.27) and single nucleotide and small indel variants called Isaac variant caller Starling 2.1.4.2 and annotated as previously described4 based on ANNOVAR.5 Copy number variants were detected as described by Trost et al.6 Individuals I‐1, I‐2, II‐4, II‐6, II‐8, and II‐9 were also genotyped using Infinium HumanOmni2.5‐8 BeadChip according to the manufacture's protocol. Homozygosity mapping was performed using Homozygosity Detector 1.0.3 module in GenomeStudio with default parameters. Informed consent was collected from all participants and this study was approved by The Hospital for Sick Children Research Ethics Board, Toronto, Canada.
Results
WES of the affected children identified eight rare autosomal variants in coding regions that were homozygous in both, none of which was considered likely pathogenic. WGS was then carried out, yielding 13 high confidence autosomal rare coding variants found to be homozygous in both affected brothers. One was a nonsense variant in exon 1 of PI4K2A, which would lead to protein truncation at amino acid 22 (chr10: 99400564C>A (hg19); c.65C>A; p.Ser22* (NM_018425)). This variant has not been observed in population databases.7, 8, 9, 10 Review of the WES data revealed that the variant had been missed by WES due to there being no coverage of this exon, likely due to high GC content. The probability of loss of function intolerance (pLI) score for PI4K2A according to the Exome Aggregation Consortium (ExAC) is 0.98, indicating high probability that the gene is intolerant to loss‐of‐function variants, even in heterozygous state.7 There are no homozygous loss of function variants reported in PI4K2A in population databases7, 8, 9, 10 or in over 10,500 individuals of Pakistani origin from a study of myocardial infarction.11 Sanger sequencing confirmed the variant and showed that it segregates in homozygous state with the disease (Fig. 1). Homozygosity mapping using SNP genotype data from individuals I‐1, I‐2, II‐4, II‐6, II‐8, and II‐9 showed only one region to be homozygous in the two affected individuals and none of the unaffected family members (chr10:95396491‐108672251 in II‐8 and chr10:95396491‐117708803 in II‐9). Unsurprisingly, PI4K2A is within this locus. No CNVs considered to be pathogenic or likely pathogenic were identified in either II‐8 or II‐9. All homozygous rare coding variants detected in II‐8 and II‐9 are listed in Table S1.
Discussion
PI4K2A encodes phosphatidylinositol 4‐kinase type 2 alpha, one of a number of phosphoinositide (PI) kinases responsible for phosphorylating phosphatidylinositol.12 Phosphatidylinositol is a membrane‐integral triglyceride with a phosphate‐linked inositol sugar. Several kinases, from two families, phosphorylate the fourth carbon of the sugar to generate the phosphoinositide (PIP) PI4P. Further phosphorylation of PI4P generates additional PIPs, all of which, including PI4P, have roles in regulation of cellular functions such as receptor signaling, vesicle trafficking, endocytosis, and cytoskeletal rearrangement. PI4Ps also play a part in ion channel regulation, synaptic vesicle recycling and neuronal cell survival, and have been proposed to have a variety of neurobiological roles.13 Accordingly, dysregulation of PIP pathways has been implicated in a range of neurological diseases.14 The several kinases that generate PI4P from phosphatidylinositol have differing subcellular localizations conferring location‐specific functions. PI4K2A is highly expressed in the brain15 and has many protein binding partners, indicating functions in addition to its catalytic activity. Pi4k2α deficient mice lacking measurable kinase activity show no obvious phenotype when young. Later, they develop tremors, spastic gait, muscle weakness, and feeding problems that worsened with age, accompanied by degeneration of long axons and Purkinje cells.16 Given the similarity with hereditary spastic paraplegia (HSP), PI4K2A was screened for mutations in a cohort of individuals with complicated HSP including other neurological features, but no pathogenic variants were identified.17
The patients described here are the first reported with complete loss of PI4K2A, with the premature stop codon being introduced after only 22 amino acids, indicating that loss of functional PI4K2A results in a neurodevelopmental phenotype with severe intellectual disability, mild epilepsy, myoclonus, and akathisia. This is in contrast to the phenotype observed in mice lacking the kinase activity of Pi4k2a, which more closely resembled adult onset motor neuron disease. The phenotypic differences observed may be the result of a residual function of the remaining noncatalytic N‐terminal portion of the protein in the mouse model or simply due to differences in human and mouse systems. However, the early regression in infancy and delayed onset of myoclonus and epilepsy could be consistent with neurodegeneration, which may further evolve to involve motor systems in future years.
The phenotype observed in this family delineates the neurodevelopmental and neurological state of the brain lacking a key phosphatidylinositol kinase. This may guide the future understanding of the basic mechanisms of the components of the PIP cascade and PI4K2A in brain formation and function, as well as implicating other members of the PI kinase family as disease genes in other related disorders. This case also demonstrates the benefits of performing WGS in clinical research as this family had evaded molecular diagnosis using WES, and indicates that there are likely further cases for which the causative variant(s) are in genomic regions not amenable to WES.
Author Contributions
Dr. Alkhater – clinical characterization; data acquisition, analysis, and interpretation; critical revision of the manuscript. Dr. Scherer – critical revision of the manuscript. Dr. Minassian – drafting and critical revision of the manuscript and study supervision. Dr. Walker – data acquisition, analysis and interpretation; drafting and critical revision of the manuscript.
Conflict of Interest
Dr. Walker and Dr. Alkhater report no disclosures. Dr. Scherer holds the GlaxoSmithKline‐CIHR Chair in Genome Sciences at the University of Toronto and The Hospital for Sick Children; is on the scientific advisory board for Deep Genomics; has served on the scientific advisory board of Population Diagnostics; has served on the editorial boards of Genomic Medicine, Genes, Genomes, Genetics, the Journal of Personalized Medicine, the Open Genomics Journal, the Hugo Journal, Genome Medicine, the Journal of Neurodevelopmental Disorders, Autism Research, PathoGenetics, Comparative and Functional Genomics, BMC Medical Genomics, and Cytogenetics and Genome Research; and has received research support from Genome Canada/Ontario Genomics Institute, Canadian Institutes of Health Research, Canadian Institute for Advanced Research, McLaughlin Centre, Canada Foundation for Innovation, government of Ontario, and NIH. Dr. Minassian holds patents for diagnostic testing of the following genes: EPM2A, EPM2B, MECP2, and VMA21; has received research support from NIH; and has received license fee payments/royalty payments from patents for diagnostic testing of the following genes: EPM2A, EPM2B, MECP2, and VMA21.
Supporting information
Figure S1: Sagittal image of the brain shows hypoplasia of the corpus callosum with loss of the gyral infolding of the cingulate gyri. There is an unusual thickened appearance to the lamina terminalis. Small pons and brainstem. Small mega cisterna magna.
Table S1: Homozygous rare coding variants detected in individuals II‐8 and II‐9. Coordinates in hg19.
Acknowledgments
We thank the family for their participation in the study and The Centre for Applied Genomics for their analytical and technical support. We thank Dr. Miriam Reuter for helpful comments. This work was supported by the Canadian Institutes of Health Research (CIHR). S.W.S. is funded by the GlaxoSmithKline‐CIHR Chair in Genome Sciences at the University of Toronto, The Hospital for Sick Children, the University of Toronto McLaughlin Centre, Genome Canada/Ontario Genomics Institute, the Canadian Institutes of Health Research (CIHR), the Canadian Institute for Advanced Research, and the Canada Foundation for Innovation. B.A.M holds the University of Toronto Michael Bahen Chair in Epilepsy Research and the University of Texas Southwestern Jimmy Elizabeth Westcott Distinguished Chair in Pediatric Neurology.
Funding Information
This work was supported by the Canadian Institutes of Health Research (CIHR). S.W.S. is funded by the GlaxoSmithKline‐CIHR Chair in Genome Sciences at the University of Toronto, The Hospital for Sick Children, the University of Toronto McLaughlin Centre, Genome Canada/Ontario Genomics Institute, the Canadian Institutes of Health Research (CIHR), the Canadian Institute for Advanced Research, and the Canada Foundation for Innovation. B.A.M holds the University of Toronto Michael Bahen Chair in Epilepsy Research and the University of Texas Southwestern Jimmy Elizabeth Westcott Distinguished Chair in Pediatric Neurology.
Funding Statement
This work was funded by Canadian Institutes of Health Research (CIHR) grant ; GlaxoSmithKline-CIHR Chair in Genome Sciences at the University of Toronto grant ; The Hospital for Sick Children grant ; University of Toronto McLaughlin Centre grant ; Genome Canada/Ontario Genomics Institute grant ; Canadian Institute for Advanced Research grant ; Canada Foundation for Innovation grant .
References
- 1. Evers C, Staufner C, Granzow M, et al. Impact of clinical exomes in neurodevelopmental and neurometabolic disorders. Mol Genet Metab 2017;121:297–307. [DOI] [PubMed] [Google Scholar]
- 2. Zech M, Boesch S, Jochim A, et al. Clinical exome sequencing in early‐onset generalized dystonia and large‐scale resequencing follow‐up. Mov Disord Off J Mov Disord Soc 2017;32:549–559. [DOI] [PubMed] [Google Scholar]
- 3. Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole‐genome sequencing as a first‐tier genetic test. Genet Med Off J Am Coll Med Genet 2018;20:435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yuen RKC, Thiruvahindrapuram B, Merico D, et al. Whole‐genome sequencing of quartet families with autism spectrum disorder. Nat Med 2015;21:185–191. [DOI] [PubMed] [Google Scholar]
- 5. Chang X, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet 2012;49:433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Trost B, Walker S, Wang Z, et al. A comprehensive workflow for read depth‐based identification of copy‐number variation from whole‐genome sequence data. Am J Hum Genet 2018;102:142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fu W, O'Connor TD, Jun G, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein‐coding variants. Nature 2013;493:216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Genomes Project Consortium . A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Akbari MR, Fattahi Z, Beheshtian M, et al. Iranome: A human genome variation database of eight major ethnic groups that live in Iran and neighboring countries in the Middle East. ASHG Annual Meeting, October 2017, Orlando‐USA.
- 11. Saleheen D, Natarajan P, Armean IM, et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature 2017;544:235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Minogue S, Anderson JS, Waugh MG, et al. Cloning of a human type II phosphatidylinositol 4‐kinase reveals a novel lipid kinase family. J Biol Chem 2001;276:16635–16640. [DOI] [PubMed] [Google Scholar]
- 13. Clayton EL, Minogue S, Waugh MG. Phosphatidylinositol 4‐kinases and PI4P metabolism in the nervous system: roles in psychiatric and neurological diseases. Mol Neurobiol 2013;47:361–372. [DOI] [PubMed] [Google Scholar]
- 14. Waugh MG. PIPs in neurological diseases. Biochim Biophys Acta 2015;1851:1066–1082. [DOI] [PubMed] [Google Scholar]
- 15. Balla A, Tuymetova G, Barshishat M, et al. Characterization of type II phosphatidylinositol 4‐kinase isoforms reveals association of the enzymes with endosomal vesicular compartments. J Biol Chem 2002;277:20041–20050. [DOI] [PubMed] [Google Scholar]
- 16. Simons JP, Al‐Shawi R, Minogue S, et al. Loss of phosphatidylinositol 4‐kinase 2alpha activity causes late onset degeneration of spinal cord axons. Proc Natl Acad Sci USA 2009;106:11535–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cleeter M, Houlden H, Simons P, et al. Screening for mutations in the phosphatidylinositol 4‐kinase 2‐alpha gene in autosomal recessive hereditary spastic paraplegia. Amyotroph Lateral Scler Off Publ World Fed Neurol Res Group Mot Neuron Dis 2011;12:148–149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Sagittal image of the brain shows hypoplasia of the corpus callosum with loss of the gyral infolding of the cingulate gyri. There is an unusual thickened appearance to the lamina terminalis. Small pons and brainstem. Small mega cisterna magna.
Table S1: Homozygous rare coding variants detected in individuals II‐8 and II‐9. Coordinates in hg19.