

Abstract
Objective
To compare fetal microchimerism (FMc) in pregnancies with uncomplicated vaginal delivery (VD) versus Cesarean delivery (CD).
Design
Prospective cohort study.
Setting
University of Washington and Fred Hutchinson Cancer Research Center, USA.
Population
Women delivering singleton pregnancies without pertinent antenatal complications with uncomplicated deliveries (n=36).
Methods
We collected maternal pre-delivery, post-delivery, and umbilical cord blood for each mother-baby pair. Following maternal and fetal genotyping, FMc was measured with quantitative PCR assays targeting fetal-specific polymorphisms. Quantification of FMc is expressed as genome equivalents (gEq) of fetal DNA/100,000 total gEq tested. FMc detection was evaluated by logistic regression while controlling for total number of cell equivalents tested and clinically relevant covariates. FMc concentrations were compared using negative binomial regression while controlling for the same covariates and pre-delivery FMc positivity.
Main Outcome Measure
Detection and concentration of FMc by mode of delivery.
Results
24 mother-baby pairs had a VD and 12 had a CD. Post-delivery FMc detection was higher following CD versus VD (58.3% vs. 16.7%, p=0.02). After controlling for covariates, the likelihood of post-delivery FMc detection was almost nine-fold higher after CD than VD (OR 8.8, 95% CI 1.6-47.6; p=0.01). With respect to post-delivery FMc concentration, the detection rate ratio for CD versus VD in the adjusted negative binomial regression model was 14.7 (95% CI, 3.2-66.8; p=0.001).
Conclusion
Post-delivery peripheral FMc detection and concentration are significantly higher after CD versus VD. As FMc is associated with long-term maternal health, our findings suggest that the mode of delivery may impact this risk.
Keywords: Cesarean delivery, maternal-fetal exchange, microchimerism, mode of delivery, pregnancy
Tweetable Abstract:
Greater fetal microchimerism found in maternal blood following Cesarean delivery compared to vaginal delivery.
INTRODUCTION
Bidirectional maternal-fetal transfer of cells and DNA occurs early in pregnancy and continues throughout gestation.(1) Fetal cells in maternal circulation and tissues, predominantly transferred transplacentally, can be present as microchimerism (Mc), and have the potential to persist for decades.(2–5) Obstetric factors seem to influence the amount of Mc transferred. In miscarriage and abortion, fetal Mc (FMc) transfer to the mother is higher among participants undergoing surgical treatment versus those treated medically.(6) Pregnancies characterized by placental dysfunction are also known to have altered fetal-maternal exchange, with preeclampsia pregnancies demonstrating greater amounts of FMc in maternal circulation.(7,8) Long-term persistence of naturally acquired cellular Mc is known to occur, and many questions regarding the role of FMc in maternal post-reproductive health remain unanswered.(9–11)
Epidemiologic data suggest that the mode of delivery may be an important factor for later life health of both the mother and neonate. Several population based studies demonstrate that Cesarean delivery (CD), compared to vaginal delivery (VD), is a risk factor for the development of childhood autoimmune diseases, including type 1 diabetes and inflammatory bowel disease.(12,13) Though less is known about the association of mode of delivery with subsequent maternal health, one large retrospective study analyzed the relationship of delivery mode with subsequent autoimmune disease (AID) in a Danish healthcare database (>1 million women). Compared to nulliparous women, the incidence of AID diagnosis was significantly greater in the first year after delivery and was higher in those who underwent a CD versus VD.(14) Because FMc is associated with certain AIDs,(15) the authors of this study speculated that a greater amount of fetomaternal interface disruption may occur during CD compared to VD, allowing for greater cellular transfer.(16)
Because obstetric factors, including mode of delivery, may impact FMc transfer to the mother with potential long-term implications, we sought to evaluate FMc transfer at VD versus CD in women with uncomplicated deliveries.
METHODS
We conducted a prospective cohort study of participants anticipated and subsequently confirmed to have an uncomplicated delivery of a pregnancy without pertinent complications. Women were recruited from the University of Washington (UW) Maternal Infant Care Clinic, Labor and Delivery unit, and the Fred Hutchinson Cancer Research Center (FHCRC). The study was approved by the Institutional Review Boards of the UW (Ref. No. 23149) and FHCRC (Ref. No. 9569). All participants provided written informed consent prior to enrollment. Patients were not directly involved in the design of this study. Funding for this study was supported by the National Institutes of Health (K08HD067221, R01HL11737, T32HD007233).
We included women ≥18 years old carrying a non-anomalous singleton fetus. Women with the following conditions were excluded as these conditions may affect FMc detection or transfer at the time of delivery: multiple gestation, conception via in vitro fertilization, preeclampsia, HELLP (hemolysis, elevated liver enzymes, low platelet) syndrome, eclampsia, fetal growth restriction (estimated fetal weight <10th percentile), external cephalic version in the current pregnancy, placental abnormalities (invasive placentation, previa, abruption), preterm labour, and maternal autoimmune disease or transplant. Women with pregestational and gestational diabetes were permitted to enroll in the study so long as their delivery was confirmed to be uncomplicated. Gestational age was determined by the last menstrual period corroborated by a first or second trimester ultrasound or the earliest ultrasound available. Mode of delivery was characterized as either VD (spontaneous and operative) or CD (scheduled and unscheduled). Initial chart review confirmed that participants were eligible for enrollment. Following delivery and collection of samples, additional detailed prenatal, intrapartum, and postpartum characteristics were reviewed to confirm an uncomplicated course for continued inclusion in the study.
Sample collection and isolation of PBMC
Maternal peripheral blood samples were collected at two time points: 1) pre-labour third trimester (36 0/7 – 42 0/7 weeks) and 2) post-delivery within approximately 2 hours of placental delivery. A cord blood sample was also collected by venipuncture of a doubly clamped section of the umbilical cord from the placenta after delivery. All blood samples were collected in acid citrate dextrose solution A-vacutainer tubes and underwent processing (unfrozen) within 24 hours. The majority of samples were processed within 12 hours. Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll Histopaque (Pharmacia Biotech, Uppsala, Sweden) gradient centrifugation at a density of 1.077 g/mL, cryopreserved in dimethylsulfoxide, and stored in the gas phase of liquid nitrogen. Genomic DNA was extracted from whole blood and PBMC using QIAmp® DNA Mini Kit (Qiagen, Hilden, Germany) or PureLink® Genomic DNA Mini Kit (Invitrogen, Thermo Fisher Scientific, Carlsbad, California).
Genotyping of mother-baby pairs
Because of extensive polymorphism in human leukocyte antigen (HLA) genes, HLA genotyping of women and their fetuses usually results in identification of a polymorphism unique to the fetus that can be targeted by quantitative polymerase chain reaction (quantitative PCR, or QPCR) to identify and quantify FMc. HLA genotyping was conducted using a Luminex-based (One Lambda, Thermo Fisher Scientific) PCR-sequence-specific oligonucleotide probe-based technique. Maternal and cord blood samples were HLA genotyped for the class II loci DRB1, DQA1, and DQB1. HLA relationships were then examined to identify non-shared HLA polymorphisms targetable to identify FMc. Because an HLA polymorphism unique to the fetus may not always be available for all mother-baby pairs, genotyping for several other polymorphic non-HLA genes was also performed as necessary. These genes included antithrombin III, thyroglobulin, and glutathione S-transferase theta 1. Genotyping for these non-HLA loci used a conventional PCR system described previously.(17) For maternal-fetal pairs without an HLA or non-HLA polymorphism targetable for FMc and with a male fetus, the SRY gene was used as a target for FMc.
Detection and quantification of FMc
After identifying a polymorphism unique to the neonate, we used the appropriate assay from a panel of QPCR assays previously developed for this purpose to test DNA extracted from maternal PBMC for FMc.(18) Sensitivity of utilized of primers was previously established via testing of each HLA-specific QPCR primer with an extended panel of well-characterized HLA cell lines. Of note, primers are designed to amplify specific HLA alleles (i.e. amplification of only the DRB1*01 allele group and no other groups such as DRB1*03, 04, etc.). Testing is performed on a cell-line known to be positive for the HLA target of interest at increasing concentrations (lowest of 0.5 cell equivalents, highest of 500 cell equivalents) amongst a background of cells known to be negative for that HLA polymorphism. Six replicates of DNA from PBMC were tested from each blood draw, with total reaction volumes of 50 µL. A calibration curve for the polymorphism-specific assay using commercially available cell lines known to be positive for the HLA target of interest was included to quantify the amount of FMc and to validate the assay for each experiment (positive control). Every sample was also tested for a nonpolymorphic gene, BGLOB. A BGLOB calibration curve (prepared from commercially available human genomic DNA [Promega, Madison, WI]) was concurrently evaluated on each plate to quantify the total number of genome equivalents (gEq) of DNA tested in each reaction. DNA quantities were reported as the DNA gEq number of FMc cells per 100,000 total gEq tested, using a conversion factor of 6.6 pg of DNA per cell.(19) A minimum of 30,000 total gEq tested per sample was assessed.
Multiple precautions were taken to minimize potential for contamination in QPCR experiments. DNA extractions and QPCR preparations were performed under an ultraviolet light-equipped safety hood cleaned with bleach and filtered tips were used during pipetting. Each experiment included multiple negative control wells to ensure the absence of contamination.
Statistical methods
A sample size calculation for this study has a high degree of uncertainty because preliminary data are limited. However, in our prior study of FMc transfer to maternal circulation in pregnancy loss (≤ 23 6/7 weeks), surgical management was associated with a detection rate ratio of 24.7 for detection of post-treatment FMc compared with medical management.(6) Based on this, inclusion of 18 participants (9 with VD and 9 with CD) would be expected to yield 90% power to detect a similar rate ratio difference at an alpha level of 0.05. Because we anticipated higher overall detection of FMc in the current study (at term versus the earlier gestational age of the prior study), which could diminish differences between groups, we sought to enroll approximately one third additional participants for the current study.
Fisher’s exact and Chi-square analyses were used to compare categorical variables as appropriate. Student’s t-test or the Mann Whitney U test was utilized for comparison of continuous variables. Logistic regression was used to evaluate the association between mode of delivery and FMc detection. Negative binomial regression was used to compare FMc concentration by mode of delivery. The use of negative binomial models for analysis of Mc concentrations has previously been tested and described.(20) Additional covariates were included in the adjusted models if they changed the coefficient for mode of delivery by 10% or more (confounding) or were significantly related to the outcome at the p ≤ 0.10 level (prediction), and included gravidity and gestational age at delivery. Because the total gEq tested is considered a crucially important covariate, this variable was forced into all adjusted models. As delivery is our obstetrical event of interest, and fetal cell transfer may have already occurred by late in the third trimester, we also included whether or not a participant was positive for FMc in the pre-delivery sample in the negative binomial models.
RESULTS
Between August 2014 and May 2016, 74 participants were prospectively recruited and had at least one sample (of the required three) collected. Following initiation of sample collection, 35 participants were excluded because one or more sample collections were missed (n=29), the participant developed preeclampsia (n=4), placenta previa was diagnosed (n=1), or a history of maternal Crohn’s disease was discovered (n=1). After completion of all sample collections and genotyping (n=39), 3 mother-baby pairs were excluded given that no HLA or non-HLA assay was informative for a unique FMc target. Ultimately, 36 mother-baby pairs were available for FMc analysis. Of these, 24 underwent VD and 12 underwent CD, including six scheduled CD and six unscheduled CD (combined for primary analysis) [Figure 1].
Figure 1:
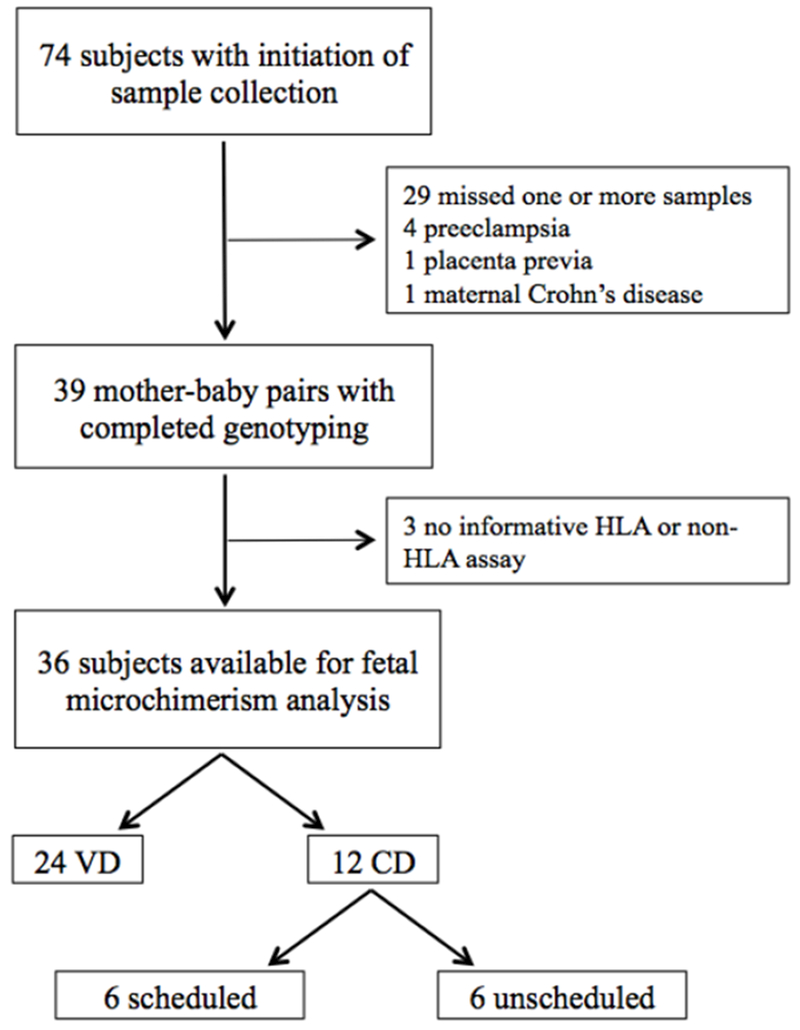
Patient recruitment flowchart.
HLA, human leukocyte antigen
VD, vaginal delivery
CD, Cesarean delivery
There were no differences in rates of induction of labor or length of labor (only including women who labored in the CD group) between the groups (Table 1). Indication for CD included scheduled repeat CD (n=6), non-reassuring fetal status (n=2), second stage arrest (n=2), first stage arrest (n=1), and desire for repeat CD after presenting in labour (n=1). Labor management and the decision to proceed with CD was at the discretion of the obstetrical providers at each delivery hospital. All placentas were delivered spontaneously without the use of manual or instrumented extraction for both VD and CD participants.
Table 1:
Demographics of study population (N=36).
| Characteristic | Vaginal Delivery (N=24) | Cesarean Delivery (N=12) | p-value |
|---|---|---|---|
| Maternal Age (years) | 32.7 (±5.4) | 33.4 (±6.3) | 0.71 |
| Maternal Race | |||
| White | 18 (75.0%) | 10 (83.3%) | 0.93 |
| Black | 2 (8.3%) | 0 (0%) | |
| Other | 4 (16.7%) | 2 (16.7%) | |
| Earliest recorded BMI (kg/m2) | 29.7 (±2.1) | 29.4 (±2.5) | 0.46 |
| Gravidity | 2 (1-5) | 2 (1-5) | 0.41 |
| Parity | 1 (0-3) | 1 (0-2) | 0.94 |
| Tobacco use | 1 (5.3%) | 0 (0%) | 0.46 |
| Gestational age at pre-delivery sample collection | 37w4d (±7.3d) | 38w2d (±7.4d) | 0.05 |
| Induction | 10 (41.7%) | 3 (33.3%) | 0.33 |
| Length of labor (min) | 312 (60-1237) | 382 (189-805)* | 0.41 |
| Gestational age at delivery | 39w3d (±10.5d) | 39w1d (±5.2d) | 0.63 |
| Estimated blood loss (mL) | 290 (±136.3) | 709.1 (±137.5) | <0.001 |
| Time from placental delivery to maternal post-delivery sample collection (min) | 96 (6-1430) | 53 (1-1200) | 0.32 |
| Neonatal birth weight (g) | 3497 (±544) | 3491 (±531) | 0.98 |
| 5 Minute Apgar score | 9 (6-9) | 9 (7-10) | 0.91 |
| Female Neonatal Sex | 8 (33.3%) | 3 (25.0%) | 0.71 |
BMI, body mass index
Data are n (%), mean (± standard deviation), or median (range).
For those women who labored prior to their Cesarean delivery.
There were no differences between the two groups with respect to demographic factors including age, race, earliest recorded BMI, gravidity, parity, use of tobacco, gestational age at delivery, birth weight, 5-minute Apgar scores, or neonatal sex. The gestational age at which the pre-delivery sample was collected tended to be slightly later in the CD group, but this difference was not statistically significant. As expected, there was significantly more estimated blood loss in the CD group compared to the VD group. No patients received a blood transfusion antepartum or postpartum. Importantly, there was no significant difference between the groups in the time from placental delivery to post-delivery maternal sample collection (median 96 minutes for VD compared to 53 minutes for CD, p=0.32) [Table 1].
The mean total number of gEq tested for FMc was similar among the pre-delivery and post-delivery samples for all participants: pre-delivery VD 139,618 gEq (± 37,320), post-delivery VD 144,571 gEq (± 43,553), pre-delivery CD 149,399 gEq (± 49,173), and post-delivery CD 142,645 gEq (± 34,148), p=0.83.
FMc detection among post-delivery samples was higher in participants following a CD versus VD (58.3% vs. 16.7%, p=0.02). In the unadjusted model, the likelihood of FMc detection in post-delivery samples was seven fold higher for participants undergoing a CD (OR 7.0, 95% CI 1.4-34.4). After controlling for the total number of gEq tested for each sample, gravidity, and gestational age at delivery, the likelihood of post-delivery FMc detection was almost nine-fold higher in those participants who had a CD versus VD (aOR 8.8, 95% CI 1.6-47.6, p=0.01) [Table 2].
Table 2:
Fetal microchimerism detection and concentration in post-delivery samples by mode of delivery.
| FMc Detection and Concentration | Unadjusted Model | Adjusted Model | ||||
|---|---|---|---|---|---|---|
| Vaginal Delivery (N=24) | Cesarean Delivery (N=12) | 95% CI | Vaginal Delivery (N=24) | Cesarean Delivery (N=12) | 95% CI | |
| Likelihood of post-delivery FMc detection (OR) | Ref | 7.0 | 1.4-34.4 | Ref | 8.8* | 1.6-47.6 |
| Detection rate ratio of post-delivery FMc (DRR) | Ref | 0.34 | 0.04-2.6 | Ref | 14.7† | 3.2-66.8 |
OR, odds ratio; CI, confidence interval; FMc, fetal microchimerism; DRR, detection rate ratio;
Ref, reference
Logistic regression model adjusted for total number of cell equivalents tested, gravidity, and gestational age at delivery.
Negative binomial regression model adjusted for total number of cell equivalents tested, gravidity, gestational age at delivery, and whether fetal microchimerism was detected in the pre-delivery sample.
Analysis of FMc concentration by negative binomial regression to determine quantitative estimates for the detection rate ratio (DRR) of FMc by mode of delivery are also shown in Table 2. In the unadjusted model, the DRR for FMc in participants who underwent a CD versus VD did not significantly differ (DRR 0.34, 95% CI 0.04-2.7; p=0.31). However, in the final model, adjusted for total gEq tested, gravidity, gestational age at delivery, and pre-delivery positivity for FMc, the DRR for FMc was higher in the CD group (aDRR 14.7, 95% CI, 3.2-66.8; p=0.001) [Table 2]. For all participants, FMc concentrations before and after delivery are shown in Figure 2. Though results from scheduled and unscheduled CDs were combined for analysis, the figure indicates subtypes of CD.
Figure 2:
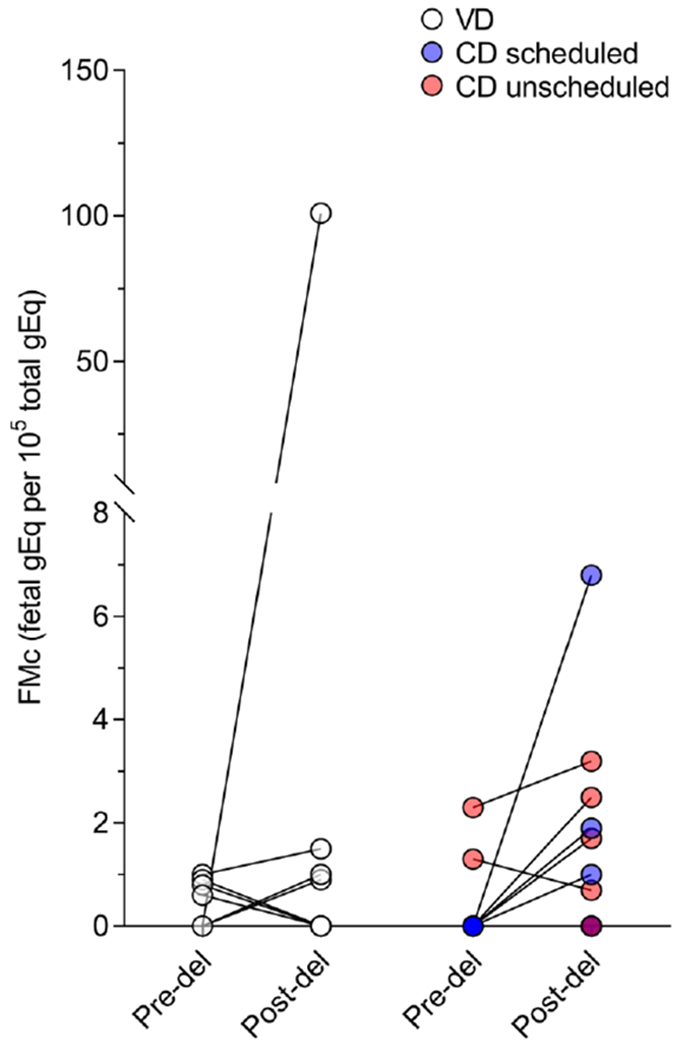
Fetal microchimerism from pre-delivery to post-delivery for each subject categorized by mode of delivery.
FMc, fetal microchimerism
VD, vaginal delivery
CD, Cesarean delivery
Although we excluded participants with preeclampsia or HELLP syndrome, one subject developed mild, transiently elevated blood pressures intrapartum; however, she was not administered magnesium sulfate and did not require antihypertensive therapy. Additionally, one subject presented with premature rupture of membranes at 36 3/7 weeks gestation in the absence of labour and underwent labour induction with cervical ripening. Given the overall uncomplicated nature of these participants’ deliveries, they were included in our analysis. Repeat analysis after removal of data from these two participants did not change our results (data not shown).
DISCUSSION
Main Findings
In this prospective study of women undergoing uncomplicated deliveries, we found that FMc transfer to maternal circulation was greater following CD versus VD. A prior study by our group demonstrated similar findings in pregnancy loss (miscarriages and terminations), with women undergoing a surgical procedure demonstrating higher FMc detection and concentration.(6) These two studies highlight the potential for obstetric treatment-related factors to influence transfer of cellular FMc to the mother. While the mechanism of increased FMc transfer in CD is unknown, surgical disruption of the maternal-fetal interface may lead to an abrupt showering of FMc into the maternal circulation as the relatively contained maternal-fetal-placental unit does not experience similar interface breaches with a normal VD. Sweeping of the intrauterine cavity during CD, commonly performed at the delivering hospitals, may also account for this difference. Additionally, other differences in uterine involution or immediate postpartum physiology after CD may contribute.
Obstetric events demonstrate transfer of differential FMc amounts(6,8) and authors of an epidemiologic study propose a potential role for FMc to explain an association with mode of delivery and development of AID.(14) Our study directly demonstrates increased detection and concentration of FMc in post-delivery maternal peripheral blood following CD compared to VD in women with uncomplicated deliveries.
Strengths and Limitations
Strengths of our study include prospective sample collection and a focus on cellular FMc with targeting of specific non-shared polymorphisms using highly sensitive assays. Our strategy for specific timed collections, pre-labour and post-delivery, allowed for comparison of FMc prior to and after delivery, permitting us to conclude that the change in FMc from pre- to post-delivery was likely acquired through delivery events, implicating the mode of delivery as an important element in directional cell transfer. Indeed, it has previously been suggested that the majority of cell transfer likely occurs later in gestation, mostly in the peripartum period.(1)
In three cases, FMc was detected in pre-delivery samples, however, was undetectable in post-delivery samples (Figure 2). All of these patients had a VD. In two of these cases, the post-delivery sample was collected at an interval that was greater than 2 standard deviations from the mean of the population, thus it is possible that circulating FMc concentrations had fallen to an undetectable level by this time point. That FMc can be detected in maternal tissues decades after delivery suggests that although these cells may clear the peripheral circulation after delivery, they continue to be harboured in the maternal system in different tissue types. All attempts were made to collect post-delivery samples within 2 hours of placental delivery. Given the variety of maternal and neonatal care needed immediately postpartum and that samples were collected from several hospitals, this was not always feasible. Removal of these two participants from the analysis did not change the findings of our study (results not shown).
Study limitations included a small sample size, and one that did not allow for granularity in CD subtypes, as we grouped scheduled and unscheduled CDs together. Future studies should make an effort to investigate these groups separately. A substantial proportion of incomplete sample sets was initially related to the unpredictable nature of labor and delivery and physician/nursing hand-offs. This became less frequent with ongoing recruitment following process changes to maximize sample collection. Though detection analysis was clear, our negative binomial regression model results to evaluate differences in FMc concentration were more complex, with no association of FMc concentration with mode of delivery in the unadjusted model. After adjustment for confounders, including gravidity, the relationship of FMc concentration was similar to the relationship with FMc detection. This discrepancy with the unadjusted negative binomial model was likely due to one outlier in our VD group with gravidity of five and a much higher post-delivery FMc concentration (100.9 gEq/105 total gEq tested) than other participants. Clinically, this participant’s delivery was otherwise unremarkable. It is possible that this participant’s higher gravidity contributed to the higher FMc concentration and thus the lack of association of FMc with mode of delivery in the unadjusted model. Higher gravidity may be a confounder as multiple pregnancies with the same partner may produce more than one offspring with the unique HLA target used for FMc detection via QPCR. Our group has previously investigated the effect of parity on FMc and maternal Mc suggesting a likely complex relationship with increasing number of potential grafts.(21)
One further limitation includes feasibility. By definition, Mc occurs at very low concentrations and thus, if more maternal cells were able to be tested, we may have found more cases of detectable FMc in both groups. This likely led to global underestimation of the true occurrence of FMc and would not be expected to differ between groups. Our focus on cellular FMc excludes other compartments important for bidirectional communication in pregnancy, including cell-free fetal DNA, exosomes, messenger RNA, microRNA, and other non-coding RNA.(22–24) Study of these particles and alterations of their kinetics by mode of delivery and other obstetric factors are warranted. We chose to focus on cellular Mc given its unique role in persistence and potential influence on subsequent maternal health.(9,23)
Interpretation
Our findings add to a growing literature on FMc transfer and its role in later life maternal health. Several studies suggest a link between the presence of FMc with either a harmful or beneficial role in AID status and outcomes.(15,18,25) FMc has been found to associate with protection from some diseases, including breast cancer,(10,11) and may be associated with an overall survival advantage.(26) On the other hand, FMc is associated with other cancers including colon cancer,(27) and possibly with cardiovascular disease.(26,28) The relationship of FMc with subsequent disease risk likely depends on numerous factors, several of which are highlighted in observations from studies of the AID rheumatoid arthritis (RA). RA is characterized by female preponderance and increasing incidence in later years of life(26,29) along with a nuanced protective association with parity,(30,31) with the association diminishing as the time interval from the last delivery increases.(25) While the mechanism underlying this effect is unclear, FMc acquisition has been hypothesized to confer temporary protection that may wane over time, similar to vaccine responses. Pregnancy and parity alone may not explain the complex relationships with AID risk. HLA relationships between maternal and FMc cells may be a contributing factor, suggesting a possible role for fetal genetic material acquired via pregnancy influencing maternal AID risk.(32) That obstetric factors, such as surgical evacuation of early pregnancy(6), preeclampsia(8), and mode of delivery, may influence FMc acquisition is of great interest as it contributes to our understanding of reproductive origins of disease and warrants consideration in relevant future basic science, epidemiologic, and clinical studies. In general, persistent FMc may influence subsequent maternal health, and further investigation of fetal-maternal cell transfer is needed to solidify our understanding of the biologic legacy of both early pregnancy events as well as peri-delivery factors.
Conclusion
In summary, we found that FMc was more readily detectable and at higher concentrations following a CD compared to VD in women undergoing uncomplicated deliveries. Anatomic disruption of the maternal-placental interface or altered immediate maternal postpartum physiology may contribute. FMc in maternal circulation may prove to be an important biomarker for later life maternal health given its association with autoimmune disease, certain malignancies, and possibly cardiovascular risk. Further research is necessary to evaluate the long-term persistence of FMc cells, their function, and subsequent consequences for long-term maternal health.
Acknowledgments
None
Funding
K08HD067221 (Gammill), R01HL11737 (Nelson), T32HD007233 (Harrington)
This work was supported by the National Institute of Health (K08HD067221, R01HL11737, T32HD007233).
Footnotes
Disclosure of Interests
JLN and SBK have ownership interests in Chimerocyte, Inc., for which highly sensitive and specific detection of allogeneic cells/DNA is its core technology. However, Chimerocyte, Inc. had no role in funding this research, providing testing material, or analyzing results. Chimerocyte was formed September 2017, well after the current work was completed. The remaining authors declare no competing financial interests. Completed disclosure of interest forms are available to view online as supporting information.
Details of Ethics Approval
Institutional Review Board approval was obtained from both the University of Washington (Date of approval 30 March 2003, Reference number 23149) and the Fred Hutchinson Cancer Research Institute (Date of approval 11 June 2003, Reference number 9569).
Contributor Information
Raj SHREE, Division of Maternal Fetal Medicine, Department of Obstetrics & Gynecology, University of Washington, Seattle, Washington.
Whitney E. HARRINGTON, Department of Pediatrics, University of Washington, Seattle Children’s Hospital, Seattle, Washington
Sami B. KANAAN, Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, Washington
Alexandra FORSYTH, Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, Washington.
Emma COUSIN, Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, Washington.
Anthony LOPEZ, Department of Obstetrics & Gynecology, University of Washington, Seattle, Washington.
J. Lee NELSON, Clinical Research Division, Fred Hutchinson Cancer Research Center, Division of Rheumatology, University of Washington, Seattle, Washington
Hilary S. GAMMILL, Division of Maternal Fetal Medicine, Department of Obstetrics & Gynecology, University of Washington, Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, Washington
REFERENCES
- 1.Adams Waldorf KM, Gammill HS, Lucas J, Aydelotte TM, Leisenring WM, Lambert NC, et al. Dynamic changes in fetal microchimerism in maternal peripheral blood mononuclear cells, CD4+ and CD8+ cells in normal pregnancy. Placenta. 2010. July;31(7):589–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gammill HS, Nelson JL. Naturally acquired microchimerism. Int J Dev Biol 2010;54(2–3):531–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bianchi DW, Zickwolf GK, Weil GJ, Sylvester S, DeMaria MA. Male fetal progenitor cells persist in maternal blood for as long as 27 years postpartum. Proc Natl Acad USA. 1996. January 23;93(2):705–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lo YM, Lau TK, Chan LY, Leung TN, Chang AM. Quantitative analysis of the bidirectional fetomaternal transfer of nucleated cells and plasma DNA. Clin Chem 2000. September;46(9):1301–9. [PubMed] [Google Scholar]
- 5.Maloney S, Smith A, Furst DE, Myerson D, Rupert K, Evans PC, et al. Microchimerism of maternal origin persists into adult life. J Clin Invest 1999. July;104(1):41–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peterson SE, Nelson JL, Guthrie KA, Gadi VK, Aydelotte TM, Oyer DJ, et al. Prospective assessment of fetal-maternal cell transfer in miscarriage and pregnancy termination. Hum Reprod 2012. September;27(9):2607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alberry MS, Maddocks DG, Hadi MA, Metawi H, Hunt LP, Abdel-Fattah SA, et al. Quantification of cell free fetal DNA in maternal plasma in normal pregnancies and in pregnancies with placental dysfunction. Am J Obstet Gynecol 2009. January;200(1):98.e1–6. [DOI] [PubMed] [Google Scholar]
- 8.Gammill HS, Aydelotte TM, Guthrie KA, Nkwopara EC, Nelson JL. Cellular fetal microchimerism in preeclampsia. Hypertension. 2013. December;62(6):1062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson JL, Furst DE, Maloney S, Gooley T, Evans PC, Smith A, et al. Microchimerism and HLA-compatible relationships of pregnancy in scleroderma. Lancet. 1998. February 21;351(9102):559–62. [DOI] [PubMed] [Google Scholar]
- 10.Gadi VK, Nelson JL. Fetal microchimerism in women with breast cancer. Cancer Res 2007. October 1;67(19):9035–8. [DOI] [PubMed] [Google Scholar]
- 11.Gadi VK. Fetal microchimerism in breast from women with and without breast cancer. Breast Cancer Res Treat 2010. May;121(1):241–4. [DOI] [PubMed] [Google Scholar]
- 12.Sevelsted A, Stokholm J, Bønnelykke K, Bisgaard H. Cesarean section and chronic immune disorders. Pediatrics. 2015. January;135(1):e92–98. [DOI] [PubMed] [Google Scholar]
- 13.Black M, Bhattacharya S, Philip S, Norman JE, McLernon DJ. Planned Cesarean Delivery at Term and Adverse Outcomes in Childhood Health. JAMA. 2015. December 1;314(21):2271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khashan AS, Kenny LC, Laursen TM, Mahmood U, Mortensen PB, Henriksen TB, et al. Pregnancy and the risk of autoimmune disease. PloS One. 2011;6(5):e19658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson JL. The otherness of self: microchimerism in health and disease. Trends Immunol 2012. August;33(8):421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Donoghue K. Pregnancy and the risk of autoimmune disease: An exploration. Chimerism. 2011. July;2(3):84–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan Z, Lambert NC, Ostensen M, Adams KM, Guthrie KA, Nelson JL. Prospective study of fetal DNA in serum and disease activity during pregnancy in women with inflammatory arthritis. Arthritis Rheum 2006. July;54(7):2069–73. [DOI] [PubMed] [Google Scholar]
- 18.Lambert NC, Erickson TD, Yan Z, Pang JM, Guthrie KA, Furst DE, et al. Quantification of maternal microchimerism by HLA-specific real-time polymerase chain reaction: studies of healthy women and women with scleroderma. Arthritis Rheum 2004. March;50(3):906–14. [DOI] [PubMed] [Google Scholar]
- 19.Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, et al. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988. January 29;239(4839):487–91. [DOI] [PubMed] [Google Scholar]
- 20.Guthrie KA, Gammill HS, Kamper-Jørgensen M, Tjønneland A, Gadi VK, Nelson JL, et al. Statistical Methods for Unusual Count Data: Examples From Studies of Microchimerism. Am J Epidemiol 2016;184(10):779–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gammill HS, Guthrie KA, Aydelotte TM, Adams Waldorf KM, Nelson JL. Effect of parity on fetal and maternal microchimerism: interaction of grafts within a host? Blood. 2010. October 14;116(15):2706–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manokhina I, Wilson SL, Robinson WP. Noninvasive nucleic acid-based approaches to monitor placental health and predict pregnancy-related complications. Am J Obstet Gynecol 2015. October;213(4 Suppl):S197–206. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell MD, Peiris HN, Kobayashi M, Koh YQ, Duncombe G, Illanes SE, et al. Placental exosomes in normal and complicated pregnancy. Am J Obstet Gynecol 2015. October;213(4 Suppl):S173–181. [DOI] [PubMed] [Google Scholar]
- 24.Mouillet J-F, Ouyang Y, Coyne CB, Sadovsky Y. MicroRNAs in placental health and disease. Am J Obstet Gynecol 2015. October;213(4 Suppl):S163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guthrie KA, Dugowson CE, Voigt LF, Koepsell TD, Nelson JL. Does pregnancy provide vaccine-like protection against rheumatoid arthritis? Arthritis Rheum 2010. July;62(7):1842–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamper-Jørgensen M, Hjalgrim H, Andersen A-MN, Gadi VK, Tjønneland A. Male microchimerism and survival among women. Int J Epidemiol 2014. February;43(1):168–73. [DOI] [PubMed] [Google Scholar]
- 27.Kamper-Jørgensen M, Biggar RJ, Tjønneland A, Hjalgrim H, Kroman N, Rostgaard K, et al. Opposite effects of microchimerism on breast and colon cancer. Eur J Cancer. 2012. September;48(14):2227–35. [DOI] [PubMed] [Google Scholar]
- 28.Kamper-Jørgensen M, Mortensen LH, Andersen A-MN, Hjalgrim H, Gadi VK, Tjønneland A. Predictors of male microchimerism. Chimerism. 2012. December;3(3):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silman A, Hochberg M. Rheumatoid Arthritis In: Silman A, Hochberg M, editors. Epidemiology of the rheumatic diseases. 2nd ed. Oxford University Press; 2001. p. 31–71. [Google Scholar]
- 30.Borchers AT, Naguwa SM, Keen CL, Gershwin ME. The implications of autoimmunity and pregnancy. J Autoimmun 2010. May;34(3):J287–299. [DOI] [PubMed] [Google Scholar]
- 31.Adams Waldorf KM, Nelson JL. Autoimmune disease during pregnancy and the microchimerism legacy of pregnancy. Immunol Invest 2008;37(5):631–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lambert NC, Lo YMD, Erickson TD, Tylee TS, Guthrie KA, Furst DE, et al. Male microchimerism in healthy women and women with scleroderma: cells or circulating DNA? A quantitative answer. Blood. 2002. October 15;100(8):2845–51. [DOI] [PubMed] [Google Scholar]