

Abstract
Glioblastoma (GBM) is a highly aggressive form of cancer that is resistant to standard therapy with concurrent radiation and temozolomide, two agents that work by inducing DNA damage. An underlying cause of this resistance may be a subpopulation of cancer stem-like cells that display a heightened DNA damage response (DDR). While this DDR represents an attractive therapeutic target for overcoming the resistance of GBMs to radiation therapy, until now, the cause of this DDR upregulation has not been understood. In this issue of Cancer Research, Dr. Ross Carruthers and colleagues investigate DNA replication stress (RS) as an underlying mechanism responsible for upregulation of the DDR and hence the radiation resistance of glioma stem-like cells (GSCs). Furthermore, the authors explore the efficacy of combined ATR (Ataxia telangiectasia and Rad3 related) kinase and PARP (Poly (ADP-ribose) polymerase) inhibitors as a strategy to leverage these mechanisms and overcome radiation resistance.
The cancer stem cell theory states that a small subpopulation of tumor cells possess unique self-renewal properties that are capable of seeding new tumors and are a source of regrowth following therapy (2). Glioblastoma stem-like cells (GSCs) are defined as CD133 positive cells that can initiate new tumors in mice (3). This subpopulation of cells was later shown to be notably radioresistant, a property attributed to an intensified DDR which could be targeted with an inhibitor of CHK1/2 (4). Subsequently, the Chalmers’ group demonstrated that enhanced G2-M checkpoint activation and DNA repair were functional consequences of an augmented DDR that treatment with ATR and PARP inhibitors could overcome to prevent radioresistance (5). Key questions prompted by these studies are what properties of GSCs lead to the enhanced basal level of DNA damage signaling and whether these mechanisms can be leveraged therapeutically to overcome the resistance of GSCs to ionizing radiation.
The underlying cause for elevated DDR in GSCs has previously been attributed to heightened levels of reactive oxygen species (ROS) leading to increased levels of PARP and SSB repair (6); however, Carruthers et al did not find evidence that ROS levels were elevated in CD133+ GSCs versus GSC-depleted cultures (1). Instead, GSCs displayed both elevated basal levels of activated ATR and CHK1, and elevated markers of RS such as foci marked with the single-stranded DNA binding protein, replication protein A (RPA) and the DNA damage markers γ-H2AX and 53BP1. Untreated GSCs also exhibited reduced replication velocities and asymmetric bidirectional DNA replication forks, indicating increased stalling of replication factories compared to non-GSC populations (1). These observations pointed to elevated levels of RS as causative of DDR activation in untreated GSCs, a hypothesis supported by the high levels of RS in GBM (7), broad activation of DDR proteins by RS (8), and the overlap between signaling in response to RS and DNA damage (9). But, can elevated RS increase radioresistance in non-GSC cultures? Carruthers and colleagues demonstrate that slowing DNA replication velocity by aphidicolin treatment imparts a radioresistant phenotype (1). Therefore, slowing DNA replication and artificially creating replication stress can lead to radioresistance. These observations lead to the next question, what is the mechanism behind elevated RS in GSCs?
Replication stress is associated with oncogene expression and is a common feature of cancers (10,11). The induction of RS by oncogenes is multifactorial and may be due to aberrant expression of genes that regulate DNA synthesis (e.g., Cyclin E), increased origin firing, depletion of deoxynucleotide pools, and formation of hard to replicate secondary structures in DNA such as G4-quadruplexes (12,13). Emerging evidence implicate a role for oncogene-driven transcription as a source of replication stress (14,15). One potential consequence of increased origin firing and elevated transcription is collision between the protein machinery for these two processes, which in turn creates abnormal replication fork structures that can be processed into DNA double stranded breaks (DSBs) (16,17).
Given the recent evidence that replication/transcription conflicts can induce RS, Carruthers and colleagues explored whether GSCs displayed altered transcription profiles compared to the bulk non-GSC populations. No evidence for altered expression of genes associated with DNA replication or genes known to be induced by RS was discovered. Instead, GSCs overexpressed a significant number of ‘very long genes’ (VLG), sequences in excess of 800 kilobases in length (1). It is interesting to note that several VLGs upregulated in GSCs are known to play important roles in neurological development, axon guidance, and synapse formation consistent with a neural progenitor phenotype (1). Some VLGs contain difficult to replicate sequences that are hotspots for forming chromosomal gaps and breaks, or common fragile sites (CFS), that are expressed under conditions of RS, such as through aphidicolin treatment (18–21).
Transcription of VLGs occurs late in the cell cycle and may not be completed until the following cell cycle (22). Therefore, replication factories will inevitably encounter the transcription of a VLG at some point during the cell cycle. Replication stalling at CFS, may further increase the incidence of replication/transcription machinery encounters (21,23). One consequence of replication/transcription collisions is the formation of stable RNA/DNA hybrids (also referred to as R Loops) which require enzymes such as RNAse H to resolve (22,24–27). R loops form when transcribed RNA hybridizes with the complementary DNA strand and displaces the nontemplate strand as single-stranded DNA (ssDNA). R loops have been detected in both bacteria and human cells and are now known to influence chromatin structure, regulation of transcription, immunoglobulin class switch recombination, and, if persistent or collide in a ‘head on’ orientation with the replication machinery, can lead to genome instability when processed into DSBs (28–34).
Slowing and/or stalling of DNA replication, such as that induced by low concentrations of aphidicolin, causes the appearance of RNA/DNA hybrids. Current evidence suggests these hybrids result from inadvertent collision of replication/transcription machinery at VLGs (22). The observation by Carruthers et al that GSCs preferentially overexpress VLGs provided novel mechanistic insight into a source for elevated RS in GSCs: enhanced transcription of multiple VLGs increases the incidence of replication/transcription conflicts resulting in DSBs, potentially at CFS (Figure 1). Indeed, this study provides evidence of DSBs, marked by γH2AX, at sites overlapping replication or transcription, marked by BrdU or RNA:DNA hybrids, respectively. Furthermore, this overlap occurred preferentially in CD133+ GSCs compared to bulk GBM cultures (1).
Figure 1. Targeting replication stress in glioblastoma stem-like cells (GSCs).
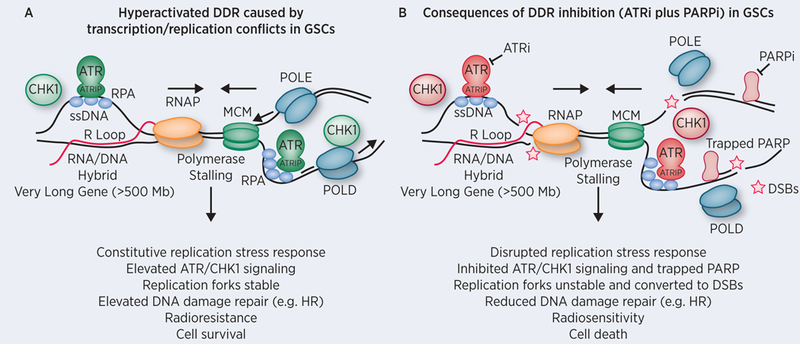
A) Carruthers et al. demonstrated that CD133+ GSCs exhibit constitutive replication stress (RS) as shown by elevated ATR and CHK1 kinase signaling (colored green). ATR is activated by binding to RPA coating extended regions of ssDNA through its partner ATR Interacting Protein (ATRIP). ATR phosphorylates and activates CHK1 thereby initiating a DNA damage response that promotes activation of the intra S and G2/M phase checkpoints, increases replication fork stability, and regulates DNA repair pathways such as homologous recombination (HR) (35). One potential source of RS in GSCs is the elevated transcription of ‘very long genes’ by RNA polymerase that may inadvertently collide with late replicating regions of the genome, activating the ATR replication stress response, which in turn promotes cell survival and radioresistance. B) Treatment of GSCs with an ATR inhibitor (colored red) is selectively toxic due to GSC dependence upon RS response signaling for survival. Inhibition of the RS response leads to increased R loop and replication fork instability that ultimately lead to DSBs following structure-specific endonuclease processing or DNA breakage. Inhibition of PARP results in base excision repair deficiency and may lead to trapping of the PARP enzyme on ssDNA breaks creating further dependence upon ATR signaling to promote stability and repair of stalled replication forks. Inhibition of ATR and PARP leads profound radiosenstization of GSCs. Abbreviations: ATR (Ataxia telangiectasia and Rad3-related), RPA (Replication Factor A), ssDNA (single-stranded DNA), RNAP (RNA polymerase II), MCM (minichromosome maintenance protein complex helicase), POLE and POLD (DNA polymerase epsilon and delta), PARP (Poly (ADP-ribose) polymerase), DSBs (Double-Stranded DNA breaks).
The poor prognosis and relative resistance of GBM to standard therapy underscores the need for more effective therapies. The major question addressed by Carruthers and colleagues is whether the heightened RS in GSCs is therapeutically actionable. The ATR kinase is a master regulator of responses to DNA damage and RS (35). ATR has a direct role in diminishing RS by promoting stabilization and restart of stalled DNA replication forks, as well as preventing aberrant replication origin firing and subsequent nucleotide exhaustion and replication stalling (35). Of relevance to the observation that transcription/replication conflicts may be a source of RS in GSCs (1), ATR activates and promotes the resolution of persistent R loops (33,36–38). Thus, inhibition of ATR may present a unique approach to attenuating constitutive DDR signaling exhibited by GSCs and reversing radioresistance.
The poly(ADP-ribose) polymerase 1 (PARP1) and PARP2 enzymes bind to ssDNA breaks and are important signal transducers within the DDR pathway. Binding to ssDNA breaks activates PARP1 and PARP2 to post-translationally modify themselves as well as other proteins by synthesizing negatively charged poly(ADP-ribose) chains. PolyADP-ribosylation recruits proteins involved in ssDNA break repair (e.g. XRCC1) and modifies chromatin structure. Distinct from ATR, PARP also functions during DNA replication and the RS response by regulating fork stabilization and restart, elongation velocity, ligation of lagging strand Okazaki fragments, and homologous recombination repair of stalled DNA replication forks (39–43). Importantly, the therapeutic activity of PARP inhibitors is in part attributed to PARP ‘trapping’, resulting from the loss of autoPARylation that facilitates removal of PARP from DNA. Trapped PARP creates obstacles that impede ongoing DNA replication. The increased abundance of trapped PARP enzymes is hypothesized to be preferentially cytotoxic to cancer cells harboring defects in homologous recombination repair (44,45).
Consistent with the complementary roles of ATR and PARP in the DDR pathway and the hypothesis that RS is a targetable feature of GSCs, Carruthers et al. tested whether the combination of ATR and PARP inhibitors is preferentially cytotoxic and radiosensitizing in GSCs (relative to bulk cells). Although PARP inhibition alone was relatively ineffective, treatment of GSCs with an ATR inhibitor inhibited stem cell-like neurosphere formation in vitro, implicating a role for ATR for viability under these conditions. This effect was even more pronounced with the combination of ATR and PARP inhibition. Importantly, the combination treatment enhanced DNA damage in GSCs and diminished the radioresistant phenotype of GSCs. It is likely that ATR and PARP inhibitors synergize by inhibiting multiple points in the DDR. The heightened levels of RS and DDR signaling in GSCs is consistent with the hypothesis that these cells have become dependent upon ATR for viability (46). Trapping PARP through the co-administration of a PARP inhibitor may further strengthen the dependence of GSCs on ATR activity for survival (Figure 1).
Of particular clinical relevance, Carruthers and colleagues found that while PARP inhibition alone was ineffective in radiosensitizing the GSC models used in this study, the combination of ATR and PARP inhibitors induces profound sensitization of GSCs to radiation, an effect that was significantly greater in CD133+ GSCs than in bulk GBM cells (1). Multiple clinical trials combining PARP inhibitors with radiation or other DDR inhibitors, such as those targeting ATR in both BRCA1/2-mutant and non-mutant cancers, are underway (45,47). The data presented in this study provide a preclinical rationale for the future clinical development of concurrent ATR and PARP inhibitors with radiation in GBMs, and potentially other cancers with a high RS burden. As an added benefit, inhibition of ATR has recently been shown to inhibit radiation-induced upregulation of Programmed death-ligand 1 (PD-L1) in tumor cells, diminish radiation-induced CD8+ T cell exhaustion, and decrease the number of tumor-infiltrating T regulatory cells to achieve a greater anti-tumor response in a mouse model of Kras-mutant cancer (48). Given the well characterized immunosuppressive tumor microenvironment associated with GBM, treatment with ATR inhibitors may present two weapons against this disease: targeting the addiction to the DDR pathway and reinvigorating T cells to attack GBM cells following radiation therapy (49,50).
Acknowledgements:
M.A. Morgan is supported by NIH U01-CA216449. C.E. Canman is supported by NIH R01-CA133046 and T32-CA009676. The authors would like to thank T.S. Lawrence for helpful comments and suggestions.
Footnotes
Conflicts of Interest: M.A. Morgan reports receiving a commercial research grant from and has received speaker’s bureau honoraria from AstraZeneca. C.E. Canman declares no conflicts of interest.
References
- 1.Carruthers RD, Ahmed SU, Ramachandran S, Strathdee K, Kurian KM, Hedley A, et al. Replication Stress Drives Constitutive Activation of the DNA Damage Response and Radioresistance in Glioblastoma Stem-like Cells. Cancer Research 2018;78:5060–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nature Reviews Clinical Oncology 2017;14:611–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature 2004;432:396–401 [DOI] [PubMed] [Google Scholar]
- 4.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444:756–60 [DOI] [PubMed] [Google Scholar]
- 5.Ahmed SU, Carruthers R, Gilmour L, Yildirim S, Watts C, Chalmers AJ. Selective Inhibition of Parallel DNA Damage Response Pathways Optimizes Radiosensitization of Glioblastoma Stem-like Cells. Cancer Research 2015;75:4416–28 [DOI] [PubMed] [Google Scholar]
- 6.Venere M, Hamerlik P, Wu Q, Rasmussen RD, Song LA, Vasanji A, et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ 2014;21:258–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartkova J, Hamerlik P, Stockhausen MT, Ehrmann J, Hlobilkova A, Laursen H, et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010;29:5095–102 [DOI] [PubMed] [Google Scholar]
- 8.Mazouzi A, Velimezi G, Loizou JI. DNA replication stress: Causes, resolution and disease. Experimental Cell Research 2014;329:85–93 [DOI] [PubMed] [Google Scholar]
- 9.Burgess Rebecca C, Misteli T. Not All DDRs Are Created Equal: Non-Canonical DNA Damage Responses. Cell 2015;162:944–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macheret M, Halazonetis TD. DNA Replication Stress as a Hallmark of Cancer. Annual Review of Pathology: Mechanisms of Disease 2015;10:425–48 [DOI] [PubMed] [Google Scholar]
- 11.Gaillard H, García-Muse T, Aguilera A. Replication stress and cancer. Nature Reviews Cancer 2015;15:276–89 [DOI] [PubMed] [Google Scholar]
- 12.Técher H, Koundrioukoff S, Nicolas A, Debatisse M. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nature Reviews Genetics 2017;18:535–50 [DOI] [PubMed] [Google Scholar]
- 13.Kotsantis P, Petermann E, Boulton SJ. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discovery 2018;8:537–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kotsantis P, Silva LM, Irmscher S, Jones RM, Folkes L, Gromak N, et al. Increased global transcription activity as a mechanism of replication stress in cancer. Nat Commun 2016;7:13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones RM, Mortusewicz O, Afzal I, Lorvellec M, Garcia P, Helleday T, et al. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 2013;32:3744–53 [DOI] [PubMed] [Google Scholar]
- 16.Macheret M, Halazonetis TD. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 2018;555:112–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stork CT, Bocek M, Crossley MP, Sollier J, Sanz LA, Chédin F, et al. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. eLife 2016;5:e17548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Tallec B, Millot Gaël A, Blin Marion E, Brison O, Dutrillaux B, Debatisse M. Common Fragile Site Profiling in Epithelial and Erythroid Cells Reveals that Most Recurrent Cancer Deletions Lie in Fragile Sites Hosting Large Genes. Cell Reports 2013;4:420–8 [DOI] [PubMed] [Google Scholar]
- 19.Wilson TE, Arlt MF, Park SH, Rajendran S, Paulsen M, Ljungman M, et al. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Research 2015;25:189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei P-C, Chang Amelia N, Kao J, Du Z, Meyers Robin M, Alt Frederick W, et al. Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells. Cell 2016;164:644–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glover TW, Wilson TE, Arlt MF. Fragile sites in cancer: more than meets the eye. Nature Reviews Cancer 2017;17:489–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helmrich A, Ballarino M, Tora L. Collisions between Replication and Transcription Complexes Cause Common Fragile Site Instability at the Longest Human Genes. Molecular Cell 2011;44:966–77 [DOI] [PubMed] [Google Scholar]
- 23.Tubbs A, Sridharan S, van Wietmarschen N, Maman Y, Callen E, Stanlie A, et al. Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse. Cell 2018;174:1127–42.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wahba L, Amon Jeremy D, Koshland D, Vuica-Ross M. RNase H and Multiple RNA Biogenesis Factors Cooperate to Prevent RNA:DNA Hybrids from Generating Genome Instability. Molecular Cell 2011;44:978–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, et al. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes & Development 2011;25:2041–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen HD, Yadav T, Giri S, Saez B, Graubert TA, Zou L. Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Molecular Cell 2017;65:832–47.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parajuli S, Teasley DC, Murali B, Jackson J, Vindigni A, Stewart SA. Human ribonuclease H1 resolves R-loops and thereby enables progression of the DNA replication fork. Journal of Biological Chemistry 2017;292:15216–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skourti-Stathaki K, Proudfoot NJ. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes & Development 2014;28:1384–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sollier J, Stork Caroline T, García-Rubio María L, Paulsen Renee D, Aguilera A, Cimprich Karlene A. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Molecular Cell 2014;56:777–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santos-Pereira JM, Aguilera A. R loops: new modulators of genome dynamics and function. Nature Reviews Genetics 2015;16:583–97 [DOI] [PubMed] [Google Scholar]
- 31.Sollier J, Cimprich KA. Breaking bad: R-loops and genome integrity. Trends in Cell Biology 2015;25:514–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.García-Muse T, Aguilera A. Transcription–replication conflicts: how they occur and how they are resolved. Nature Reviews Molecular Cell Biology 2016;17:553–63 [DOI] [PubMed] [Google Scholar]
- 33.Hamperl S, Bocek MJ, Saldivar JC, Swigut T, Cimprich KA. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017;170:774–86.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lang KS, Hall AN, Merrikh CN, Ragheb M, Tabakh H, Pollock AJ, et al. Replication-Transcription Conflicts Generate R-Loops that Orchestrate Bacterial Stress Survival and Pathogenesis. Cell 2017;170:787–99.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nature Reviews Molecular Cell Biology 2017;18:622–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hodroj D, Recolin B, Serhal K, Martinez S, Tsanov N, Abou Merhi R, et al. An ATR‐dependent function for the Ddx19 RNA helicase in nuclear R‐loop metabolism. The EMBO Journal 2017;36:1182–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kabeche L, Nguyen HD, Buisson R, Zou L. A mitosis-specific and R loop–driven ATR pathway promotes faithful chromosome segregation. Science 2018;359:108–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen HD, Leong WY, Li W, Reddy PNG, Sullivan JD, Walter MJ, et al. Spliceosome Mutations Induce R Loop-Associated Sensitivity to ATR Inhibition in Myelodysplastic Syndromes. Cancer Research 2018;78:5363–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, et al. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J 2009;28:2601–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 2010;37:492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 2012;72:2814–21 [DOI] [PubMed] [Google Scholar]
- 42.Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, Bartek J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018;559:279–84 [DOI] [PubMed] [Google Scholar]
- 43.Hanzlikova H, Kalasova I, Demin AA, Pennicott LE, Cihlarova Z, Caldecott KW. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Molecular Cell 2018;71:319–31.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med 2016;8:362ps17. [DOI] [PubMed] [Google Scholar]
- 45.Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017;355:1152–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lecona E, Fernandez-Capetillo O. Targeting ATR in cancer. Nature Reviews Cancer 2018;18:586–95 [DOI] [PubMed] [Google Scholar]
- 47.Forment JV, O’Connor MJ. Targeting the replication stress response in cancer. Pharmacology & Therapeutics 2018;188:155–67 [DOI] [PubMed] [Google Scholar]
- 48.Vendetti FP, Karukonda P, Clump DA, Teo T, Lalonde R, Nugent K, et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell–dependent antitumor activity following radiation. The Journal of Clinical Investigation 2018;128:3926–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woroniecka K, Chongsathidkiet P, Rhodin KE, Kemeny HR, Dechant CA, Farber SH, et al. T Cell Exhaustion Signatures Vary with Tumor Type and are Severe in Glioblastoma. Clinical Cancer Research 2018; 24:4175–4186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clinical Cancer Research 2018;24:3792–802 [DOI] [PMC free article] [PubMed] [Google Scholar]