

Abstract
Among the four known mechanisms of intron removal, three are reputedly catalyzed by RNA molecules. In the fourth mechanism, a protein endonuclease removes introns from nuclear tRNA and all archaeal RNAs. Three strictly conserved residues of the splicing endonuclease, a histidine, a lysine, and a tyrosine, were predicted to catalyze the intron cleavage reaction in a manner similar to that of the catalytic triad of ribonuclease A. Single-turnover kinetic parameters were obtained for the wild-type enzyme and two triad mutants. Mutation of histidine to alanine produced an at least ~28-fold reduction; mutation of tyrosine to phenylalanine produced an at least ~7-fold reduction in activity, while a histidine and tyrosine double mutation abolished cleavage. The single mutation of lysine to glutamic acid abolished RNA cleavage activity in the absence of a divalent metal but maintained a substantial level of activity in the presence of specific divalent metals. These data support important functional roles already proposed for the catalytic triad and suggest an intriguing hypothesis in which the splicing endonuclease is an intermediate in the transition from the RNA to the RNP world.
The splicing endonuclease is an evolutionarily conserved RNA cleavage enzyme required for the removal of intervening sequences (introns) from nuclear tRNA and all archaeal RNAs (see ref 1 and references cited therein). Of the four known splicing mechanisms, this is the only one catalyzed by protein enzymes; the other mechanisms are known to be or are putatively mediated by RNA enzymes. Therefore, studies of the splicing endonuclease’s catalytic properties will contribute to our understanding of RNA cleavage mechanisms and the evolution of intron removal processes.
RNA cleavage enzymes are known to accelerate RNA cleavage by four strategies, with proton transfer and transition state stabilization providing the most significant rate enhancements (2, 3) followed by placement of the “in-line” conformation (4). On the basis of the conserved catalytic residues and reaction products, the cleavage chemistry of the splicing endonuclease is believed to be similar to that of the SN2-like reaction of ribonuclease A, where a general base deprotonates the adjacent 2’-OH group and a positively charged residue stabilizes the transition state. consistent with the SN2-like phosphodiester transfer mechanism, cleavage by the splicing endonuclease produces 5’-hydroxyl and 2’,3’- cyclic phosphate termini (5, 6). However, unlike RNase A which recognizes unstructured RNA, the splicing endonucleases recognizes a folded RNA motif that may influence the cleavage chemistry. This motif is the “bulge-helix-bulge” (BHB) motif, and it consists of two three-nucleotide bulges separated by a 4 bp helix (5). Each three-nucleotide bulge hosts one cleavage site. A cocrystal structure of the splicing endonuclease from Archaeoglobus fulgidus bound to a BHB RNA substrate (7) revealed that at both cleavage sites, three universally conserved amino acids are clustered around the scissile phosphate group: a histidine (H257), a tyrosine (Y246), and a lysine (K287) (Figure 1A). The Nɛ atom of K287 is ~2.2 Å from the pro-Sp nonbridging oxygen, presumably poised for stabilization of the transition state. The Nδ atom of H257 is 3.5 Å from the leaving 5’-oxygen and is thus a candidate for the general acid. H257 is also believed to participate in aromatic stacking with the third bulge nucleotide, presumably to stabilize the nucleotide in its flipped down position. The hydroxyl group of Y246 is 2.2 Å from the key ribose’s 2’-hydroxyl and is predicted to be part of a proton shuttling network that deprotonates the attacking nucleophile (Figure 1B). It is also worth noting that the opposing catalytic subunits of the endonuclease significantly facilitate placement of the scissile phosphates into each other’s active sites by flipping out the first bulge nucleotides (7). This base flipping twists the phosphate backbone into a nearly perfect in-line geometry with a τ angle of ~170° (τ is the angle in space formed among the nucleophilic 2’-oxygen, the scissile phosphate, and the 5’- leaving oxygen, and 180° is a perfectly in-line τ angle). RNAs are known to more readily self-cleave when they are in this conformation (4). These structural results (7) combined with computational results (8) suggest that the splicing endonuclease also accelerates reaction by facilitating the inline conformation. A computed energy profile with respect to the pseudorotation angle around the scissile phosphate bond showed that the endonuclease provides ~ 10- 12-fold stabilization of the RNA structure (8).
Figure 1:
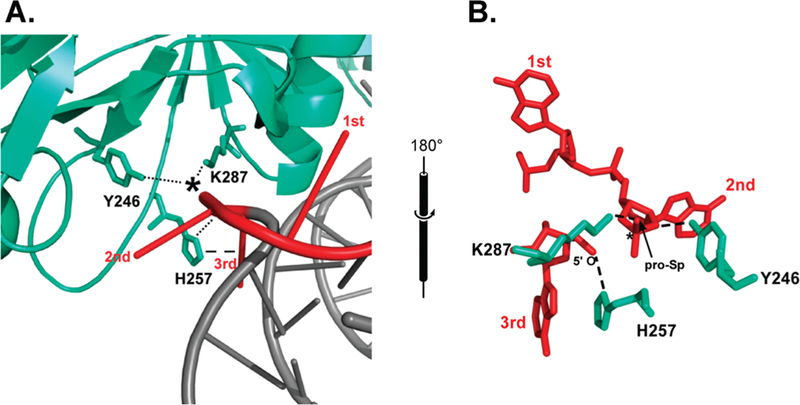
Conserved triad residues clustered around the scissile phosphate. (A) The enzyme (cyan) is bound to the RNA substrate (gray) specifically at the bulge nucleotides (red). The scissile phosphate is indicated by an asterisk, and putative interactions are indicated by dotted and dashed lines. Residues responsible for the positioning of the first bulge nucleotide are not shown. (B) Opposing view stick model illustrating the triad around the scissile phosphate. Specific atomic distances in the text are indicated by dashed lines [Protein Data Bank (PDB) entry 1GJW (7)].
We wanted to confirm our structural data with kinetic studies to demonstrate the importance of the conserved triad residues to cleavage rate enhancement. To this end, we conducted single-turnover kinetic experiments using the A. fulgidus (AF) splicing endonuclease and its conserved triad mutants in cleavage assays on a model BHB RNA substrate. In this work, we explore in a very broad sense the importance of this conserved triad to cleavage. We establish that mutating these residues reduces cleavage rates, almost entirely in the case of K287. In addition, we show that the deficient catalytic activity of the K287 mutants was rescued by addition of specific divalent metal ions, suggesting the critical importance of the positive charge of K287 in the RNA cleavage reaction, a role similar to that played by metal ions in RNA cleavage by ribozymes.
RESULTS
Mutation of the Catalytic Triad.
We expressed and purified four His257, two Tyr246, and three Lys287 mutant AF splicing endonucleases. These are H257A, H257C, H257D, H257N, Y246H, Y246F, K287E, and K287R. The purification process was designed to eliminate any wild-type contamination. Detailed methods of mutagenesis and purification are described in Materials and Methods. Figure 2A provides a crude analysis of the purity of the mutant proteins and a calibration of their relative concentrations. Purified mutants were first tested by simple cleavage assays at 1 μM with incubation for 20 min at 50 °C with 3 nM substrate RNA to determine prospects for further kinetic analysis. Of these, H257A, 257C, H257N, and Y246F exhibited reasonable cleavage activity while others exhibited no readily visible cleavage (Figure 2B).
Figure 2:
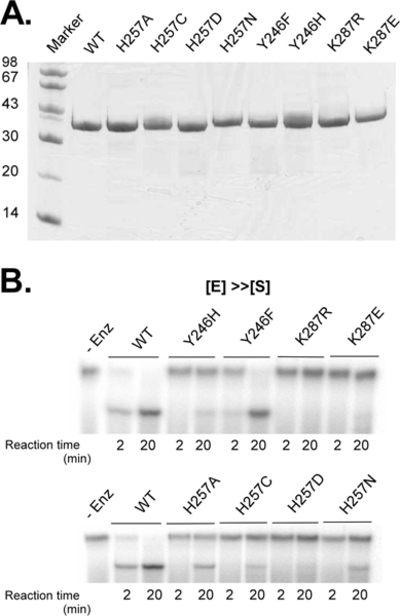
Evaluation of mutants. (A) Sodium dodecyl sulfatepolyacrylamide gel electrophoresis (SDS-PAGE) analysis of protein samples used in kinetic trials. Ten microliters of the 10 μM sample was loaded for each mutant. (B) Cleavage tests of mutants. One micromolar enzyme samples were reacted with substrate, and aliquots were removed at 2 and 20 min time points for all samples.
Rate Enhancement by the Wild-Type AF Splicing Endonuclease.
Single-turnover kinetic conditions were established in which the enzyme concentrations ([E]) were at least 10fold greater than the substrate concentration ([S]) such that substrate binding and release processes could be separated from product binding and release (9). In addition, at saturating enzyme concentrations, the binding step would in theory be overridden and the rate of chemical cleavage could be directly measured. A limitation to this method is that if there is a rate-limiting step other than cleavage (not including product release), it will not be readily discernible. This method is commonly used by kineticists working on RNA ribozymes where the quantity of enzyme is not limited. It was demonstrated that Michaelis-Menten-like kinetic parameters k2 and KM obtained at high enzyme concentrations are valid and similar to those derived from steady state kinetics (10). The k2 and KM parameters provide a basic understanding of the splicing endonuclease with regard to its RNA binding affinities and catalytic properties.
To establish a baseline of substrate self-cleavage under the conditions used in our assay, we made use of the established relationship between the in-line conformation and the self-cleavage rate by Soukup and Breaker (4). We measured the t angles and 2’-O-P attack distances (4) of both cleavage site linkages from the NMR structure of the free BHB motif (11). Both linkages were constrained by the natural shape of the molecule to a non-in-line conformation and would theoretically have a self-cleaving rate of 1 × 10−7 min−1 at 23 °C. Our cleavage assays were performed at 50 °C, so we estimated the self-cleavage rates of these linkages at 50 °C to be 7 × 10−6 min−1 (see Materials and Methods). This baseline rate of self-cleavage corresponds to a free energy barrier of ~29 kcal/mol, which indicates that this substrate is not very likely to self-cleave at a rapid rate.
We then analyzed the dependence of the single-turnover rate constant on enzyme concentration with the goal of gaining basic information regarding the reaction rate constant and apparent binding affinities between reactants. Protein concentrations at least 10-fold greater than substrate concentrations were used to establish pseudo-first-order conditions, and 4 μM was the highest protein concentration we used because earlier trials demonstrated that >4 μM samples produced progressively lower cleavage activity (data not shown). We observed two distinct phases in the wild-type time course experiments (Figure 3A), suggesting the presence of an alternative substrate. To improve our understanding of the biphasic feature, we repeated the single-turnover experiments with elevated substrate concentrations (from 3 to 30 nM), which yielded time courses with excellent fits to a single exponential at various enzyme concentrations (compare panels E and F of Figure 3). The concentration- dependent biphasic feature is consistent with a fast depletion of substrate followed by a slow formation (two-strand association) and cleavage of the substrate. Consistent with this explanation, the rate of the slow phase is independent of enzyme concentration and ranged from 0.0002 to 0.003 min−1 (Figure 3D, inset). In contrast, the rate of the fast phase depends on enzyme concentration by a hyperbolic relationship (Figure 3D). The kinetic behavior of the fast phase is consistent with a reversible reaction producing the activated enzyme-substrate complex (a second-order process) followed by cleavage (a first-order process), presumably irreversible once the activated E-S complex is formed. Such a scenario is described by the following kinetic scheme:
Figure 3:
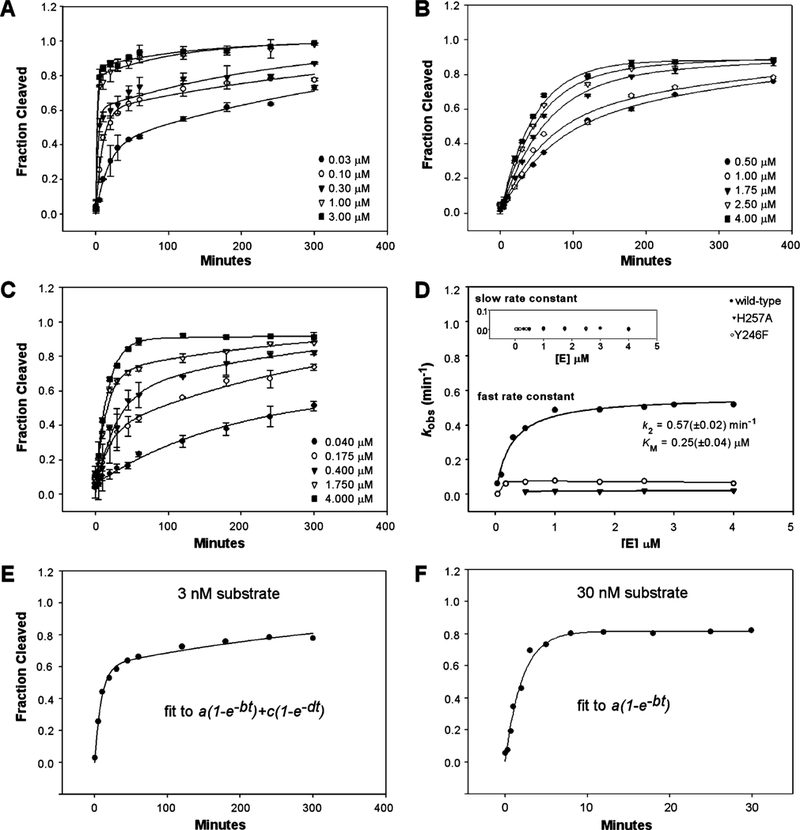
Kinetic parameters for the wild-type splicing endonuclease and time course curves for H257A and Y246F. (A) Measurement of single-turnover rate constants for the wild-type (WT) enzyme. Shown are selected time course data ranging over the concentrations used. All time courses were fit to a double-exponential equation as described in Materials and Methods to produce corresponding rates for the predominant fast phase (kobs) in each reaction. (B) Measurement of single-turnover rate constants for H257A. (C) Measurement of singleturnover rate constants for Y246F. Shown are selected time course data ranging over the concentrations used. (D) Dependence of the single-turnover rate constants on enzyme concentration for the WT enzyme. The rate constants for the fast phase (kobs) are plotted as a function of enzyme concentration and fit to a model for a second-order reaction followed by an irreversible first-order reaction as described previously (13). The inset shows the dependence of the rate constants for the slow phase for the wild type, H257A, and Y246F. (E and F) Comparison of enzyme cleavage progress curves between a low (E) and high (F) substrate concentration at the same enzyme concentration (100 nM). The equations to which each progress curve was fitted are indicated in the two panels.
From this scheme, we gleaned k2, the cleavage rate constant for the fast phase at saturating enzyme concentrations, and KM, the apparent dissociation constant between enzyme and substrate, by plotting the fast kobs values as a function of enzyme concentration and fitting the data as described in a similar experiment (12). Manual pipetting prevented us from obtaining accurate early cleavage fractions at higher concentrations, which might have skewed our results toward lower limits. Despite this, all of our data fit reasonably well to this kinetic scheme, and we obtained a rate constant of 0.57 min−1 for the fast phase of the reaction and a KM of 0.25 μM (Figure 3D). This k2 is in the ~1 min−1 rates seen for hammerhead ribozymes (9) and RNase III (12), another nuclease specific for double-stranded RNA substrates. While it pales in comparison to enzymes with cleavage rates of hundreds to thousands per second such as degradative RNases, this rate reflects a 8 × 104-fold rate enhancement over uncatalyzed cleavage.
Histidine 257.
Given that this histidine is absolutely conserved and has no obvious near neighbors available to compensate for its loss, we anticipated that at the very least it should provide the optimal shape and electrostatic environment for substrate conformation and proton transfer. Correspondingly, all four histidine mutations disrupt the RNA cleavage activity of the enzyme. However, the fact that H257D completely abolished activity under the singleturnover conditions used in our assay, while H257A, H257C, and H257N appeared to retain detectable cleavage activity (Figure 2B), argues against an essential role of protonation provided by H257 in the entire phosphoryl transfer reaction. We focused on H257A for kinetic analysis.
Similar to those of the wild-type enzyme, H257A time courses were also biphasic. The rate of the fast phase was significantly reduced, ranging narrowly from 0.0156 to 0.0214 min-1. The slow rates remained around 0.002 min−1, similar to those of the wild-type enzyme. Because the fast and slow rates are likely to be very closely coupled early in the reaction, our methods might not have allowed us to accurately separate the two rates for this mutant. Consequently, extraction of the enzyme concentration dependence of the fast rates was difficult. However, from an upper limit of the fast cleavage rate of 0.02 min−1, the reduction in rate of cleavage by the H257A mutation is estimated to be ~ 28-fold from the k2 of the WT enzyme.
Tyrosine 246.
Two Y246 mutants were obtained and assessed for their RNA cleavage activities. Y246H had severely impaired activity, but Y246F retained significant activity (Figure 2B), suggesting that stereochemistry, rather than the hydroxyl group of this residue, accounts for its importance with respect to cleavage. The Y246F time course data also could fit well to a double-exponential equation (Figure 3C). Fast phase rates ranged from 0.002 to 0.08 min−1, with a slight apparent hyperbolic curve in their dependence on enzyme concentration (Figure 3D). Slow rates ranged from 0.0005 to 0.004 min−1, again with no discernible dependence on enzyme concentration. Again, due to the close coupling between the two rate processes, no kinetic parameters were computed from the kobs versus [E] plot. As with the H257A mutant, the Y246F fast phase rates provided an upper limit of cleavage of 0.08 min−1, a reduction of ~7- fold from the apparent k2 of the WT enzyme.
Y246F/H257A Double Mutant.
Since cleavage still occurs relatively well in the absence of either Y246 or H257 individually, it would appear that the remaining two conserved triad residues are sufficient to enable activity. To determine the effects on cleavage if both H257 and Y246 were missing, we created an H257A/Y246F double mutant. With all reaction conditions the same as those of wild-type reactions, a 5 h incubation of the double mutant enzyme with the RNA substrate yielded no detectable RNA activity (Figure 4). Given the relatively small rate reductions by individually mutated residues, the abolished activity of the double mutant indicates the likelihood that H257 and Y246 compensate for each other. Without both residues, the enzyme loses its ability to align the substrate in the in-line conformation and to facilitate proton transfer. This result provides the strongest evidence in this work that these residues are essential to the cleavage process.
Figure 4:
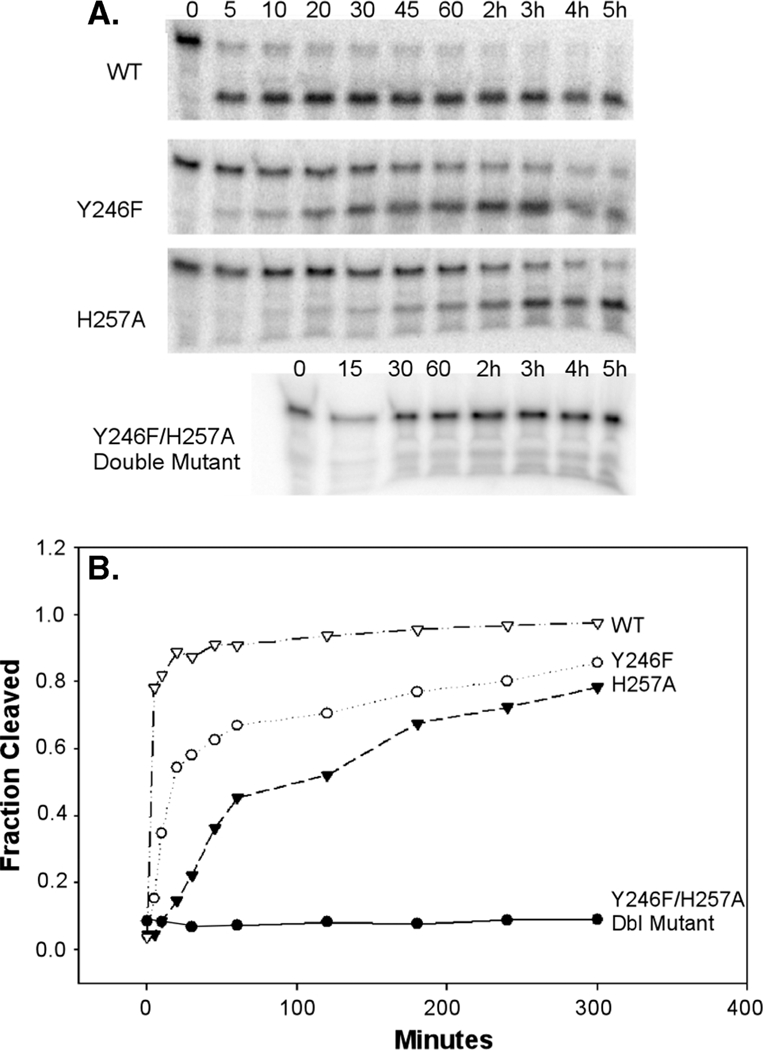
Evaluation of the Y246F/H257A double mutant. (A) Representative gels of cleavage tests using the wild-type enzyme and samples of each mutant. (B) Time course plot showing the relative activities of the protein samples over a 5 h incubation (hours are indicated; otherwise, the units are minutes). The Y246F/H257A double mutant concentration is 2.5 μM, and the concentrations of all other samples are 1 μM.
Lysine 287.
We initially tested cleavage activities of two lysine mutants, K287E and K287R, and found that they had severely impaired activity (Figure 2B), which indicates an essential role for this residue in RNA cleavage. The K287E loss of activity was expected, but the degree to which K287R lost activity was surprising. A possible explanation for K287R’s lack of inherent activity might be that its side chain is too large to function readily in the same capacity as K287. To determine that K287’s positive charge is specifically required for cleavage (by potentially stabilizing the transition state), we created a new K287Q mutant to serve as a negative control for K287E and tested K287E, K287Q, and K287R in cleavage with a battery of metal ions. We wondered if the loss of positive charge of Lys287 is the most crucial factor for the loss of enzyme activity and, if so, whether this function can be replaced by positively charged metal ions.
First, we determined the effects of Mg2+ alone on cleavage of the BHB RNA. In Figure 5A, our results show that mixing up to 100 mM Mg2+ with BHB RNA did not result in any visible cleavage of the RNA. Furthermore, the splicing endonuclease is shown to be independent of metal ions by cleavage in the presence of 25 mM EDTA (ethylenediamine- tetraacetic acid).
Figure 5:
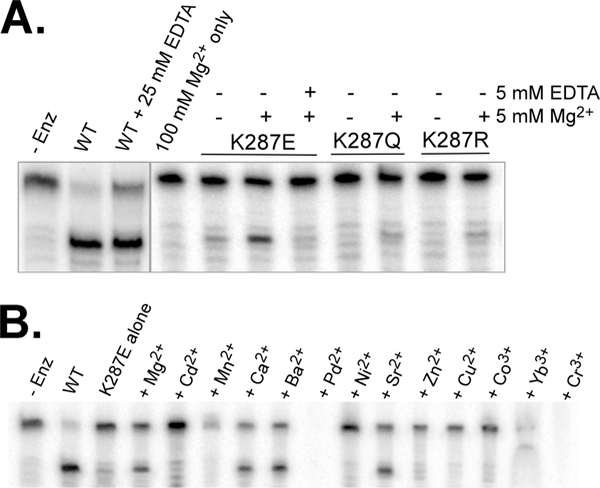
Magnesium rescue assays. In all cases, protein concentrations are 2.5 mM. (A) Cleavage in the presence of high and low Mg2+ concentrations. (B) Rescue by members of group 2A in the periodic table. Metal concentrations are all 25 μM. There was no explanation for why no cleavage product was observed in lanes containing Pb2+ and Cr3+.
We then added 5 mM Mg2+ to cleavage reaction mixtures with the K287 mutants. The metal ions rescued the activity of the K287E mutant quite well, and a low level of activity by the K287R and K287Q mutants was also rescued. When we conducted this experiment with 25 mM Mg2+, we found that rescue was more prominent in all three mutant samples (data not shown) but K287E rescue was always the most significant. The presence of specific cleavage in rescue lanes suggests that the K287 mutants were otherwise performing normally.
Next, we explored whether any other metal ions would produce the same effect. Figure 5B shows that divalent ions from group 2A of the periodic table, but not monovalent (cleavage buffer contains 100 mM NaCl), trivalent, or even divalent transition metal ions, rescued the deficient RNA cleavage activity of K287E. Through hard metal-hard ligand interactions with the K287E side chain, it is possible that the glutamic acid residue provides the best ligand to position divalent ion in the vicinity of the scissile phosphate group within the same space required by K287. The coordinated metal ion provides a positive charge for stabilizing the negative transition state in a mechanism reminiscent of that of metal-dependent ribozymes.
DISCUSSION
In eukaryotes, the RNA splicing endonuclease removes introns from precursor tRNA molecules. This enzyme is strongly conserved among eukaryotes and archaea but is not present in bacteria, where introns in tRNAs are often self-splicing (13, 14). We have confirmed through the study of an archaeal homologue that the conserved catalytic triad of the splicing endonuclease does play an important role in cleavage. A possible deficit in this work is that the observed rate of cleavage at saturating enzyme concentrations does not necessarily correspond to the rate of chemistry, which complicates interpretation of the activity of active site mutants. However, our data are sufficient to support the qualitative nature of our conclusions, including a relationship between two of the conserved triad residues which has emerged from this work. Mutating Y246 or H257, each of which is thought to participate in proton transfer, definitely reduced cleavage activity. When residues homologous to Y246 or H257 are mutated in RNase A, the rate reductions are in the range of 105-fold, although RNase A has significantly greater overall rate enhancement (1011-fold) compared to the splicing endonuclease (8 × 104-fold). Therefore, it perhaps is not surprising that the upper limit rate reductions of these two catalytic triad residues are in the range of 7—28-fold. However, in the absence of both Y246 and H257, activity is completely lost, indicating a synergistic effect between the two catalytic residues. Given the observed interactions of Y246 and H257 with RNA substrate, they may play important roles in placement of the substrate into the in-line conformation and possibly take part in proton shuttling. Different from degradative ribonucleases such as RNase A, the splicing endonuclease depends completely on the RNA structure. Here, the catalysis is believed to rely upon achieving the in-line conformation, a process initiated by a pair of residues from the opposing catalytic subunit and fine-tuned in the active site by H257 and Y246. Once this conformation is achieved, the Y246 hydroxyl oxygen, putatively charge stabilized by a neighboring arginine (7), is in the optimal position to take the hydrogen from the labile 2’-OH group. From there, as the nucleophilic 2’-O attacks the scissile phosphate, a proton shuttling network possibly involving solvent waters (8) and the scissile phosphate’s pro-Rp nonbridging oxygen direct a proton to the 5’-leaving group. H257’s role would be to help create an optimal electrostatic and conformational environment for protons to reach the leaving 5’-oxygen. The role of K287 would be to stabilize the charge on the scissile phosphate to lower the activation energy. Support for K287’s function in this capacity is found in the metal ion rescue experiments where the severely impaired K287E mutant was activated by the addition of divalent metal ions. Substitution of K287 with glutamic acid in effect changes the splicing endonuclease from being metal-independent to being divalent metal-dependent. Mg2+ is well-known to help maintain RNA structure and activate nucleophiles in RNA hydrolysis, and many phosphodiesterases make use of metal ions, including both protein and RNA-catalyzed reactions (15-18). Perhaps the splicing endonuclease’s cleavage mechanism is metalindependent because it intrinsically contains residues capable of interacting with and folding the RNA into the required conformation for catalysis to occur without Mg2+. This is a reasonable explanation given that archaeal organisms can have very high cellular concentrations of metals, including Mg2+ [as high as 135 mM in A.fulgidus (19)]. The organisms might have evolved to have RNA that remains extremely stable in such an abundance of metals, such that the only way to cleave it is through metal-independent processes. It is interesting to note that archaeal splicing endonucleases are often considered to be the ancestors of the eukaryotic enzymes, which retain metal-independent cleavage ability. On the other hand, it has been proposed previously that tRNA introns were originally self-cleaving (20) and that the role of the splicing endonuclease is to “improve the efficiency and preciseness of an inherent pre-tRNA self-cleavage activity” (21). The results presented here support the possibility that the splicing endonuclease acts as an accessory protein similar to those upon which the group I and II selfsplicing introns rely for stabilizing their autocatalytic conformations. From either scenario, it is intriguing to consider the prospect that the splicing endonuclease could be among the evolutionary links between the active protein catalyst and the RNA world.
MATERIALS AND METHODS
Protein Expression and Purification.
Primers designed to generate site specific mutations of the catalytic residues were ordered from IDT (Coralville, IA). Mutations were generated on the plasmid coding the AF splicing endonuclease using the Quikchange mutagenesis kit (Stratagene, La Jolla, CA), and sequences were confirmed by DNA sequencing. Using methods published previously (7, 22), the proteins were overexpressed in Escherichia coli and purified to homogeneity through use of Ni-NTA columns followed by gel filtration chromatography. To prevent possible cross contamination, each mutant was purified using a separate Ni-NTA column and a thoroughly washed gel filtration column. Normal elution profiles from the gel filtration purification step were observed for all mutants. Samples were concentrated and stored at −80 °C. Protein concentrations were obtained using Bradford assay reagent (Bio-Rad, Hercules, CA) and absorbance at 595 nm. Each mutant was prepared at least twice; separate batches of proteins were assayed independently.
RNA Preparation.
Synthetic RNA oligos were ordered from Dharmacon (Boulder, CO) and deprotected and/or desalted according to the manufacturer’s protocols. The 21mer three-nucleotide bulge oligo sequences are the same as those used previously (23). Strands were phosphorylated using T4 polynucleotide kinase (New England Biolabs, Beverly, MA) and [γ−32P]ATP (MP Biomedical, Irvine, CA). Concentrations were calculated on the basis of the assumption that 80% of the RNA stock with a known concentraion is phosphorylated. Phosphorylated strand A was annealed to a 100-fold excess of the nonlabeled B strand for 15 min each at 70, 25, and 4 °C. In all but the Mg2+ rescue experiments, annealing buffer contained 30 mM cacodylic acid (pH 7.5), 1 mM spermidine, and 2.5 mM MgCl2.
Calculations establishing that the BHB substrate is constrained to a non-in-line conformation were conducted by applying methods used by Soukup and Breaker (4) to scissile phosphate bonds in the NMR free BHB RNA structure by Diener and Moore (11). The resulting τ measurements were then used to calculate in-line fitness factors for the BHB scissile phosphate bonds. Optimal in-line fitness values approximate a value of 1, and lower values reflect deviation from optimal conditions. Uncatalyzed self-cleavage rates of the BHB RNA were calculated using the Arrhenius equation:
| (1) |
Kinetic Cleavage Assays.
Cleavage assays were carried out essentially as previously described (23) with minor changes to sample mixing methods, higher reaction volumes, temperature, 6 h of cleavage time, and carefully selected protein concentrations since cleavage activity often decreased when enzyme concentrations were in excess of 4 μM. Mutants were modified for concentration and time such that for all samples, ~70% or more of the substrate was cleaved and the time course curves plateaued. Prepared substrates were not frozen and were stored at 4 °C for no more than 48 h for maintenance of its proper structures. The upper limit of the RNA substrate reaction concentration was 3 nM in all cases, and the total reaction volume was 130 μL. Assays were conducted at 50 °C in a water bath, with a mercury thermometer for monitoring the bath temperature. A tube rack was secured to a metal block and placed in the water bath. Rack sections were filled with water from the bath such that the water levels inside and outside the tube rack were the same. This setup equilibrated for 30 min. Prior to the enzyme and substrate being mixed, all samples equilibrated in the water bath for 4 min and 5 μL uncut RNA aliquots were removed. Reaction aliquots (10 μL) were taken at each time point and extinguished in 40 μL of denaturing loading buffer. Samples (50 μL) were loaded onto a 10% denaturing polyacrylamide gel containing 7 M urea and 1 x TBE. All gel and buffer reagents were prepared in advance for all reactions to ensure consistency. Gels were run for 30 min at 700 V and exposed to phosphorimaging screens. Cleavage products were separated on a 10% (w/v) denaturing acrylamide gel and visualized by phosphorimaging using the Molecular Dymanics Storm 860 Scanner and ImageQuant (GE Healthcare). Local average background was corrected, and fraction cleaved was calculated by Catalytic Mechanism of the Splicing Endonuclease
| (2) |
Analysis of Kinetic Data.
Initial time course curves were fit by Sigma Plot 8 to data points using the doubleexponential equation
| (3) |
where y is the fraction of substrate cleaved, a and c are the final amplitudes of the fast and slow reaction phases, respectively, b and d are the rate constants for the fast and slow reaction phases, respectively, and t is the reaction incubation time. Amplitude parameters a and c were constrained such that a + c = 1 based on the assumption that given enough time, the reactions would eventually reach completion.
To obtain kinetic parameters k2 and KM, the various kobs values for the WT enzyme were plotted as a function of enzyme concentration ([E]) and fit to a hyperbolic function as described previously (12). The hyperbolic function is described as
| (4) |
where kobs is the pseudo-first-order rate constant, [E] is enzyme concentration, k2 is the cleavage rate constant at a saturating enzyme concentration, and KM = (k2 + k-1)/k1.
Metal Ion Rescue Experiments.
These assays were conducted as described in the cleavage assay section at half the final volume, with no aliquot removal, incubation times of 15-30 min, and Mg2+ removed from all of the buffers involved. Enzyme storage buffers contained only Na+ ions. It is unclear why both Pb2+ and Cr3+ repeatedly led to degradation of substrate RNA. The effects of these two metal ions on RNA cleavage activity rescue were, thus, not observed.
ACKNOWLEDGMENT
We thank Dr. Olke Uhlenbeck, Dr. Michael Harris, Dr. Brian Miller, and the members of the Li laboratory for helpful discussions.
† This work was supported by National Science Foundation Grants 0817638 and 00517300 (H.L.). K.C. and X.S. are American Heart Association Florida/Puerto Rico Affiliate pre-doctoral (0415091B) and post-doctoral fellows (0725583B), respectively.
REFERENCES
- 1.Calvin K, and Li H (2008) RNA-splicing endonuclease structure and function. Cell. Mol. Life Sci. 65, 1176–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Emilsson GM, Nakamura S, Roth A, and Breaker RR (2003) Ribozyme speed limits. RNA 9, 907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Breaker RR, Emilsson GM, Lazarev D, Nakamura S, Puskarz IJ, Roth A, and Sudarsan N (2003) A common speed limit for RNA-cleaving ribozymes and deoxyribozymes. RNA 9, 949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soukup GA, and Breaker RR (1999) Relationship between internucleotide linkage geometry and the stability of RNA. RNA 5, 1308–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson LD, Brandon LD, Nieuwlandt DT, and Daniels CJ (1989) Transfer RNA intron processing in the halophilic archaebacteria. Can. J. Microbiol. 35, 36–42. [DOI] [PubMed] [Google Scholar]
- 6.Peebles CL, Gegenheimer P, and Abelson J (1983) Precise excision of intervening sequences from precursor tRNAs by a membrane-associated yeast endonuclease. Cell 32, 525–536. [DOI] [PubMed] [Google Scholar]
- 7.Xue S, Calvin K, and Li H (2006) RNA recognition and cleavage by a splicing endonuclease. Science 312, 906–910. [DOI] [PubMed] [Google Scholar]
- 8.Min D, Xue S, Li H, and Yang W (2007) “In-line attack” conformational effect plays a modest role in an enzyme-catalyzed RNA cleavage: A free energy simulation study. Nucleic Acids Res. 35, 4001–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stage-Zimmermann TK, and Uhlenbeck OC (1998) Ham-merhead ribozyme kinetics. RNA 4, 875–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hiromi K (1979) Kinetics of fast enzyme reactions, Kodansha, Ltd, Tokyo. [Google Scholar]
- 11.Diener JL, and Moore PB (1998) Solution structure of a substrate for the archaeal pre-tRNA splicing endonucleases: The bulge-helix-bulge motif. Mol. Cell 1, 883–894. [PubMed] [Google Scholar]
- 12.Campbell FE Jr., Cassano AG, Anderson VE, and Harris ME (2002) Pre-steady-state and stopped-flow fluorescence analysis of Escherichia coli ribonuclease III: Insights into mech-anism and conformational changes associated with binding and catalysis. J. Mol. Biol. 317, 21–40. [DOI] [PubMed] [Google Scholar]
- 13.Cech TR (1990) Self-splicing of group I introns. Annu. Rev. Biochem. 59, 543–568. [DOI] [PubMed] [Google Scholar]
- 14.Dai L, and Zimmerly S (2002) Compilation and analysis of group II intron insertions in bacterial genomes: Evidence for retroelement behavior. Nucleic Acids Res. 30, 1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gan J, Tropea JE, Austin BP, Court DL, Waugh DS, and Ji X (2006) Structural insight into the mechanism of double-stranded RNA processing by ribonuclease III. Cell 124, 355–366. [DOI] [PubMed] [Google Scholar]
- 16.McConnell TS, Herschlag D, and Cech TR (1997) Effects of divalent metal ions on individual steps of the Tetrahymena ribozyme reaction. Biochemistry 36, 8293–8303. [DOI] [PubMed] [Google Scholar]
- 17.Nowotny M, Gaidamakov SA, Crouch RJ, and Yang W (2005) Crystal structures of RNase H bound to an RNA/DNA hybrid: Substrate specificity and metal-dependent catalysis. Cell 121, 1005–1016. [DOI] [PubMed] [Google Scholar]
- 18.Lee TS, Silva Lopez C, Giambasu GM, Martick M, Scott WG, and York DM (2008) Role of Mg2+ in hammerhead ribozyme catalysis from molecular simulation. J. Am. Chem. Soc. 130, 3053–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lapaglia C, and Hartzell PL (1997) Stress-Induced Production of Biofilm in the Hyperthermophile Archaeoglobus fulgidus. Appl. Environ. Microbiol. 63, 3158–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lykke-Andersen J, Aagaard C, Semionenkov M, and Garrett RA (1997) Archaeal introns: Splicing, intercellular mobility and evolution. Trends Biochem. Sci. 22, 326–331. [DOI] [PubMed] [Google Scholar]
- 21.Weber U, Beier H, and Gross HJ (1996) Another heritage from the RNA world: Self-excision of intron sequence from nuclear pre-tRNAs. Nucleic Acids Res. 24, 2212–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, and Abelson J (2000) Crystal structure of a dimeric archaeal splicing endonuclease. J. Mol. Biol. 302, 639–648. [DOI] [PubMed] [Google Scholar]
- 23.Calvin K, and Li H (2007) Achieving specific RNA cleavage activity by an inactive splicing endonuclease subunit through engineered oligomerization. J. Mol. Biol. 366, 642–649. BI801141Q [DOI] [PMC free article] [PubMed] [Google Scholar]