

Abstract
Cytokines and cytokine signaling pathways are crucial for regulating cellular functions, including cell growth, proliferation, differentiation, and cell death. Cytokines regulate physiological processes such as immune responses and maintain immune homeostasis, and they also mediate pathological conditions such as autoimmune diseases and cancer. Hence, the precise control of the expression of cytokines and the transduction of cytokine signals is tightly regulated at transcriptional and post-transcriptional levels. In particular, post-transcriptional regulation at the level of mRNA stability is critical for coordinating cytokine expression and cytokine signaling. Numerous cytokine transcripts contain AU-rich elements (AREs), whereas transcripts encoding numerous components of cytokine signaling pathways contain GU-rich elements (GREs). AREs and GREs are mRNA decay elements that mediate rapid mRNA degradation. Through ARE- and GRE-mediated decay mechanisms, immune cells selectively and specifically regulate cytokine networks during immune responses. Aberrant expression and stability of ARE- or GRE-containing transcripts that encode cytokines or components of cytokine signaling pathways are observed in disease states, including cancer. In this review, we focus on the role of AREs and GREs in regulating cytokine expression and signal transduction at the level of mRNA stability.
Keywords: cytokines, cytokine signaling, post-transcriptional regulation, mRNA stability, mRNA decay, ARE, GRE, immune responses, cancer
INTRODUCTION
Cytokines are small secretory proteins produced by a variety of cells, including immune cells, and play vital roles in many cellular processes, such as cell growth, differentiation, proliferation, and apoptosis. Cytokines are indispensable regulators in development, immune responses, inflammation, and often play important roles in mediating diseases. They act through receptors on targets cells, triggering cascades of intracellular signaling to upregulate or downregulate certain genes to modulate the behavior of the affected cells. Cytokine levels are highly dynamic and are precisely regulated by several mechanisms, both transcriptional and post-transcriptional [1–4]. Dysregulated expression of cytokines and components of cytokine signaling pathways are often associated with human diseases, including autoimmune diseases, inflammatory diseases, and cancer [5–7].
The expression of cytokine genes is tightly regulated at multiple different levels, such as transcription, RNA localization, RNA turnover, and translation. In particular, post-transcriptional regulation at the level of mRNA stability plays important roles in the regulation of cytokine expression and cytokine signaling cascades. The cellular mRNA decay machinery works to coordinate the expression of cytokine genes during immune or inflammatory responses by controlling the stability and lifespan of cytokine mRNAs. Altered mRNA stability accounts for 20–50% of the changes in gene expression in mammalian cells upon stimulation, highlighting the importance of mRNA stability in determining gene expression [8]. A well-known mechanism for selectively regulating mRNA stability is through the interaction between cisacting elements and trans-acting factors [9, 10]. Cis-acting elements are sequences often found in the untranslated regions of mRNAs, which can be recognized by trans-acting factors, such as RNA-binding proteins (RBPs) or microRNAs (miRNAs). Several trans-acting factors may compete for the same cis-acting element, or work synergistically, and the net effect of their interaction determines the stability of the mRNAs that harbor the cisacting element [11, 12]. Post-transcriptional regulation of cytokine expression and signaling is mainly achieved by the cis-elements within mRNA transcripts encoding cytokines or cytokine signaling proteins [13].
Regulation of ARE-containing cytokine transcripts
The ARE is an extensively studied cis-element that was first identified in 1986 as a conserved sequence present in the 3’ untranslated region (UTR) of both human and mouse tumor necrosis factor (TNF) transcripts [14]. Later studies identified additional AREs in a variety of cytokine and early-response transcripts, and AREs were subsequently categorized into clusters based on sequence characteristics and decay kinetics [15–18]. A subset of AREs, characterized by overlapping AUUUA pentamers, is enriched in secreted proteins such as cytokines and chemokines. Notably, ARE-containing transcripts account for 5–8% of the human transcriptome [19], but they comprise approximately 80% of the transcripts within cytokine networks [4]. The importance of AREs in regulating cytokine expression was first demonstrated by the mutation of AREs in cytokine mRNAs, such as TNF-α or IFN-γ, resulting in autoimmune-like inflammatory syndrome due to increased expression of these cytokines [20, 21].
AREs regulate mRNA stability by interacting with a variety of ARE-binding proteins (AREBPs). Some AREBPs, such as Tristetraprolin (TTP, also known as ZFP36), promote transcript decay by recruiting cellular enzymes for deadenylation and degradation [22, 23], while other AREBPs, such as human antigen R (HuR, also known as ELAV-like protein 1), stabilize transcripts when they bind to the AREs [24], possibly by preventing the binding of decaypromoting AREBPs [25]. Some ARE-containing transcripts are targeted for degradation by cellular ribonucleases directly. For example, ZC3H12A initiates rapid degradation of several cytokine transcripts, such as TNFα, IL1b, IL2, IL6 and IL12b, by cleaving the stem loop mRNA structure that is proximal to the AREs [26]. HuR can counteract the decay function of ZC3H12A by binding to AREs in the 3’UTR of these transcripts, promoting transcript stabilization and increased mRNA and protein levels [27–31]. In addition, some microRNAs can directly bind to AREs and influence the half-life of the ARE-containing transcripts [32]. Thus, AREBPs and microRNAs can work in concert to determine the stability of the ARE-harboring transcripts.
ARE-mediated mRNA decay is indispensable in the resolution of immune responses by shutting off the expression of ARE-containing immune activation genes such as cytokine genes [2, 13, 33]. In activated T lymphocytes, the transcription of several ARE-containing cytokine transcripts, such as IL2, IL4, IFN-γ, and TNF-α, is induced to initiate immune responses. During the immune resolution phase, these cytokine transcripts are marked for degradation through the AREs to limit their expression [34, 35]. In self-reactive T cells, AREs are responsible for the post-transcriptional silencing of cytokine mRNAs, causing these cells to exhibit anergic phenotypes [36]. Overall, AREs are critical for regulating cytokine expression during immune responses.
Aberrant stabilization of ARE-containing cytokine transcripts in cancer
Unlike normal immune cells, where cytokine mRNA decay is tightly regulated in a dynamic way during an immune response, in malignant cells this dynamic regulation is lost. For example, ARE-containing cytokine transcripts that are normally unstable in primary cells can become constitutively stable in malignant cells, leading to overexpression of cytokines [37, 38]. The aberrant stabilization of these ARE-containing transcripts may be due to abnormal expression, post-translational modification or altered localization of AREBPs or irregular microRNA interactions in malignant cells [39, 40]. Two AREBPs in particular, HuR and TTP, have been extensively studied with regard to their role in ARE-mediated mRNA decay in malignant compared to normal cells. These two proteins potentially compete for thousands of overlapping ARE-binding sites, but they have antagonistic effects [41, 42]. HuR facilitates transcript stabilization and up-regulation upon binding to AREs [43], while TTP promotes degradation and down-regulation of ARE-containing transcripts [41]. During immune responses, binding by HuR to ARE-containing cytokine transcripts mediates transient cytokine expression, whereas activation-induced expression of TTP mediates subsequent rapid transcript degradation, allowing the resolution of immune responses [25].
Remarkably, the balance between these HuR and TTP function is disturbed in malignant cells. In many types of cancer, the function and expression of HuR is elevated but the function of TTP is nearly abolished, leading to increased production of cytokines that promote malignant phenotypes [44–48]. In fact high HuR/TTP ratio is associated with high levels of mitosis-related ARE-containing transcripts in many solid cancers [39], and single nucleotide polymorphisms in these two proteins often correlate with poor prognosis [49, 50]. In cancer cells, increased cytoplasmic HuR levels and HuR phosphorylation are strongly associated with aberrant cell growth, proliferation, and chemo-resistance [51–54]. Also, HuR increases the stability and expression of pro-angiogenic and pro-inflammatory cytokine transcripts through the AREs within their 3’UTRs, further promoting tumor growth [47, 48].
In contrast to HuR overexpression in malignant cells, the decay-promoting protein, TTP, is often down-regulated or even absent in various cancers, which makes the cancerous cell unable to degrade ARE-containing mRNAs that are normally targeted by TTP [55]. For example, TTP down-regulation or deficiency in many tumors accounts for the overexpression of pro-inflammatory and tumorigenic cytokines and growth factors, including IL1, 2, 6, 8, 10, 16, 17, and 23, IFNγ, TNFα/β, and VEGF [56–59]. When TTP is exogenously overexpressed, the progression of tumor growth and metastasis is decreased in several cancer cell lines [60]. In fact, the cancer drug Sorafenib, an inducer of TTP re-expression in melanoma cells, can reduce the expression of pro-angiogenic cytokines that contain AREs in their mRNA 3’UTRs [61].
Overall, abnormal expression of HuR and TTP seems to contribute to malignant phenotypes in cancer cells. Future studies may focus on developing agents that can correct the imbalance of these two proteins to achieve favorable post-transcriptional regulation of ARE networks in cancer cells. Such agents could potentially serve as therapies for a variety of cancer types.
Regulation of GRE-containing signal transduction transcripts
The GRE, another cis-element that mediates rapid mRNA decay, was recently identified to be enriched in the 3’ UTRs of short-lived transcripts expressed in primary human T cells. The consensus GRE sequence has been defined as UGUU(/G)UGUU (/G)UGU, which functions as a mediator of mRNA decay. Insertion of a GRE into the 3’ UTR of a beta-globin reporter transcript caused the transcript to decay more rapidly compared to the same reporter without GRE insertion [62, 63]. The GRE binds to the RNA-binding protein CUGBP and Etr3-Like Factor 1 (CELF1) and together they mediate rapid degradation of the GRE-containing transcripts [64]. Genome-wide RNA-immunoprecipitation studies in primary cells or cell lines reveal that GREs are enriched in CELF1-target transcripts involved in a variety of biological functions such as cell growth, development, apoptosis, and cytokine signal transduction [65, 66]. The proposed mechanism for GRE/CELF1-mediated mRNA decay is that binding by CELF1 to GRE-containing transcripts leads to recruitment of components of the mRNA decay machinery to the transcripts, resulting in deadenylation and subsequent degradation of the target transcripts [67]. Numerous transcripts encoding the protein components of cytokine signaling cascades contain GREs, suggesting that the downstream effects of cytokines are regulated through GREs. Figure 1 shows an example where GRE-containing transcripts encode multiple components of IL1 signaling cascades (grey nodes). Presumably, the presence of GREs in transcripts encoding multiple cytokine signaling components is responsible for rapid decay of these transcripts, limiting their expression, and allowing them to be regulated during immune responses. For example, phosphorylation of CELF1 occurs following the activation of primary human T lymphocytes, leading to decreased binding by CELF1 to GRE sequences and consequently, increased stability and expression of GRE-containing transcripts, including numerous transcripts encoding components of cytokine signaling pathways [68].
Figure 1. ARE- and GRE-containing transcripts are enriched in the IL1 signaling pathway.
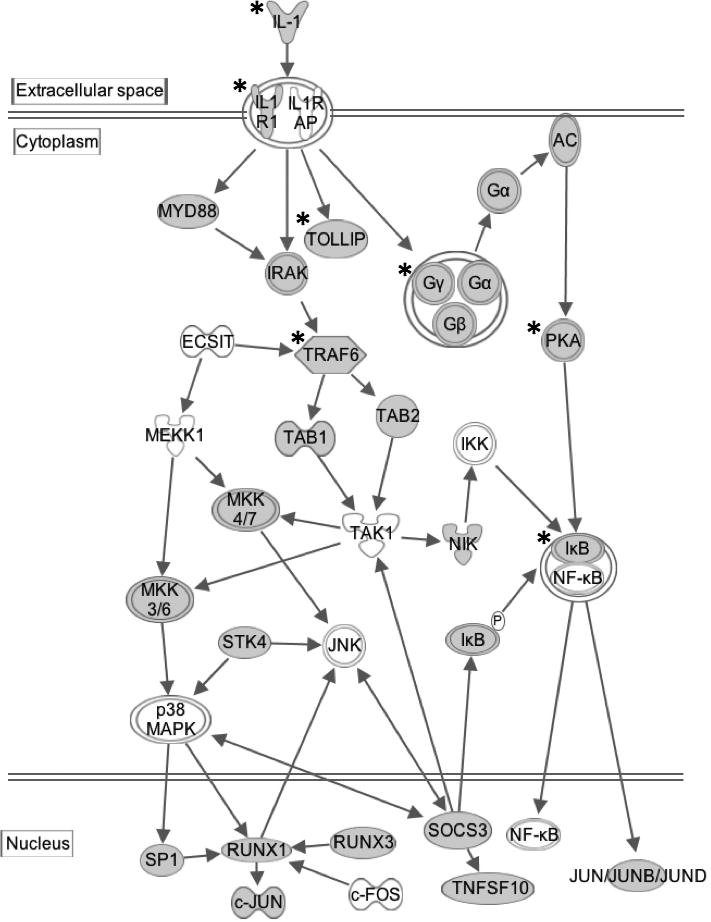
Transcripts shown in grey nodes contain GREs in their 3’UTRs. Transcripts shown in grey nodes and labeled with an asterisk (*) contain AREs in their 3’UTRs. This pathway diagram was built using Ingenuity Pathway Assistant Software.
Aberrant GRE-mediated decay of cytokine signaling transcripts in cancer
The rapid decay of many GRE-containing transcripts observed in normal immune cells was not observed in malignant T cells [69]. In malignant T cells, CELF1 lost its ability to bind to a subset of GRE-containing transcripts that were targeted in normal T cells. The loss of binding was probably due to hyper-phosphorylation of CELF1 since the target transcripts were present in both normal and malignant cells and the CELF1 expression level was similar in both cell types. The lack of CELF1 binding resulted in the stabilization and upregulation of GRE-containing target transcripts in these malignant cells, including transcripts encoding transcription factors (such as JUN, STAT5) and transcripts encoding cytokine signaling molecules that control cell growth [69]. Notably, while hyperphosphorylation of CELF1 in malignant cells blocked its ability to bind to a subset of GRE-containing transcripts, this hyper-phosphorylation caused CELF1 to gain the ability to bind to and mediate decay of another subset of GRE-containing transcripts. These transcripts encoded proteins involved in suppressing proliferation such as SOCS5, TNFRSF4, and PIAS1 [69]. The mechanism by which CELF1 hyperphosphorylation leads to switching target binding preferences is not clear, and more experiments are needed to investigate the impact of post-translational modification of CELF1 on GRE/CELF1-mediated mRNA decay in cancer.
Coordinated regulation of cytokine expression and signaling transduction by AREs and GREs
While AREs regulate the stability of many cytokine transcripts, GREs control the stability of transcripts encoding numerous components of cytokine signaling pathways, indicating that cytokine function is coordinately regulated by both AREs and GREs. The ARE-binding protein HuR and GRE-binding protein CELF1 share many target transcripts, because HuR can also bind to GU-rich or poly U sequences in addition to AREs. Therefore, HuR and CELF1 may compete for the GU- or U- rich binding sites and exert opposite stabilizing effects on target transcripts [70, 71]. The cytoplasmic abundance and binding affinity of these two proteins vary in different cell types or activation states, thus the stability of a given target transcript varies depending on whichever protein is predominant in the cell. Particularly, in cancer cells, cytoplasmic HuR overexpression and phosphorylation, plus CELF1 hyper-phosphorylation, push GU- or U-rich mRNAs to undergo stabilization by HuR [4, 69, 72, 73]. Therefore, a plausible model is that in normal cells, CELF1 targets numerous cytokine signaling transcripts for degradation through GRE-mediated decay. Whereas in malignant cells, CELF1 is hyperphosphorylated and loses its ability to bind to certain GRE-containing transcripts. In the meantime, HuR is overexpressed and phosphorylated in malignant cells such that its binding affinity increases to both AREs and GREs, resulting in increased stability and expression of some ARE and GRE-containing transcripts, as we have seen in malignant cells [74, 75]. More evidence is required to confirm this model.
The crosstalk between ARE and GRE networks is evident in many cytokine signaling pathways. For instance, both HuR and CELF1 target transcripts are involved in the TNF signaling pathway that regulates apoptosis, including members of the BCL2 superfamily [76, 77]. HuR and CELF1 also co-regulate the IL1 receptor signaling transduction through targeting AREs or GREs in transcripts that encode components of this pathway [29] (Figure 1). As another example, transcripts encoding cytokines of the interferon type I, II and III family all contain AREs and are controlled by the ARE/AREBP-mediated decay, whereas the interferon receptors harbor GREs in their mRNAs and undergo GRE/CELF1-mediated decay. Moreover, many components of the interferon signaling pathway contain ARE, GRE or both and are subject to their regulation [78]. Therefore, coordination of ARE and GRE networks is required for effective interferon responses.
SUMMARY
Both ARE and GRE networks coordinate post-transcriptional regulation of cytokine expression and signaling in health and disease [79]. AREs modulate the expression of many cytokines as well as components of the cytokine signaling pathways, whereas GREs are overrepresented in the mRNAs encoding cytokine receptors and cytokine signaling proteins and modulate their transcript stability. Malfunctions in ARE- and GRE-binding proteins, such as HuR, TTP, and CELF1, are highly correlated with dysregulated cytokine production and signaling, and play roles in mediating autoimmune diseases and cancer. Understanding the mechanism of ARE- and GRE-mediated mRNA decay and the interactions between ARE and GRE networks may shed light on the development of therapeutic strategies for these diseases.
Acknowledgments
This work was supported by NIH grants AI057484 and AI072068 to P. R. B. L. G. was funded through NIH grant T32 AI83196. I.V-S. was funded through a fellowship from the Lymphoma Research Foundation and supported by start-up funds from the Department of Medicine at the University of Minnesota.
Footnotes
CONFLICT OF INTEREST STATEMENT
There are no conflicts of interest.
References
- 1.Anderson P. Nat. Immunol. 2008;9(4):353–9. doi: 10.1038/ni1584. [DOI] [PubMed] [Google Scholar]
- 2.Ivanov P, Anderson P. Immunol. Rev. 2013;253(1):253–72. doi: 10.1111/imr.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palanisamy V, Jakymiw A, van Tubergen EA, D’Silva NJ, Kirkwood KL. J. Dent. Res. 2012;91(7):651–8. doi: 10.1177/0022034512437372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vlasova-St Louis I, Bohjanen PR. Cytokine Growth Factor Rev. 2017;33:83–93. doi: 10.1016/j.cytogfr.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seruga B, Zhang H, Bernstein LJ, Tannock IF. Nat. Rev. Cancer. 2008;8(11):887–99. doi: 10.1038/nrc2507. [DOI] [PubMed] [Google Scholar]
- 6.Saharinen P, Eklund L, Pulkki K, Bono P, Alitalo K. Trends Mol. Med. 2011;17(7):347–62. doi: 10.1016/j.molmed.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 7.Seko Y, Cole S, Kasprzak W, Shapiro BA, Ragheb JA. Autoimmun. Rev. 2006;5(5):299–305. doi: 10.1016/j.autrev.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 8.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Nature. 2011;473(7347):337–42. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 9.Blackinton JG, Keene JD. Semin. Cell Dev. Biol. 2014;34:44–54. doi: 10.1016/j.semcdb.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morris AR, Mukherjee N, Keene JD. Wiley Interdiscip Rev. Syst. Biol. Med. 2010;2(2):162–80. doi: 10.1002/wsbm.54. [DOI] [PubMed] [Google Scholar]
- 11.Keene JD. Nat. Rev. Genet. 2007;8(7):533–43. doi: 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- 12.Beisang D, Bohjanen PR. Wiley Interdiscip Rev. RNA. 2012;3(5):719–31. doi: 10.1002/wrna.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vlasova-St Louis I, Bohjanen PR. J. Interferon Cytokine Res. 2014;34(4):233–41. doi: 10.1089/jir.2013.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caput D, Beutler B, Hartog K, Thayer R, Brown-Shimer S, Cerami A. Proc. Natl. Acad. Sci. USA. 1986;83(6):1670–4. doi: 10.1073/pnas.83.6.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen CY, Shyu AB. Trends Biochem. Sci. 1995;20(11):465–70. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 16.Bakheet T, Williams BR, Khabar KS. Nucleic Acids Res. 2003;31(1):421–3. doi: 10.1093/nar/gkg023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bakheet T, Williams BR, Khabar KS. Nucleic Acids Res. 2006;34(Database issue):D111–4. doi: 10.1093/nar/gkj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halees AS, El-Badrawi R, Khabar KS. Nucleic Acids Res. 2008;36(Database issue):D137–40. doi: 10.1093/nar/gkm959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bakheet T, Frevel M, Williams BR, Greer W, Khabar KS. Nucleic Acids Res. 2001;29(1):246–54. doi: 10.1093/nar/29.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hodge DL, Berthet C, Coppola V, Kastenmuller W, Buschman MD, Schaughency PM, Shirota H, Scarzello AJ, Subleski JJ, Anver MR, Ortaldo JR, Lin F, Reynolds DA, Sanford ME, Kaldis P, Tessarollo L, Klinman DM, Young HA. J. Autoimmun. 2014;53:33–45. doi: 10.1016/j.jaut.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacob CO, Hwang F, Lewis GD, Stall AM. Cytokine. 1991;3(6):551–61. doi: 10.1016/1043-4666(91)90481-r. [DOI] [PubMed] [Google Scholar]
- 22.Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. Mol. Cell Biol. 1999;19(6):4311–23. doi: 10.1128/mcb.19.6.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen CY, Gherzi R, Ong SE, Chan EL, Raijmakers R, Pruijn GJ, Stoecklin G, Moroni C, Mann M, Karin M. Cell. 2001;107(4):451–64. doi: 10.1016/s0092-8674(01)00578-5. [DOI] [PubMed] [Google Scholar]
- 24.Brennan CM, Steitz JA. Cell Mol. Life Sci. 2001;58(2):266–77. doi: 10.1007/PL00000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raghavan A, Robison RL, McNabb J, Miller CR, Williams DA, Bohjanen PR. J. Biol. Chem. 2001;276(51):47958–65. doi: 10.1074/jbc.M109511200. [DOI] [PubMed] [Google Scholar]
- 26.Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, Satoh T, Kato H, Tsujimura T, Nakamura H, Akira S. Nature. 2009;458(7242):1185–90. doi: 10.1038/nature07924. [DOI] [PubMed] [Google Scholar]
- 27.Fan J, Ishmael FT, Fang X, Myers A, Cheadle C, Huang SK, Atasoy U, Gorospe M, Stellato C. J. Immunol. 2011;186(4):2482–94. doi: 10.4049/jimmunol.0903634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopez de Silanes I, Zhan M, Lal A, Yang X, Gorospe M. Proc. Natl. Acad. Sci. USA. 2004;101(9):2987–92. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukherjee N, Lager PJ, Friedersdorf MB, Thompson MA, Keene JD. Mol. Syst. Biol. 2009;5:288. doi: 10.1038/msb.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stellato C, Gubin MM, Magee JD, Fang X, Fan J, Tartar DM, Chen J, Dahm GM, Calaluce R, Mori F, Jackson GA, Casolaro V, Franklin CL, Atasoy U. J. Immunol. 2011;187(1):441–9. doi: 10.4049/jimmunol.1001881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winzen R, Gowrishankar G, Bollig F, Redich N, Resch K, Holtmann H. Mol. Cell Biol. 2004;24(11):4835–47. doi: 10.1128/MCB.24.11.4835-4847.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.von Roretz C, Gallouzi IE. J. Cell Biol. 2008;181(2):189–94. doi: 10.1083/jcb.200712054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson P. Nat. Rev. Immunol. 2010;10(1):24–35. doi: 10.1038/nri2685. [DOI] [PubMed] [Google Scholar]
- 34.Khabar KS. J. Leukoc Biol. 2007;81(6):1335–44. doi: 10.1189/jlb.0207109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schott J, Stoecklin G. Wiley Interdiscip Rev. RNA. 2010;1(3):432–56. doi: 10.1002/wrna.13. [DOI] [PubMed] [Google Scholar]
- 36.Villarino AV, Katzman SD, Gallo E, Miller O, Jiang S, McManus MT, Abbas AK. Immunity. 2011;34(1):50–60. doi: 10.1016/j.immuni.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vlasova IA, McNabb J, Raghavan A, Reilly C, Williams DA, Bohjanen KA, Bohjanen PR. Genomics. 2005;86(2):159–71. doi: 10.1016/j.ygeno.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 38.Chu PC, Kulp SK, Chen CS. Carcinogenesis. 2013;34(12):2694–705. doi: 10.1093/carcin/bgt251. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Hitti E, Bakheet T, Al-Souhibani N, Moghrabi W, Al-Yahya S, Al-Ghamdi M, Al-Saif M, Shoukri MM, Lanczky A, Grepin R, Gyorffy B, Pages G, Khabar KS. Cancer Res. 2016;76(14):4068–80. doi: 10.1158/0008-5472.CAN-15-3110. [DOI] [PubMed] [Google Scholar]
- 40.Al-Ahmadi W, Al-Ghamdi M, Al-Haj L, Al-Saif M, Khabar KS. Nucleic Acids Res. 2009;37(11):3612–24. doi: 10.1093/nar/gkp223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukherjee N, Jacobs NC, Hafner M, Kennington EA, Nusbaum JD, Tuschl T, Blackshear PJ, Ohler U. Genome Biol. 2014;15(1):R12. doi: 10.1186/gb-2014-15-1-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raghavan A, Ogilvie RL, Reilly C, Abelson ML, Raghavan S, Vasdewani J, Krathwohl M, Bohjanen PR. Nucleic Acids Res. 2002;30(24):5529–38. doi: 10.1093/nar/gkf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Calaluce R, Gubin MM, Davis JW, Magee JD, Chen J, Kuwano Y, Gorospe M, Atasoy U. BMC Cancer. 2010;10:126. doi: 10.1186/1471-2407-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kotta-Loizou I, Giaginis C, Theocharis S. Med. Oncol. 2014;31(9):161. doi: 10.1007/s12032-014-0161-y. [DOI] [PubMed] [Google Scholar]
- 45.Sanduja S, Blanco FF, Young LE, Kaza V, Dixon DA. Front Biosci. (Landmark Ed.) 2012;17:174–88. doi: 10.2741/3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suswam E, Li Y, Zhang X, Gillespie GY, Li X, Shacka JJ, Lu L, Zheng L, King PH. Cancer Res. 2008;68(3):674–82. doi: 10.1158/0008-5472.CAN-07-2751. [DOI] [PubMed] [Google Scholar]
- 47.Nabors LB, Gillespie GY, Harkins L, King PH. Cancer Res. 2001;61(5):2154–61. [PubMed] [Google Scholar]
- 48.Nabors LB, Suswam E, Huang Y, Yang X, Johnson MJ, King PH. Cancer Res. 2003;63(14):4181–7. [PubMed] [Google Scholar]
- 49.Griseri P, Bourcier C, Hieblot C, Essafi-Benkhadir K, Chamorey E, Touriol C, Pages G. Hum. Mol. Genet. 2011;20(23):4556–68. doi: 10.1093/hmg/ddr390. [DOI] [PubMed] [Google Scholar]
- 50.Upadhyay R, Sanduja S, Kaza V, Dixon DA. Int. J. Cancer. 2013;132(3):E128–38. doi: 10.1002/ijc.27789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blanco FF, Jimbo M, Wulfkuhle J, Gallagher I, Deng J, Enyenihi L, Meisner-Kober N, Londin E, Rigoutsos I, Sawicki JA, Risbud MV, Witkiewicz AK, McCue PA, Jiang W, Rui H, Yeo CJ, Petricoin E, Winter JM, Brody JR. Oncogene. 2016;35(19):2529–41. doi: 10.1038/onc.2015.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lafarga V, Cuadrado A, Lopez de Silanes I, Bengoechea R, Fernandez-Capetillo O, Nebreda AR. Mol. Cell Biol. 2009;29(16):4341–51. doi: 10.1128/MCB.00210-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gabai VL, Meng L, Kim G, Mills TA, Benjamin IJ, Sherman MY. Mol. Cell Biol. 2012;32(5):929–40. doi: 10.1128/MCB.05921-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang J, Modi Y, Yarovinsky T, Yu J, Collinge M, Kyriakides T, Zhu Y, Sessa WC, Pardi R, Bender JR. Am. J. Pathol. 2012;180(4):1751–60. doi: 10.1016/j.ajpath.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carrick DM, Blackshear PJ. Arch. Biochem. Biophys. 2007;462(2):278–85. doi: 10.1016/j.abb.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 56.Lee HH, Yang SS, Vo MT, Cho WJ, Lee BJ, Leem SH, Lee SH, Cha HJ, Park JW. Mol. Cells. 2013;36(6):571–6. doi: 10.1007/s10059-013-0268-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patial S, Curtis AD, 2nd, Lai WS, Stumpo DJ, Hill GD, Flake GP, Mannie MD, Blackshear PJ. Proc. Natl. Acad. Sci. USA. 2016;113(7):1865–70. doi: 10.1073/pnas.1519906113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ross CR, Brennan-Laun SE, Wilson GM. Ageing Res. Rev. 2012;11(4):473–84. doi: 10.1016/j.arr.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Tubergen E, Vander Broek R, Lee J, Wolf G, Carey T, Bradford C, Prince M, Kirkwood KL, D’Silva NJ. Cancer. 2011;117(12):2677–89. doi: 10.1002/cncr.25859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brennan SE, Kuwano Y, Alkharouf N, Blackshear PJ, Gorospe M, Wilson GM. Cancer Res. 2009;69(12):5168–76. doi: 10.1158/0008-5472.CAN-08-4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bourcier C, Griseri P, Grepin R, Bertolotto C, Mazure N, Pages G. Am. J. Physiol. Cell Physiol. 2011;301(3):C609–18. doi: 10.1152/ajpcell.00506.2010. [DOI] [PubMed] [Google Scholar]
- 62.Vlasova IA, Tahoe NM, Fan D, Larsson O, Rattenbacher B, Sternjohn JR, Vasdewani J, Karypis G, Reilly CS, Bitterman PB, Bohjanen PR. Mol. Cell. 2008;29(2):263–70. doi: 10.1016/j.molcel.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rattenbacher B, Beisang D, Wiesner DL, Jeschke JC, von Hohenberg M, St Louis-Vlasova IA, Bohjanen PR. Mol. Cell Biol. 2010;30(16):3970–80. doi: 10.1128/MCB.00624-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vlasova-St Louis I, Dickson AM, Bohjanen PR, Wilusz CJ. Biochim. Biophys. Acta. 2013;1829(6–7):695–707. doi: 10.1016/j.bbagrm.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vlasova IA, Bohjanen PR. RNA Biol. 2008;5(4):201–7. doi: 10.4161/rna.7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Le Tonqueze O, Gschloessl B, Legagneux V, Paillard L, Audic Y. Genom Data. 2016;8:97–103. doi: 10.1016/j.gdata.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moraes KC, Wilusz CJ, Wilusz J. RNA. 2006;12(6):1084–91. doi: 10.1261/rna.59606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beisang D, Rattenbacher B, Vlasova-St Louis IA, Bohjanen PR. J. Biol. Chem. 2012;287(2):950–60. doi: 10.1074/jbc.M111.291658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bohjanen PR, Moua ML, Guo L, Taye A, Vlasova-St Louis IA. RNA. 2015;21(10):1757–69. doi: 10.1261/rna.049940.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu L, Ouyang M, Rao JN, Zou T, Xiao L, Chung HK, Wu J, Donahue JM, Gorospe M, Wang JY. Mol. Biol. Cell. 2015;26(10):1797–810. doi: 10.1091/mbc.E14-11-1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lebedeva S, Jens M, Theil K, Schwanhausser B, Selbach M, Landthaler M, Rajewsky N. Mol. Cell. 2011;43(3):340–52. doi: 10.1016/j.molcel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 72.Zhu Z, Wang B, Bi J, Zhang C, Guo Y, Chu H, Liang X, Zhong C, Wang J. Tumour Biol. 2013;34(4):2299–308. doi: 10.1007/s13277-013-0774-3. [DOI] [PubMed] [Google Scholar]
- 73.Kuyumcu-Martinez NM, Wang GS, Cooper TA. Mol. Cell. 2007;28(1):68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vlasova-St Louis I, Bohjanen PR. Cells. 2016;5(1):4. doi: 10.3390/cells5010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Simone LE, Keene JD. Curr. Opin. Genet. Dev. 2013;23(1):35–43. doi: 10.1016/j.gde.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang L, Lee JE, Wilusz J, Wilusz CJ. J. Biol. Chem. 2008;283(33):22457–63. doi: 10.1074/jbc.M802803200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dean JL, Wait R, Mahtani KR, Sully G, Clark AR, Saklatvala J. Mol. Cell Biol. 2001;21(3):721–30. doi: 10.1128/MCB.21.3.721-730.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Khabar KS, Young HA. Biochimie. 2007;89(6–7):761–9. doi: 10.1016/j.biochi.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Khabar KS. J. Interferon Cytokine Res. 2014;34(4):215–9. doi: 10.1089/jir.2013.0151. [DOI] [PMC free article] [PubMed] [Google Scholar]