

ABSTRACT
The field of interstitial lung disease (ILD) has undergone significant evolution in recent years, with an increasing incidence and more complex, ever expanding disease classification. In their most severe forms, these diseases lead to progressive loss of lung function, respiratory failure and eventually death. Despite notable advances, progress has been challenged by a poor understanding of pathological mechanisms and patient heterogeneity, including variable progression. The diagnostic pathway is thus being continually refined, with the introduction of tools such as transbronchial cryo lung biopsy and a move towards genetically aided, precision medicine. In this review, we focus on how to approach a patient with ILD and the diagnostic process.
KEYWORDS: Cryoscopic lung biopsy, idiopathic pulmonary fibrosis, interstitial lung disease
Introduction
Interstitial lung disease (ILD) is an umbrella term for over 200 different diseases that display considerable variation in terms of clinical course, treatment and prognosis. Broadly speaking, they can be subdivided into those with an identifiable cause and those without; the latter being referred to as idiopathic interstitial pneumonias. Clinical assessment aims to identify a possible cause; screening for features of systemic disease (eg connective tissue disease) or environmental triggers. Relevant exposures include pneumotoxic drugs, radiation therapy, occupational exposures (eg asbestosis) or implicated allergens (hypersensitivity pneumonitis).
Distinguishing the various forms of pulmonary fibrosis is critical for determining correct management and for predicting prognosis; however, this is often obfuscated by the fact the lung has a limited repertoire in response to injury and, consequently, a finite number of disease patterns. In essence, all ILD is characterised by variable degrees of inflammation and fibrosis, not only between diseases, but also among individuals with the same disease (Fig 1). In inflammation dominant disease, the histology is that of organising pneumonia or non-specific interestitial pneumonitis, while in fibrosis dominant disease, one would expect to see usual interstitial pneumonitis (UIP) – characterised by fibroblastic foci and only mild to moderate inflammation. These histological patterns are associated with specific radiological features, the recognition of which may abrogate the need for a formal biopsy and tissue diagnosis.
Fig 1.

Schematic classification of interstitial lung diseases according to aetiology. The finding of histological usual interstitial pneumonitis in a patient with an idiopathic interstitial pneumonia leads to the specific diagnosis of idiopathic pulmonary fibrosis. NSIP = non-specific interstitial pneumonitis.
Idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is the most common of the idiopathic interstitial pneumonias, with an incidence of approximately 6,000 cases per year in the UK, affecting mainly older males. Median survival is worse than many cancers at just 3 years and the disease accounts for 5,300 deaths each year in the UK.1 IPF is a growing problem, with an annual increase in incidence of 11% between 1991 and 2003, a rise that is only partly explained by an ageing population.2
As already alluded to, distinguishing IPF from other ILDs, including other idiopathic forms, is important for multiple reasons. IPF does not respond to immunosuppressive therapy; in fact, immunomodulation may worsen outcomes.3 By contrast there is evidence, particularly in systemic sclerosis associated ILD,4 of benefit from cyclophosphamide and mutliple case reports suggest a potential role for rituximab5 as salvage therapy in connective tissue disease-ILD. In addition, there are now two drugs, pirfenidone and nintedanib, approved by the National Institute for Health and Care Excellence (NICE) for IPF; however, at an annual cost of around £26,000 per patient and the potential for significant side effects, accurate disease identification is essential. Finally, IPF has a worse prognosis than other ILD; therefore, a definitive diagnosis allows for timely involvement of palliative care physicians and consideration of lung transplantation.
Pathogenesis of idiopathic pulmonary fibrosis
The pathogenesis of IPF is complex and poorly understood, but involves aberrant wound healing in the context of repetitive alveolar injury. This results in abnormal fibroblast proliferation, differentiation and activation, which in turn drives expansion of the extracellular matrix with loss of normal lung architecture. Inflammation plays a less dominant role. This pathogenesis is illustrated schematically in Fig 2.
Fig 2.

The pathogenesis of idiopathic pulmonary fibrosis. 1 – In an initiating phase, there is lung alveolar epithelial damage with loss of the normal lung architecture and disruption of the basement membrane across which gas exchange takes place. With further epithelial damage and apoptosis, comes upregulation of epithelial integrins, such as αvβ6, and a phase of fibroproliferative repair dominates – driven by high levels of TGF-β. Released in an inactive form, this cytokine requires an activation step facilitated by integrins that bind the Arg-Gly-Asp (arginine-glycine-aspartic acid; RGD) motif of pro-TGFβ and promote its cleavage and activation. 2 – Locally activated TGF-β drives the recruitment of fibroblasts and a feed-forward cycle of further TGF-β production. 3 – Under these conditions, fibroblasts differentiate into myofibroblasts that express high levels of integrin αvβ6, are resistant to apoptosis and lay down a collagen matrix. 4 – Once collagen has been laid down in a lung, the architecture of which is already distorted, gas exchange is no longer efficient. There is a change in the vasculature of the lung parenchyma with both fall-out of blood vessels and neo-angiogenesis driven by local production of vascular endothelial and platelet derived growth factors (VEGF and PDGF). At this final phase, the lung is irreversibly scarred. AEC = alveolar epithelial cell; PDGF = platelet-derived growth factor; TGF-β = transforming growth factor beta; TNF-α = tumour necrosis factor alpha; VEGF = vascular endothelial growth factor.
Genetics in diagnosis and management of idiopathic pulmonary fibrosis
Although the initiating events in IPF are poorly understood, the disease is likely to be the result of environment exposures in genetically susceptible individuals. Certainly, it is estimated that approximately 20% of idiopathic interstitial pneumonias have a genetic component and familial cases (referred to as familial interstitial pneumonias) were first described in the 1950s. The majority of these familial interstitial pneumonias are autosomal dominant with incomplete penetrance, but some may arise de novo. The most commonly affected genes are those involved in surfactant processing and telomere biology. At present, routine genetic testing is not recommended; however, ILD patients with at least one affected first degree relative should be offered the opportunity to enrol in the UK-wide 100,000 Genomes Project, whereby they undergo whole genome sequencing.
Genetics also have a proven role in sporadic IPF. Polymorphisms in the promoter for the gene encoding the salivary mucin, 5b (MUC5B) and for the Toll-interacting protein (TOLLIP) are both associated with an increased risk of developing IPF, although both result in a relatively mild phenotype. These genetic variants provide the first possible targets for precision medicine in IPF, with a post hoc analysis of the PANTHER data establishing a variable response to N-acetyl cysteine depending on the individual's TOLLIP genotype.6
Diagnostic work up in interstitial lung diseases
Initial investigations include blood tests to detect the presence of autoantibodies (Table 1), precipitating immunoglobulins against organic antigens and serum angiotensin-converting enzyme. These tests alone rarely confirm the diagnosis and there is potential for both false positive (particularly autoantibodies in older patients) and false negative (failure to identify an antigen and IgG does not exclude hypersensitivity pneumonitis) results; however, they can be useful in helping direct further diagnostics.
Table 1.
Autoantibodies in connective tissue ILDs
| Antibody | Associated connective tissue disease |
|---|---|
| ANA (>1:320) | Many |
| RF (>60 IU/mL) Anti-CCP | RA, Sjögren's syndrome, SLE RA |
| Anti-centromere | Systemic sclerosis |
| Anti-nuclear ANA | Systemic sclerosis |
| Anti Ro (SS-A) | SLE, Sjögren's syndrome and others |
| Anti La (SS-B) | SLE, Sjögren's syndrome |
| Anti-RNP | SLE, MCTD |
| Anti-dsDNA | SLE |
| Anti-Smith | SLE |
| Anti tRNA synthetase | Poly-/dermatomyositis (anti-synthetase syndrome) |
| Anti-PM-Scl | Systemic sclerosis/myositis overlap |
| Anti-Th/To | Systemic sclerosis |
| Anti-U3 RNP | Systemic sclerosis |
| ANCA panel | Vasculitides |
| Anti-topoisomerase (Scl-70) | Systemic sclerosis |
ANA = antinuclear antibody; ANCA = Anti-neutrophil cytoplasmic antibodies; Anti-CCP = anti-cyclic citrullinated peptide antibody; anti-dsDNA = anti-double stranded DNA; anti-PM-Scl = anti polymyositis-scleroderma; anti-RNP = anti-ribonucleoprotein; ILD = interstitial lung disease; MCTD = mixed connective tissue disease; RA = rheumatoid arthritis; RF = Rheumatoid Factor; SLE = systemic lupus erythematosus
Box 1.
The ATS/ERS consensus statement for the diagnosis of IPF set out criteria for the optimal HRCT technique for evaluation of ILD1
| Optimal HRCT technique for evaluation of ILD |
|---|
|
ATS = American Thoracic Society; ERS = European Respiratory Society; HRCT = high resolution computerised tomography; ILD = interstitial lung disease; IPF = idiopathic pulmonary fibrosis
Pulmonary function tests are key in appraising these patients and while they rarely refine the specific diagnosis in individuals with proven ILD, they inform on disease severity at baseline and response to treatment over follow up.
Radiological work up
Chest radiographs
A chest X-ray is often the first radiological investigation in ILD patients and while it is rarely sufficient to make a confident diagnosis, X-ray can play a role in establishing disease chronicity and progression.
High-resolution computerised tomography
High-resolution computerised tomography (HRCT) of the thorax has revolutionised the diagnosis and classification of ILD and, in many cases, removes the need for invasive diagnostic procedures. However, the quality of the images is dependent on the scanning protocol employed (Box 1).
The American Thoracic Society/European Respiratory Society (ATS/ERS) 2011 consensus statement7 provides criteria for a definite UIP pattern on HRCT (Fig 3), with the presence of these conferring a sensitivity of approximately 40%, but a specificity of 95% for histological UIP. The main discriminating feature for UIP is the presence of honeycombing; however, typical CT appearances are only present in two thirds of patients and it is in the remaining third of cases that biopsy may have a role.
Fig 3.
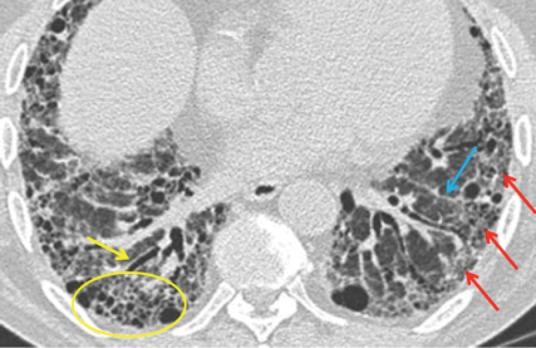
Diagnostic criteria for a definite usual interstitial pneumonitis pattern on high resolution computerised tomography. 1 – subpleural, basal predominance (red arrows); 2 – reticular abnormality (blue arrow); 3 – honeycombing with or without traction bronchiectasis (yellow arrow); 4 – absence of features inconsistent with usual interstitial pneumonitis pattern.
Bronchoscopy in the assessment of interstitial lung disease
Bronchoalveolar lavage, endobronchial ultrasound and transbronchial biopsy
There is much variation in practice surrounding the use of bronchoalveolar lavage (BAL), particularly between European centres who, in contrast to their North American colleagues, often perform this investigation routinely. Clearly, there is value in excluding infection, which may be a differential diagnosis; however, BAL alone is rarely diagnostic, with perhaps one of the difficulties being a lack of consistency in terms of how samples are taken and processed. Under optimal circumstances, BAL reflects cellular traffic in the alveolar space and the cell differential may provide supplemental information to help refine, rather make a diagnosis. In particular, an excess of lymphocytes should call into question a presumptive IPF diagnosis, with Ohshimo et al8 describing a BAL lymphocytosis of >30% in 6 of 74 patients with definite UIP features on HRCT. In all six cases, further investigations led to a final diagnosis of chronic hypersensitivity pneumonitis.
Transbronchial biopsy (TBB) with standard forceps is a minimally invasive technique, but does not always provide adequate lung tissue to establish a final diagnosis. The biopsies are small, subject to crush artefact and may not be representative in spatially heterogeneous disease.9,10 However, such biopsies may be helpful in the diagnosis of sarcoidosis and organising pneumonia.
Transbronchial cryo lung biopsy
Transbronchial cryo lung biopsy (TBCLB) was first described in 2008.11 It has since been shown to be a safe, minimally invasive and effective diagnostic tool for the histological diagnosis of ILD, with a diagnostic yield of up to 74–80%.11–15 The advantage of TBCLB over TBB lies in the larger specimen size, with a mean size of 9–64 mm2.11–20 In addition, the technique avoids crush or bleeding artefact, which can distort the tissue architecture (Fig 4). The published data on TBCLB shows a safety profile that is comparable to TBB, with bleeding post biopsy in around 10% of cases, all of which was controlled bronchoscopically. The mean rate of pneumothorax requiring chest drain insertion is around 4%, although there is a wide variation between centres. Exacerbations of ILD are rare (0.5%) and only one mortality has been reported (0.2%).11–20
Fig 4.
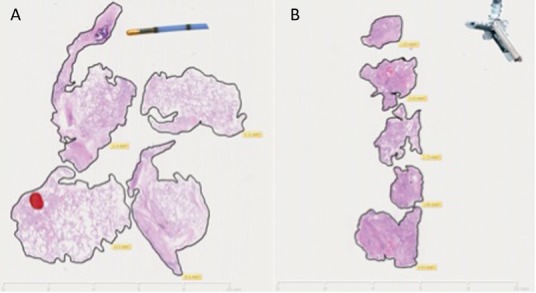
Comparison between transbronchial cryo lung biopsy (A) and traditional forceps transbronchial biopsy (B) performed on the same patient at two different sittings. Transbronchial cryo lung biopsy shows preserved architecture of parenchymal tissue with a total biopsy area of 46.81 mm2 and mean biopsy area of 11.7 mm2. Traditional forceps transbronchial biopsy is characterised by crush and haemorrhagic artefact with a total biopsy area of 14.11 mm2 and mean biopsy area of 2.8 mm2.
Pajares et al14 conducted a prospective randomised trial comparing TBCLB with TBB and demonstrated a mean specimen size for TBCLB samples of 14.7 mm2 (versus 3.3 mm2 for TBB biopsy, p<0.001), resulting in a histological diagnosis in 74.4% of patients versus 34.2% in the TBB group (p<0.001). When comparing this technique with surgical lung biopsy there are various potential advantages. General anaesthesia is not necessary and the procedure can be performed as a day case with uncomplicated cases returning home the same day. Future clinical trials and an increase in real world experience of TBCLB is likely to cement its use for selected cases, potentially reducing the number of surgial lung biopsies performed.
Surgical lung biopsy
Surgical lung biopsies are the current gold standard for obtaining histological material in the diagnosis of clinically and radiologically unclassifiable ILD. It is usually performed via the less invasive video-assisted thoracoscopic surgical (VATS) approach. As previously described, using the criteria set out in the 2011 ATS/ERS consensus statement, about two thirds of IPF cases can be diagnosed on the basis of typical clinical and radiological findings of UIP (Fig 5); however, only 7.5–12% of suspected IPF patients undergo surgical lung biopsy in the UK.21 This reflects clinicians’ reluctance to refer patients for a procedure associated with a significant mortality and morbidity.
Fig 5.

Diagnostic algorithm for idiopathic pulmonary fibrosis, in part adapted from the ATS/ERS consensus statement. HRCT = high-resolution computerised tomography; ILD = interstitial lung disease; IPF = idiopathic pulmonary fibrosis; UIP = usual interstitial pneumonitis
The average hospital stay associated with VATS biopsy is 2–4 days,22 with mortality rates of 3–4% and an overall complication rate of up to 16%.23 Common complications include persistent air leak, exacerbations of underlying ILD due to mechanical stress of single lung ventilation, bleeding and delayed wound healing. In addition, 57% of patients report pain at the incision site 6–12 months after surgery.24 It is also worth remembering that surgical lung biopsy does not guarantee a definite pathological diagnosis, with diagnosis rates ranging from 34–100%.22,23
Multidisciplinary team
Taking into consideration the various investigations involved in ILD diagnosis it is clear that no single diagnostic test can provide a confident answer. A consensus approach by a multidisciplinary team (MDT) with expertise in ILD is thus considered the gold standard (Fig 6). The MDT integrates all available data at several stages of the work up. This not only improves inter-observer agreement and diagnostic confidence, but may also prevent unnecessary surgical biopsies, while identifying patients in whom a biopsy may effectively contribute to the diagnosis.25 Current NICE guidelines recommend that IPF should only be diagnosed by MDT consensus and stipulates a minimum MDT composition.26
Fig 6.

The role of specialist multidisciplinary teams and specialist referral centres in the diagnosis and management of interstitial lung disease. CTD = connective tissue disease; ILD = interstitial lung disease; IPF = idiopathic pulmonary fibrosis; LAM = lymphangioleiomyomatosis; LCH = Langerhans cell histiocytosis; MDT = multidisciplinary team; NSIP = non-specific interstitial pneumonitis
Novel therapies in idiopathic pulmonary fibrosis
There has been a dramatic increase in clinical trial activity in IPF in recent years, with the discovery and approval of two new anti-fibrotic drugs (pirfenidone and nintedanib) heralding a new era in the disease. While these novel anti-fibrotic agents have been shown to slow the decline in forced vital capacity (FVC), they neither halt progression nor reverse existing fibrosis. In part because of considerable cost, their use is restricted by NICE to patients fulfilling certain criteria – namely a predicted FVC of 50–80%, thereby excluding patients at each extreme of the disease process and those with spuriously maintained FVC due to concurrent emphysema. Given these restrictions, as well as the limitations of these therapies, the importance of non-pharmacological therapy, such as pulmonary rehabilitation, plus the enrolment of patients into clinical trials (Fig 7) should not be underestimated.
Fig 7.

Schematic interstitial lung disease treatment algorithm. *No robust evidence for managing exacerbations with variation between centres, should be discussed with specialist centre if possible. BAL = bronchoalveolar lavage; GOR = gastro-oesophageal reflux; HRCT = high-resolution computerised tomography; ILD = interstitial lung disease; IPF = idiopathic pulmonary fibrosis; MDT = multidisciplinary team; PFT = pulmonary function test; PHT = pulmonary hypertension
Novel biomarkers
The need to distinguish the different idiopathic interstitial pneumonias has driven the search for novel diagnostic biomarkers. In addition, there are marked survival differences even within specific groups, such as IPF. Biomarkers that can identify these phenotypes are needed for clinical decision making, but they also have the potential to aid cohort enrichment in clinical trials. Previous landmark studies have beautifully illustrated this need, with variable rates of decline in placebo arms leading to inconsistent results and a delay in drugs being approved.21
To date, efforts have focused on serum biomarkers27 that are relatively easy to access and novel imaging modalities that potentially inform on disease activity within the lung. In particular, positron emission tomography (PET) allows non-invasive measurement of cellular metabolism in vivo. The 18F-fluorodeoxyglucose (18F-FDG) PET signal is consistently raised in ILD28 and is both stable and reproducible (Fig 8). In a population of over 200 ILD patients, we have shown that baseline measures of 18F-FDG uptake on PET are related to patient survival in both IPF and other idiopathic interstitial pneumonias (unpublished data). High pulmonary 18F-FDG uptake is associated with poor survival, giving additional information to pulmonary function testing and, thus, could become a valuable part of the initial work up in newly diagnosed patients.
Fig 8.
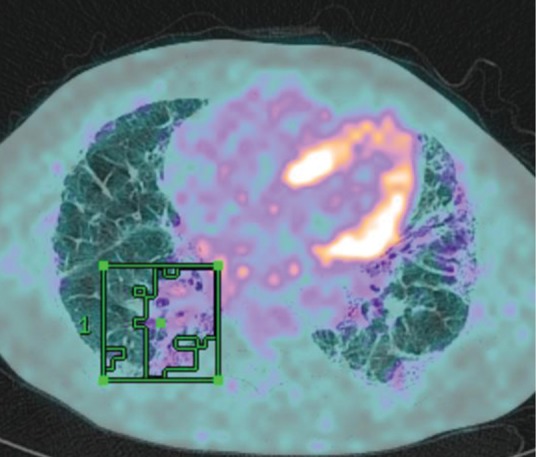
Positron emission tomography signal superimposed on high-resolution computerised tomography scan of a patient with usual interstitial pneumonitis. A region of interest has been drawn around the area of highest standardised uptake value.
Conclusion
The diagnosis of ILD is a challenging and involved process. It relies on detailed history taking and the integration of various investigations and specialties. The relative rarity of these diseases makes distinguishing subtypes even more difficult for clinicians with a mixed respiratory case workload and, thus, infrequent exposure to ILD. Having said this, the incidence of ILD is increasing and there is potential for specialist centres to become overwhelmed with patients, putting a greater than ever emphasis on collaboration with referring centres and a concerted effort to employ a hub and spoke model. This has the added advantage of facilitating a more patient-centred approach, minimising the need for unnecessary travel and facilitating access to ancillary local services, such as pulmonary rehab, oxygen providers and palliative care services.
Irrespective of expertise, uncertainty is inherent in the diagnosis of these diseases, although arguably encountering ILD on a frequent basis allows the physician to become more comfortable with these uncertainties, thus embracing the concept of continuous diagnostic review. The hope remains that, in time, reliable, non-invasive biomarkers will identify disease subtypes, predict prognosis and potentially replace the need for biopsy. Much of the heterogeneity seen in IPF may be explained by the existence of endotypes, in other words, mechanistically different disease subtypes, which consequently exhibit very different responses to therapy. Therefore, future treatments have the potential to be greatly influenced by identifying these groups through the use of genetic testing and a move towards personalised disease management.
Conflicts of interest
The authors have no conflicts of interest to declare.
Note
This article was originally published in the 2016 Clinical Medicine supplement Horizons in Medicine 28. All articles in this supplement are available online at www.clinmed.rcpjournal.org/content//16/Suppl_6
References
- 1.British Lung Foundation Lung disease in the UK – big picture statistics. https://statistics.blf.org.uk/lung-disease-uk-big-picture. [Accessed 19 October 2016]
- 2.Gribbin J. Hubbard RB. Le Jeune I, et al. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006;61:980–5. doi: 10.1136/thx.2006.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Idiopathic Pulmonary Fibrosis Clinical Research Network Raghu G. Anstrom KJ, et al. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968–77. doi: 10.1056/NEJMoa1113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tashkin DP. Elashoff R. Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354:2655–66. doi: 10.1056/NEJMoa055120. [DOI] [PubMed] [Google Scholar]
- 5.Keir GJ. Maher TM. Ming D, et al. Rituximab in severe, treatment-refractory interstitial lung disease. Respirology. 2014;19:353–9. doi: 10.1111/resp.12214. [DOI] [PubMed] [Google Scholar]
- 6.Oldham JM. Ma S-F. Martinez FJ, et al. TOLLIP, MUC5B, and the response to N-acetylcysteine among Individuals with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2015;192:1475–82. doi: 10.1164/rccm.201505-1010OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raghu G. Collard HR. Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohshimo S. Bonella F. Cui A, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:1043–7. doi: 10.1164/rccm.200808-1313OC. [DOI] [PubMed] [Google Scholar]
- 9.Wall CP. Gaensler EA. Carrington CB. Hayes JA. Comparison of transbronchial and open biopsies in chronic infiltrative lung diseases. Am Rev Respir Dis. 1981;123:280–5. doi: 10.1164/arrd.1981.123.3.280. [DOI] [PubMed] [Google Scholar]
- 10.Shim HS. Park MS. Park IK. Histopathologic findings of transbronchial biopsy in usual interstitial pneumonia. Pathol Int. 2010;60:373–7. doi: 10.1111/j.1440-1827.2010.02528.x. [DOI] [PubMed] [Google Scholar]
- 11.Babiak A. Hetzel J. Krishna G, et al. Transbronchial cryobiopsy: a new tool for lung biopsies. Respiration. 2009;78:203–8. doi: 10.1159/000203987. [DOI] [PubMed] [Google Scholar]
- 12.Fruchter O. Fridel L. El Raouf BA, et al. Histological diagnosis of interstitial lung diseases by cryo-transbronchial biopsy. Respirology. 2014;19:683–8. doi: 10.1111/resp.12296. [DOI] [PubMed] [Google Scholar]
- 13.Casoni GL. Tomassetti S. Cavazza A, et al. Transbronchial lung cryobiopsy in the diagnosis of fibrotic interstitial lung diseases. PLoS One. 2014;9:e86716. doi: 10.1371/journal.pone.0086716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pajares V. Puzo C. Castillo D, et al. Diagnostic yield of transbronchial cryobiopsy in interstitial lung disease: a randomized trial. Respirology. 2014;19:900–6. doi: 10.1111/resp.12322. [DOI] [PubMed] [Google Scholar]
- 15.Griff S. Schönfeld N. Ammenwerth W, et al. Diagnostic yield of transbronchial cryobiopsy in non-neoplastic lung disease: a retrospective case series. BMC Pulm Med. 2014;14:171. doi: 10.1186/1471-2466-14-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fruchter O. Fridel L. Rosengarten D, et al. Transbronchial cryobiopsy in lung transplantation patients: first report. Respirology. 2013;18:669–73. doi: 10.1111/resp.12037. [DOI] [PubMed] [Google Scholar]
- 17.Kropski JA. Pritchett JM. Mason WR, et al. Bronchoscopic cryobiopsy for the diagnosis of diffuse parenchymal lung disease. PLoS One. 2013;8:e78674. doi: 10.1371/journal.pone.0078674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pajares V. Torrego A. Puzo C, et al. [Transbronchial lung biopsy using cryoprobes] Arch Bronconeumol. 2010;46:111–5. doi: 10.1016/j.arbres.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 19.Jabbardarjani H. Kiani A. Karimi M. Fakhri M. Cryobiopsy shows better safety profile and diagnostic yield as compared to conventional forceps for interstitial lung disease. Eur Respir J. 2014;42(Suppl 57):P2301. [Google Scholar]
- 20.Hagmeyer L. Theegarten D. Wohlschläger J, et al. Transbronchial cryobiopsy and open lung biopsy in interstitial lung disease: Comparison of two diagnostic tools. Eur Respir J. 2014;42(Suppl 57):P2303. [Google Scholar]
- 21.Noble PW. Albera C. Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377:1760–9. doi: 10.1016/S0140-6736(11)60405-4. [DOI] [PubMed] [Google Scholar]
- 22.Morris D. Zamvar V. The efficacy of video-assisted thoracoscopic surgery lung biopsies in patients with interstitial lung disease: a retrospective study of 66 patients. J Cardiothorac Surg. 2014;9:45. doi: 10.1186/1749-8090-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaarteenaho R. The current position of surgical lung biopsy in the diagnosis of idiopathic pulmonary fibrosis. Respir Res. 2013;14:43. doi: 10.1186/1465-9921-14-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maguire MF. Ravenscroft A. Beggs D. Duffy JP. A questionnaire study investigating the prevalence of the neuropathic component of chronic pain after thoracic surgery. Eur J Cardiothorac Surg. 2006;29:800–5. doi: 10.1016/j.ejcts.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Flaherty KR. Andrei A-C. King TE, et al. Idiopathic interstitial pneumonia: do community and academic physicians agree on diagnosis? Am J Respir Crit Care Med. 2007;175:1054–60. doi: 10.1164/rccm.200606-833OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lynch DA. Godwin JD. Safrin S, et al. High-resolution computed tomography in idiopathic pulmonary fibrosis: diagnosis and prognosis. Am J Respir Crit Care Med. 2005;172:488–93. doi: 10.1164/rccm.200412-1756OC. [DOI] [PubMed] [Google Scholar]
- 27.Jenkins RG. Simpson JK. Saini G, et al. Longitudinal change in collagen degradation biomarkers in idiopathic pulmonary fibrosis: an analysis from the prospective, multicentre PROFILE study. Lancet Respir Med. 2015;3:462–72. doi: 10.1016/S2213-2600(15)00048-X. [DOI] [PubMed] [Google Scholar]
- 28.Win T. Thomas BA. Lambrou T, et al. Areas of normal pulmonary parenchyma on HRCT exhibit increased FDG PET signal in IPF patients. Eur J Nucl Med Mol Imaging. 2014;41:337–42. doi: 10.1007/s00259-013-2514-8. [DOI] [PMC free article] [PubMed] [Google Scholar]