

Abstract
Physiologically, retinal pigment epithelium (RPE) expresses high levels of CD73 in their membrane, converting AMP to immune suppressive adenosine, mediates an anti-inflammatory effect. However, after being exposed to inflammatory factors, RPE rapidly becomes CD73-negative cells, which render RPE’s immune suppressive function and accelerate local inflammation. Here, we investigated the mechanism leading to the loss of membrane CD73 in RPE. We found the controversy that when membrane CD73 was significantly diminished in inflammatory RPE, Cd73 mRNA levels were not changed at all. It was further verified that, matrix metalloproteinase-9 (MMP-9) mediated the shedding of CD73 from the cell membrane of inflammatory RPE by catalyzing its K547/F548 site. However, MMP-9 could not catalyze uncomplexed CD73, the interaction of CD73 with adenosine receptor A1 subtype (ARA1) is necessary for being catalyzed by MMP-9. After being treated by LPS and TNF-α, the formation of CD73/ARA1 complex in RPE was verified by co-immunoprecipitation and FRET-based assays. It was also revealed that CD73 need to be localized in lipid rafts to be capable of interacting with ARA1, since CD73/ARA1 interaction and CD73 shedding were completely blocked by the addition of lipid raft synthesis inhibitor. As a conclusion, multiple steps are involved in CD73 shedding in RPE, including up-regulation of MMP-9 activity, localization of CD73 in lipid rafts, and the formation of CD73/ARA1 complex. Lipid rafts committed CD73 with high mobility, shuttled CD73 to ARA1 to form a complex, which was capable of being recognized and catalyzed by MMP-9.
Electronic supplementary material
The online version of this article (10.1007/s11302-018-9628-1) contains supplementary material, which is available to authorized users.
Keywords: CD73, Shedding, Lipid rafts, MMP-9, Adenosine receptor A1
Introduction
Ecto-5′-nucleotidase (CD73) is a critical enzyme involved in the modulation of purinergic signaling. Under pathological conditions, such as hypoxia and inflammation, adenosine triphosphate (ATP) was released out of cells. If existed two membrane located enzymes, CD39 and CD73, could quickly convert pro-inflammatory ATP to anti-inflammatory adenosine. Firstly, CD39 converts ATP via ADP to AMP; then, CD73 converts AMP to adenosine. Consequently, CD73 participate in immune regulation by controlling adenosine production in tissues and organs [1–4]. Physiologically, CD73 is located on the surface of some immune regulatory cells, including regulatory T (Treg) cells, dendritic cells, and T cell receptor (TCR)γδ T cells [5–7], to create an environment with relatively high adenosine concentration for maintaining immune homeostasis. Under pathological conditions, CD73-positive cells may change to CD73-negative; and which is considered as a pro-inflammatory transformation. It hindered the immune suppressive effect of adenosine by decreasing its local production.
We had previously reported that CD73 is highly expressed on the surface of naïve retinal pigment epithelium (RPE) cells. However, when local inflammation existed or upon the treatment of inflammatory factors, RPE cells lost surface CD73, leading to significantly inhibited local adenosine production. Decreased environmental adenosine concentration hindered its immune suppressive effect and promoted experimental autoimmune uveitis (EAU) [8]. Clarifying the mechanism inducing the loss of membrane CD73 in RPE cells could help us understanding local immune disorders as well as finding novel therapeutic target. This may also benefit the study of other fields, since in addition to its role in autoimmune diseases [9–12], CD73 also has great impact in oncology as well [13–16].
Materials and methods
Materials
Female C57BL/6 (B6) and ARA1−/− and CD73−/− mice on a B6 background (all 12 to 14 weeks old) were purchased from the National Resource Center for Mutant Mice of China (NRCMM) (Nanjing, Jiangsu, China). The mice were housed and maintained in the animal facilities of Tianjin Medical University. Institutional approval was obtained, and institutional guidelines regarding animal experimentation were followed. Phycoerythrin (PE)-labeled anti-mouse RPE65 and fluorescein isothiocyanate (FITC)-labeled anti-mouse CD73 were purchased from eBioscience (ThermoFisher Scientific, Beijing, China). Phosphatidylinositol-specific phospholipase C (PI-PLC) inhibitor U-73122 was purchased from Medkoo Bioscience, Inc. (MedKoo, USA), protease inhibitor cocktail (PIC) and a Biotrak MMP-9 activity assay system, the product of GE Healthcare (code RPN2634), were purchased from Sigma (Sigma-Aldrich, USA). OptiPrep Density gradient medium was purchased from STEMCELL Technologies (Shanghai, China). Unlabeled anti-mouse CD73 (ab175396) and anti-mouse ARA1 polyclonal antibody (ab151523) were purchased from Abcam (Shanghai, China), and labeled with enhanced green fluorescent protein 2 (EGFP2) or red fluorescent protein (RFP) by GL Biochem Ltd. (Shanghai, China).
RPE isolation and culture
Using a previously published method [17], primary mouse RPE cells were isolated from naïve (either wild-type or Cd73−/−) mice with B6 genetic background and experimental autoimmune uveitis (EAU)-induced B6 mice. Briefly, after the anterior segment of the eye and lens were removed, eyecups were incubated in 0.5% (w/v) trypsin containing 0.25% (w/v) ethylenediaminetetraacetic acid (EDTA) at 37 °C for 1 h. Cells were harvested through centrifugation, and the supernatant was carefully aspirated. For RNA isolation, the cells were directly suspended in TRIzol reagent. For further in vitro culture, single-cell suspensions were seeded onto culture dishes containing Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 100 μg/ml primocin. Cells were passaged at 80–90% confluence, and the cell phenotype was confirmed by RPE65 staining [18]. EAU induction was performed according to a previously published method. Briefly, following an intraperitoneal (i.p.) injection of 300 ng of pertussis toxin (PTX), B6 mice were subcutaneously injected with a 200 μl emulsion containing 200 μg IRBP1-20 in complete Freund’s adjuvant (CFA) at six spots on the flank.
CD73 modification of Cd73−/− RPE
Firstly, Wt Cd73 cDNA was cloned into a His-tagged expression vector of pCMV6-AN-His to obtain a recombinant vector of pCMV-His-wtCd73. Mutation to the CD73 coding sites of K547-F548 was introduced by site-directed mutagenesis, to obtain another recombinant construct of pCMV-His-mutCd73. Both pCMV-His-wtCd73 and pCMV-His-mutCd73 were transfected into cultured Cd73−/− RPE by Lipofectamine 2000. Transfected Cd73−/− RPE cells with stably expressed Wt- and Mut-CD73 were screened by G418 antibiotic selection [19]; the expression of His-tagged CD73 was verified by western blotting using an anti-CD73 polyclonal antibody from Abcam (ab175396).
CD73 detection in RPE cells and their culture medium
Primary cultured RPE cells were treated with lipopolysaccharide (LPS; 50 ng/ml) and tumor necrosis factor (TNF)-α (2000 ng/ml) separately or in combination, with or without PLC or MMPs inhibitors in the presence. Cell surface expressed CD73 was tested by flow cytometry analysis (FACS) and western blotting. For FACS assay, cultured RPE cells were harvested after EDTA treatment, washed twice with phosphate-buffered saline (PBS), stained with PE-labeled anti-mouse RPE65 and FITC-labeled anti-mouse CD73 antibodies for 1 h at room temperature. The cells were then washed twice, suspended in PBS, and subjected to flow cytometry analysis in a BD FACSCalibur. For western blotting, cell membrane protein was extracted; 100 μg total proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) separation, nitrocellulose membrane transfer, and antibody blotting. Na+/K+ ATPase in the same samples were blotted as well, served as reference. Cd73 mRNA was isolated from differently treated RPE cells, detected by real-time qPCR with Gapdh as a reference. Meanwhile, the physical existence and enzymatic activity of CD73 in the medium of cultured RPE cells, in particular the LPS/TNF-α-treated RPE, was detected by western blot. Medium samples were concentrated by ultra-centrifuge firstly; an aliquot of concentrate corresponding to 1/3 volume of the original medium was subjected to SDS-PAGE and western blot.
CD73 activity assay
The conversion of AMP to adenosine was measured to assess the enzymatic activity of CD73 in RPE cells and concentrated mediums. Adenosine was detected by a HPLC-based method which was published by us before [8]; and all the detection was performed with EHNA (50 μM), a potent adenosine deaminase (ADA) inhibitor, in the presence to exclude possible adenosine degradation. For cell samples, 5 × 104 cells were used to catalyze AMP substrate at the concentration of 1 mM in 100 μl DPBS buffer; after 1 h incubation at 37 °C, adenosine in the reaction was detected by HPLC. CD73 activity were represented as “generated adenosine (μmol)/105 cells/per hour.” For evaluating the enzymatic activity of solubilized CD73, the medium samples were concentrated by ultracentrifuge. Concentrated medium containing same amount of CD73 as normal RPE cells was subjected to the activity assay. The activities of two forms of solubilized CD73 in the medium, MMP-9 derived C-terminal truncated CD73 phosphatidylinositol specific phospholipase C (PI-PLC) released intact form of solubilized CD73 were compared.
mRNA expression and active form of MMP-9 detection
Inside cell expression and out of cell activity of MMP-9 were evaluated by real-time PCR and biotrack activity assay. Total RNA was extracted from 1X106 RPE cells; 1.0 μg total RNA was reverse transcribed to cDNA and its relative amount was determined by real-time PCR with Gapdh as the reference. The activity of secreted MMP-9 in the medium was evaluated by a MMP-9 Biotrak activity system (GE Healthcare, RPN2634) following manufacture’s protocol. Medium samples were two times diluted with assay buffer and tested in triplicate. Briefly, 100 μl of sample was added to anti-MMP-9 antibody coated 96-well microplate, kept at 4 °C overnight, washed and added 100 μl of detection enzyme solution containing pro form of an inactive detection enzyme that can be activated by the captured active MMP-9, and incubated at 37 °C for 2 h. Then MMP-9-activated detection enzyme was measured using a specific chromogenic peptide substrate, and the resultant color is read at 405 nm. The concentration of active MMP-9 in each sample was determined by interpolation from a standard curve.
Co-immunoprecipitation and identification of CD73-associated protein
For co-immunoprecipitation, Mut-CD73-modified Cd73−/− RPE cells were treated with LPS/TNF-α or left untreated. Cells were harvested, and the membrane protein was extracted. The membrane extract was washed with cold PBS, suspended in lysis buffer (20 mM Tris HCl pH 8.0, 137 mM NaCl, 0.5% Nonidet P-40, 2 mM EDTA, and proteinase inhibitor cocktails), and maintained constant agitation for 2 h at 4 °C. After being centrifuged for 1 h, 100,000×g at 4 °C, the supernatant was aspirated to a new tube. The supernatant was incubated with anti-His antibodies at 4 °C for 6 h under gentle agitation. Protein A/G agarose beads were added to the solution and incubated at 4 °C for another 4 h under agitation. The mixture was washed with PBS and centrifuged. The pellet was suspended in SDS-PAGE loading buffer and subjected to SDS-PAGE analysis. The gel was stained with Coomassie brilliant blue R250. The unknown band which was pulled down with CD73 in LPS/TNF-α-treated RPE cells was transferred to a polyvinylidene fluoride (PVDF) membrane and send out for peptide sequencing. The protein was fractioned by enzyme digestion prior to sequencing. BLAST results indicated that the unknown protein was likely to be ARA1. Western blotting against ARA1 was performed for additional confirmation.
Lipid raft isolation
Lipid rafts were isolated from RPE cells following published methods [20] with or without detergent in the presence; and all procedures were carried out on ice. RPE cells in two D150 cell culture dishes were washed, treated with EDTA, and scraped into preparation buffer (20 mM Tris-HCl and 250 mM sucrose, pH 7.8 with freshly added 1 mM CaCl2, and 1 mM MgCl2). For detergent free lipid rafts isolation, the cells were pelleted by centrifugation for 2 min at 250×g and resuspended in 1 ml preparation buffer containing proteinase inhibitors cocktail (0.2 mM aminoethyl-benzene sulfonyl fluoride, 1 μg/ml aprotinin, 10 μM bestatin, 10 μg/ml leupeptin, 2 μM pepstatin, and 50 μg/ml calpain inhibitor I). The cells were then lysed by passage through a 22 gauge needle repeatedly. For detergent method, cell pellet was suspended in the buffer as above with the addition of 1% Triton X-100, kept on ice for 30 min, and then lysed by relatedly gauge passage. All the lysates were centrifuged at 1000×g for 10 min, the supernatant was collected to a separate tube. An equal volume of preparation buffer containing 50% OptiPrep was mixed with the supernatant to obtain a 25% final OptiPre concentration and placed in the bottom of a 12-ml centrifuge tube, and an 8 ml gradient of 0 to 20% OptiPrep in preparation buffer was poured on top. The gradients were centrifuged for 90 min at 52,000×g in a Beckman ultracentrifuge. After centrifugation, a diffuse band was observed approximately one third of the way down the gradient, and a distinct band was apparent at the interface of the 20% end of the gradient and the 25% OptiPrep bottom layer. Gradients were fractionated into 0.67-ml fractions, most lipid raft markers and glycosylphosphatidylinositol (GPI)-anchored proteins were expected to be enriched in fractions 2–3.
FRET detection of CD73-ARA1 interactions
The mechanism for detecting CD73 and ARA1 interactions by FRET is illustrated in Fig. 8. RPE cells were seeded in 96-well cell culture plates and differentially treated, and FRET test was kinetically performed at different times posterior to the treatment. Briefly, EGFP2-labeled anti-CD73 and RFP-labeled anti-ARA1 antibodies were added to the culture to a final concentration of 10 ng/ml of each, maintained in an incubator for 3 h, and thoroughly washed with cold PBS to remove free fluorescence. The intensity of red fluorescence was detected by a fluorometer when the wells were excited at different wavelengths, 483, 506, and 555 nM. The cells were also stained with 4′ 6-diamidino-2-phenylindole (DAPI) to reveal the nuclei when EGFP2, RFP, and FRET were visualized at different excitation wavelength under fluorescence microscopy.
Fig. 8.

Illustration of FRET for detecting CD73 ARA1 association. Anti-CD73 antibody, labeled with EGFP2, was used as the donor fluorescent molecule (excitation maximum/emission maximum, 483/506 nM). Anti-ARA1 antibody, labeled with RFP, was used as the receptor fluorescent molecule (555/584 nM). When excited at 483 nM, EGFP2 generates green fluorescence with an emission wavelength spectrum ranging from 470 to 600 nm, which overlaps RFP’s excitation wavelength spectrum ranging from 470 to 610 nM. In untreated RPE cells, ARA1 is poorly expressed, and CD73 is inert without being localized to lipid rafts. These proteins are separated in the cell membrane. When the donor fluorescence excitation wavelength (483 nM) was applied, the emission energy from the donor could not be utilized by the receptor due to relatively large distance, and red fluorescence could not be visualized. Following LPS/TNF-α treatment, ARA1 expression was significantly increased, and the lipid rafts provided CD73 with high mobility. These facilitated the association of CD73 with ARA1. Once closely interacted, donor-emitted energy can be transferred to the receptor, and excited red fluorescence emission. It could be determined whether CD73 and ARA1 were closely interacted by detecting FRET’s fluorescence intensity
Results
Diminished membrane localization but unchanged mRNA expression of CD73 in treated RPE
Western blotting results in Fig. 1a showed that, when compared with naïve RPE cells, EAU-RPE cells exhibited significantly decreased amounts of CD73 in their membrane. Figure 1b further confirmed the possibility of losing membrane CD73 in RPE. Naïve RPE initially had high levels of membrane CD73, and which changed to almost undetectable after LPS/TNF-α treatment. However, qPCR result in Fig. 1c revealed an unchanged Cd73 mRNA expression in EAU-RPE vs naïve-RPE or LPS/TNF-α-treated vs untreated RPE.
Fig. 1.

CD73 expression in differently treated RPE cells. RPE cells were isolated from naïve and EAU-induced B6 mice. Amount of CD73 in membrane extract was determined by western blotting with Na+/K+ ATPase as reference (a). Naïve RPE cells were cultured, treated with LPS (50 ng/ml) and TNF-α (2000 ng/ml). Membrane CD73 was determined by western blot (b); Cd73 mRNA was detected by qPCR (c). a, b A representative western blotting result (left column) and optical density analysis (right column, n = 6). c qPCR determined Cd73 mRNA in freshly isolated RPE and cultured RPE (n = 6)
MMP catalyzes CD73 shedding
FACS results in Fig. 2a indicated that LPS/TNF-α induced CD73 diminish from RPE membrane was largely inhibited by the addition of proteinase inhibitor cocktail (PIC). Figure 2b revealed that LPS/TNF-α treatment or the addition of PIC din not change Cd73 mRNA expression. Two potential targets responsible for CD73 shedding were graphically shown in Fig. 2c. One is the GPI anchor which could be catalyzed by PI-PLC; and then released CD73. The other site is the 547th and 548th amino acid residues in CD73 molecule, where there is an anticipated MMP catalyzing site. Figure 2d indicated that the shedding of CD73 from LPS/TNF-α-treated RPE could be completely blocked by the addition of a general MMPs inhibitor, but not a PLC inhibitor.
Fig. 2.
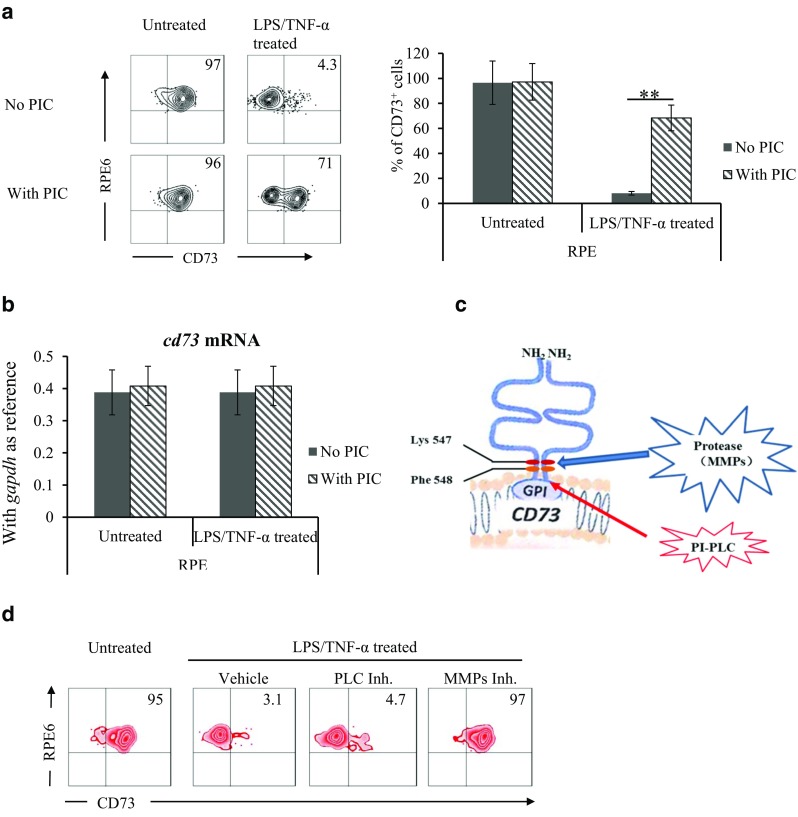
Matrix metalloproteinase catalyzes CD73. In vitro cultured RPE cells were treated with LPS/TNF-α, with protease inhibitor cocktail (PIC) in the presence or absence. Surface CD73 and Cd73 mRNA expression were tested by flow cytometry analysis (a) and qPCR (b). Effects of PI-PLC and MMP inhibitors on LPS/TNF-α-induced CD73 shedding was compared (d). a A representative flow cytometry result (left column), statistical analysis of the percentage of CD73+ cells (right column, n = 6). b Cd73 mRNA in isolated RPE and cultured RPE cells (n = 6). c Potential catalyzing sites for the GPI anchored CD73. d Effects of PI-PLC and MMP inhibitors on CD73 shedding
MMP-9 catalyzes CD73 shedding at the K547/F548 site
The upper panel of Fig. 3a clearly showed that either selective MMP-9 inhibitor or semi-selective MMP-2/9 was capable of blocking the catalysis of CD73, while selective MMP-2 inhibitor could not. The lower panel of Fig. 3a showed that MMP-9 inhibitor blocked CD73 shedding in a dose-dependent manner. Figure 3b indicated the difference of amino acid residues in Wt- and Mut-CD73; those were expressed in Cd73−/− RPE. Western blotting results in Fig. 3c confirmed membrane localization of exogenously expressed CD73 (Wt- and Mut-). Figure 3d indicated that following LPS/TNF-α treatment Wt-CD73 was diminished from Cd73−/− RPE; however, the membrane localization of Mut-CD73 was completely maintained.
Fig. 3.
MMP-9 catalyzes CD73 shedding at the K547-F548 site. Selective MMP-9, MMP-2 inhibitors, and semi-selective MMP2/9 inhibitors were added to LPS/TNF-α-treated RPE cells, their effects on regulating membrane-expressed CD73 were analyzed by flow cytometry (a). Mutant and wild-type CD73 (b) were expressed in Cd73−/− RPE cells (c), their responses to LPS/TNF-α treatment were tested (d). a Effects of selective MMP-2, MMP-9 inhibitors on protecting CD73 shedding (upper panel), and the dose-dependent effect of selective MMP-9 inhibitors (lower panel). b Coding sequence and amino acid residues at 547–548 sites in wild-type and mutant CD73. c Expression of Wt- and mutant-CD73 in Cd73−/− RPE cells was verified by western blotting. d Responses of Wt- and mutant-CD73 to LPS/TNF-α treatment were tested by flow cytometry
Inactive truncated CD73 in the medium
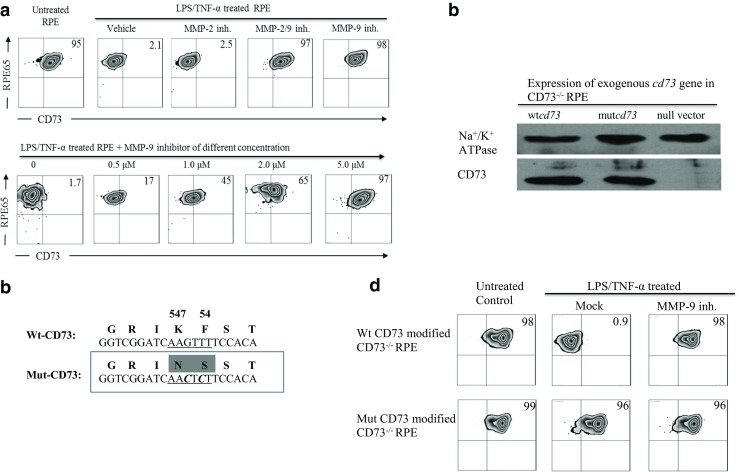
To further confirm the existence of CD73 shedding, the detection of solubilized CD73 in the medium of LPS/TNF-α-treated RPE was performed. Results from the samples of membrane extract (Fig. 4a, upper panel) and concentrated medium (lower panel) revealed that LPS/TNF-α treatment significantly decreased membrane CD73, and generated detectable CD73 in the medium. This was blocked by the addition of MMP-9 inhibitor. Figure 4b showed that LPS/TNF-α-treated RPE had significantly diminished enzymatic activity of converting 5’-AMP to adenosine; this was completely restored by the addition of MMP-9 inhibitor. Although Fig. 4a (lower panel) showed the existence of CD73 molecule in concentrated medium from LPS/TNF-α-treated RPE, its enzymatic activity remained undetectable (Fig. 4c). Our supplemental result indicated that different from MMP-9-derived truncated CD73, PI-PLC-released intact form of solubilized CD73 was verified of obviously detectable enzymatic activity.
Fig. 4.
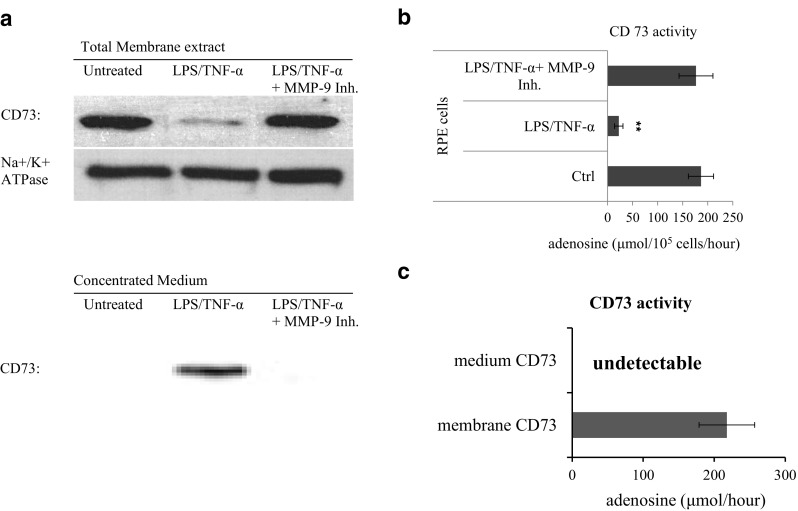
CD73 in the medium of treated RPE. Medium was collected from differently treated RPE cells and concentrated by ultra-centrifuge. Physical existence of CD73 molecule was detected by western blotting (a). CD73’s enzymatic activities were compared between live cells (b) and concentrated medium (c), accessed by their ability of converting AMP to adenosine. a Western blotting assay of CD73 in membrane extract (upper) and concentrated medium (lower). b Cd73’s enzymatic activity in different treated RPE cells (n = 3). c Cd73’s enzymatic activity in concentrated medium (n = 3)
Up-regulated MMP-9 in treated RPE
To clarify why CD73 shedding only occurred to LPS/TNF-α-treated but not untreated RPE cells, we evaluated the regulatory effect of LPS and TNF-α on Mmp-9 expression and the amount of active MMP-9 in the medium. Results shown in Fig. 5a revealed that either LPS or TNF-α could individually up-regulate Mmp-9 expression to some extent, and an additive effect was detected. In consistence with elevated mRNA expression, LPS and TNF-α treatment significantly increased the amount of active MMP-9 in the medium (Fig. 5b) as well. When exogenous recombinant MMP-9 was added to mmp-9−/− RPE, the interesting results in Fig. 5c indicated that the addition of MMP-9 could only induce CD73 shedding in LPS/TNF-α-treated but not untreated mmp-9−/− RPE cells.
Fig. 5.

Up-regulated MMP-9 is not uniquely responsible for CD73 shedding. LPS and TNF-α, individually or in combination, on regulating MMP-9 expression extracellular activity were detected by qPCR (a) and a MMP-9 Biotrak activity assay (b). Effect of exogenous MMP-9 on CD73 shedding in Mmp9−/− RPE cells received LPS/TNF-α treatment or not (c). a qPCR results of Mmp-9 expression in RPE cells (n = 6). b Amount of active MMP-9 in the medium of RPE cells. c Effect of exogenous recombinant MMP-9 on regulating CD73 shedding; a representative flow cytometry result (left column) and statistical analysis of FACS determined percentage of CD73 positive cells (right column)
The association of ARA1 and CD73 is necessary for CD73 shedding
To determine whether a specific protein is associated with CD73 after LPS/TNF-α treatment, we performed co-immunoprecipitation. An anti-His antibody was used as the capture antibody to capture His tagged Mut-CD73. The SDS-PAGE result in Fig. 6a clearly showed an extra band was pulled down together with Mut-CD73 in LPS/TNF-α-treated RPE. Control test, no addition of anti-His antibody, was performed in parallel, and neither CD73 band nor unknown band could be visualized in SDS-PAGE (data not shown). The amino acid sequencing blast results, shown in Fig. 6b, indicated that this unknown CD73-associated protein was likely to be adenosine receptor A1 subtype (ARA1). The western blotting results (Fig. 6c) showed that the unknown protein could be detected by anti-ARA1 antibody. Although the CD73-ARA1 association was confirmed in LPS/TNF-α-treated RPE cells, it is still unclear whether ARA1 plays critical role in regulating CD73 shedding. Fig. 6d showed LPS are effective on up-regulating Ara1 mRNA expression in RPE cells, and it was confirmed in Fig. 6e that LPS/TNF-α-induced CD73 shedding was completely abolished in Ara1−/− RPE cells.
Fig. 6.
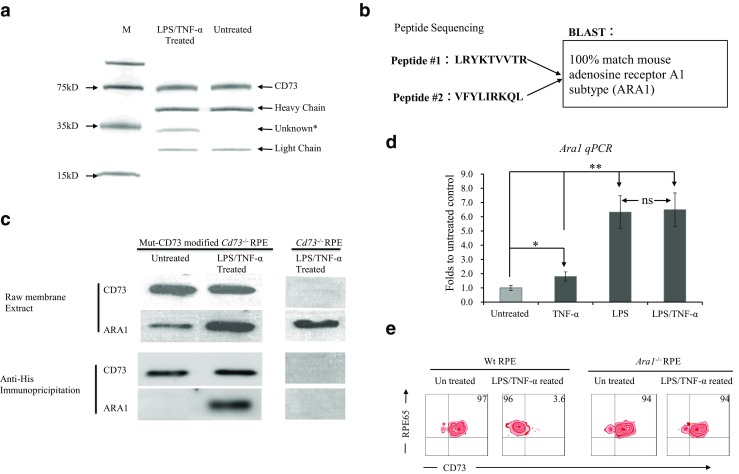
ARA1 is necessary for CD73 shedding. An anti-His antibody, recognizing His tagged recombinant CD73, was used to pull down CD73 and its associated proteins. Co-immunoprecipitate from mutant CD73-modified Cd73−/− RPE cells was analyzed by SDS-PAGE (a). The CD73-associated protein was fractioned and subjected to amino acid sequencing of its N terminal; the sequencing result was blasted (b). The pull-down protein was identified by western blotting (c). Ara1 mRNA expression in LPS- and TNF-α-treated RPE cells was determined by qPCR (d). CD73 shedding in Ara1−/− RPE was tested by flow cytometry (e). a SDS-PAGE result of the co-immunoprecipitate from mutant CD73-modified Cd73−/− RPE. b Amino acid sequencing and BLAST results of fractioned CD73-associated protein. c Western blot identification of CD73-associated protein. d Ara1 mRNA expression in RPE cells tested by qPCR (n = 6). e Flow cytometry analysis of CD73 shedding in Ara1−/− RPE
Lipid rafts enabled CD73-ARA1 interaction
CD73 should be localized to lipid rafts as other GPI anchored proteins; however, it needs to be clarified whether ARA1 was also co-localized to lipid rafts when it was associated with CD73. Lipid rafts were isolated from RPE cells. The existence of CD73 in lipid rafts fractions (CAV-1 positive) and non-lipid rafts fractions (Na+/K+ ATPase positive) were detected by western blot. Mevastatin, a lipid rafts synthesis blocker, was used to test whether lipid rafts synthesis is necessary for the formation of CD73-ARA1 complex and the shedding of CD73. Figure 7a showed that in normal RPE cells (left panel), both CD73 and ARA1 were mostly existed in non-lipid rafts fractions. LPS/TNF-α treatment induced CD73 shedding, and only tiny amount of CD73 could be detected in lipid rafts fractions. Meanwhile, ARA1 changed its major location from non-lipid rafts fractions to lipid rafts fractions (middle panel). When CD73 shedding was blocked by the addition of MMP-9 inhibitor, large amount of CD73 was visualized in lipid rafts fractions together with ARA1 (right panel). As far as the existences of CD73 and ARA1 were concerned, same results were got from the lipid rafts prepared by either detergent free or detergent resistant methods. Figure 7b, as a representation, showed the co-existence of CD73 and ARA1 in lipid rafts fractions isolated from LPS/TNF-α and MMP-9 inhibitor-treated RPE cells by the detergent resistant method. Figure 7c showed that without the existence of CD73 (left column) ARA1 could not be recruited to lipid rafts fractions. With the expression of exogenous CD73 (right column), ARA1 was located to lipid rafts fractions in response to LPS/TNF-α treatment. Figure 7d indicated that mevastatin largely, if not completely, blocked CD73 shedding without any influence to MMP-9 activity and without the presence of MMP-9 inhibitor. Figure 7e showed when lipid rafts synthesis was blocked by mevastatin, both CAV-1 and CD73 were localized to non-lipid rafts but not lipid raft fractions; and ARA1 could not be pulled down together with CD73 although they were all existed in membrane (Fig. 7f).
Fig. 7.

Lipid raft localization is necessary for CD73 shedding. Cell membrane extract was primarily isolated from differently treated RPE cells, further separated into lipid raft and non-lipid raft fractions by either detergent free (a) or detergent resistant methods (b). All the fractions were western blotting for CAV-1 as the marker of lipid rafts fractions and Na+/K+ ATPase as the marker of non-lipid fractions. The effect of mevastatinon CD73 shedding (c), CD73 lipid rafts localization (e) and ARA1 co-IP (f) were evaluated by western blotting. The amount of active MMP-9 in the medium was detected (D). a The existence of CD73 and ARA1 in lipid raft and non-lipid raft fractions, determined by western blotting. b The co-existence of CD73 and ARA1 in lipid raft fractions prepared by detergent resistant methods. c Localization of ARA1 in Cd73−/− RPE, with or without Wt-CD73 modification. d The effect of mevastatin on CD73 shedding was determined by western blotting (upper panel). Active MMP-9 in the medium was determined by MMP-9 Biotrak activity assay (lower panel, n = 6). e The effect of mevastatin on CD73 localization, determined by western blotting. f The effect of mevastatin on the Co-IP of CD73 and ARA1
FRET confirmed CD73-ARA1 interaction
As schematically illustrated in Fig. 8, the close interaction between CD73 and ARA1 might be detected by FRET. Figure 9a provides substantial information. LPS/TNF-α treatment augmented ARA1 expression in RPE membrane which was visualized as red fluorescence. Meanwhile, membrane CD73 was visualized as green fluorescence and which decreased significantly when RPE cells were treated by LPS/TNF-α. When CD73 shedding was blocked by the addition of MMP-9 inhibitor or mevastatin, same intensity of green fluorescence was visualized in LPS/TNF-α-treated and untreated control RPE. However, MMP-9 inhibitor and mevastatin showed significantly different effects on FRET results. FRET-induced red fluorescence could be largely visualized in MMP-9 inhibitor-treated cells, but hardly detected in mevastatin-treated RPE. Figure 9b kinetically measured and compared red fluorescence in Wt- and Cd73−/− RPE cells. When 555 nM (the maximum excitation wavelength of RFP) and 506 nM (the emission wavelength of EGFP2) were being used as the excitation wave length, they induced same intensity of red fluorescence in Wt- and Cd73−/− RPE. Following LPS/TNF-α treatment FRET generated red fluorescence of low intensity could be detected in Wt-RPE (upper panel); and a peak fluorescence was observed approximately 60 h after treatment. While, in Cd73−/− RPE cells (lower panel), FRET did not generated any fluorescence. In Figure 9c, FRET induced red fluorescence was compared between LPS/TNF-α-treated and untreated Wt-RPE cells, with or without the addition of MMP-9 inhibitor or mevastatin. RPE cells received LPS/TNF-α treatment with the addition MMP-9 inhibitor yielded the highest intensity of red fluorescence. As shown in Fig. 9d, FRET-induced red fluorescence was also detected in Cd73−/− RPE cells with Wt- and Mut-CD73 modifications. Mut-CD73-modified Cd73−/− RPE cells, received LPS/TNF-α treatment, yield significantly higher intensity of red fluorescence.
Fig. 9.
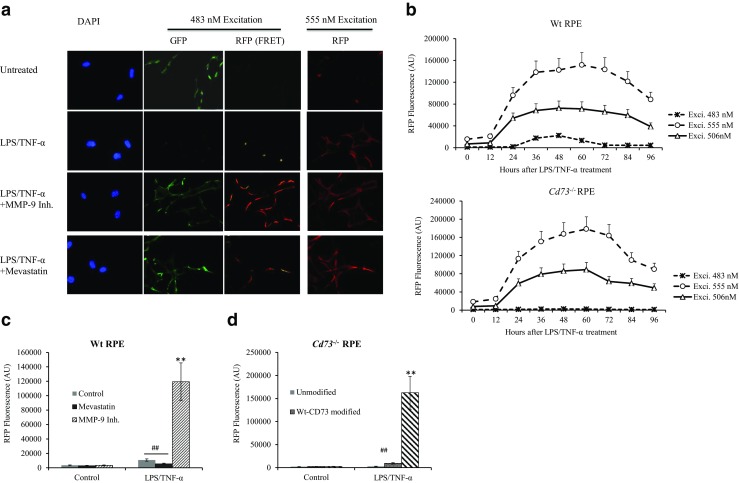
FRET assay for determining associated CD73-ARA1. In vitro cultured wild-type RPE and Cd73−/− RPE modified with Wt- and Mut-CD73 were differentially treated and subjected to FRET assay. Briefly, EGFP2-labeled anti-CD73 and RFP-labeled anti-ARA1 antibodies were added to RPE cells; the fluorescence from FRET receptor of GFP was detected by fluorescence microscopy (a) and a fluorometer (b–d). a Wt-RPE cells were treated with LPS/TNF-α and other factors. Green and red fluorescence were investigated by fluorescence microscopy. b The fluorescence intensity of RFP, excited by varied wavelengths at different times after LPS/TNF-α treatment, was evaluated by a fluorometer (n = 6) in Wt- (upper panel) and Cd73−/− RPE (lower panel) cells. c FRET-induced fluorescence of RFP in Wt-RPE at 48 h after treatment (n = 6). d FRET-induced fluorescence of RFP in Cd73−/− RPE modified with Wt- and Mut-CD73 at 48 h after treatment (n = 6)
Discussion
ATP and its metabolites have strong immune and inflammation regulatory effects. ATP is generally regarded as a pro-inflammatory factor, while, adenosine is verified of having strong anti-inflammatory function. In eye diseases study, ATP was reported of inducing photoreceptor death, aggravating glaucoma, and facilitating neuronal injury in the retina [21–23], while, adenosine showed protective effect [24–26]. By controlling the conversion of AMP to adenosine, CD73 intensively participate in the regulation of immune response, including autoimmune diseases and tumor genesis [27–30].
Not only for regulating immune response, ATP and adenosine also have functions of modulating the physiological and pathological response of multiple ocular tissues. In 2014, Sanderson reviewed the function of purines in the eye [31]. ATP and adenosine act in an autocrine or paracrine fashion to maintain homeostasis as well as respond to insults [32]. Adenosine modulates intraocular pressure (IOP) by modifying both inflow and outflow pathways of aqueous humor through the activation of adenosine A3 receptor (ARA3) in non-pigmented ciliary epithelial [33]. Conversion of released ATP into adenosine by CD39 and CD73 influenced the production of lipofuscin and lipid oxidation in RPE cells [34]. All this findings supported the importance of adenosine and CD73 in the maintenance of ocular function. Previously, we reported that RPE cells physiologically express CD73 on their surface and converts ATP to adenosine for maintaining immune homeostasis of retina. When inflammation happened, RPE cells lost their membrane-localized CD73. This inhibited the conversion of AMP to adenosine, significantly down-regulated adenosine’s immune suppressive function, and promoted local inflammation [8].
As shown in Fig. 1, membrane-localized CD73 was significantly decreased in EAU-RPE vs naïve-RPE, or in LPS/TNF-α-treated RPE vs untreated RPE. However, Cd73 mRNA expression remained unchanged between comparisons. Thus decreased membrane CD73 must be controlled by some mechanisms other than expression regulation. It was reported as early as in 1997 that there was the shedding of membrane localized CD73 in lymphocyte which significantly affected the cells’ function [35]. The latest reports also supported the existence of membrane released solubilized CD73 circulating in human vitreous [36]. Thus, it needs to be clarified whether enzyme controlled CD73 shedding accounted for RPE’s CD73 lose; two kinds of enzymes were investigated. PI-PLC may catalyze GPI to release its anchored molecules [37, 38], and proteinase may hydrolyze the K547/F548 site inside CD73. Results in Fig. 2 confirmed that proteinase inhibitors cocktail, but not PI-PLC inhibitor, completely blocked CD73 diminish in LPS/TNF-α-treated RPE cells; and it was MMP-9 catalyzed CD73’s K547/F548 site to induce CD73 shedding.
To support the idea of CD73 shedding, the physical existence of CD73 molecule and CD73’s enzymatic activity are expected to be detected in the medium of LPS/TNF-α-treated RPE cells. Although the existence of CD73 molecule in this medium was successfully verified by western blot in Fig. 4a, no CD73’s enzymatic activity could be detected (Fig. 4c). This result was not in consistence with several published reports supporting the enzymatic activity of solubilized CD73 (sCD73) [39, 40]. In their articles, the sCD73 was released from cell surface due to PI-PLC-mediated digestion of GPI anchor, and those are intact form of CD73. MMP-9 catalyzed K547/F548 sites to release a C terminal-truncated CD73. To answer the question whether intact and truncated sCD73 are significantly different at their enzymatic activities, we compared PI-PLC- and MMP-9-released CD73 from RPE cells. The supplemental results indicated that when same amount of sCD73 was evaluated, PI-PLC-released sCD73 showed high enzymatic activity, while, the activity of MMP-9-released CD73 were still undetectable. It was likely to be concluded that the enzymatic activity of MMP-9-released truncated CD73 was completely abolished or at least largely decreased. Further study need to be done to illustrate whether this truncated CD73 has other bio functions except of ecto-5′-nucleotidase activity.
As MMP-9 is critical for LPS/TNF-α-induced CD73 shedding, it need to know whether LPS and TNF-α regulate MMP-9’s expression and activity. Figure 5a, b confirmed that LPS and TNF-α had synergistic effect on elevating Mmp-9 expression in RPE cells as well as increasing the amount of active MMP-9 in cell culture medium. Thus, it is reasonable to consider that LPS and TNF-α induce CD73 shedding by enhancing MMP-9’s activity. However, the results in Fig. 5c indicated that the addition of exogenous recombinant MMP-9 only leads to CD73 shedding in LPS/TNF-α-treated but not untreated Mmp-9−/− RPE. This means LPS/TNF-α treatment must bring other necessary changes to RPE besides enhancing MMP-9 activity. Based on two reasons, we proposed that following LPS/TNF-α treatment, CD73 interacted with other molecules to form a complex, and this complexed CD73 was capable of being recognized and catalyzed by MMP-9. The first reason: GPI-anchored proteins have high membrane mobility and tend to form complexes with other molecules [41, 42]. The second reason: previously published studies already indicated that CD73 is likely to interact with other proteins, especially adenosine receptors, to form functional complex [43, 44]. A pull-down assay was performed in Mut-CD73 modified Cd73−/− RPE cells to verify this proposal. Sustained membrane localization of Mut-CD73 following LPS/TNF-α treatment ensured the associated protein could be effectively pulled down. Figure 6a–c identified ARA1 as the CD73 associated protein in LPS/TNF-α-treated RPE cells.
LPS/TNF-α treatment augmented the function of MMP-9 and induced the formation of CD73-ARA1 complex. Then it needs to be clarified how the CD73-ARA1 complex was formed following LPS/TNF-α treatment. Figure 6d showed that LPS had strong ability of up-regulating ARA1 expression, and it was verified in Fig. 6e that ARA1 is indispensable for CD73 shedding. Till now, it is reasonable to conclude that LPS/TNF-α up-regulated ARA1 expression facilitating the formation of CD73-ARA1complex. But there is still another question. Both Fig. 6c, d showed there was basal ARA1expression in untreated RPE cells, why this basally expressed ARA1 could not form complex with CD73.
Lipid rafts are involved in a number of cellular processes and are important for membrane trafficking [45]. A variety of proteins, include GPI-anchored proteins, are selectively enriched in lipid rafts [46–48]; both LPS and TNF-α were reported having function of recruiting membrane factors to lipid rafts [49, 50]. Whether CD73 is also involved in the synthesis of lipid rafts and by which facilitates its trafficking, complex formation, and shedding. Lipid rafts were isolated from RPE cells by detergent free (Fig. 7a) and detergent resistant (Fig. 7b) method. The co-localization of CD73 and ARA1 in lipid rafts fractions was confirmed in LPS/TNF-α-treated RPE cells, and it was CD73 that further introduced ARA1 to the lipid rafts (Fig. 7c). When mevastatin, a lipid rafts formation inhibitor, was added LPS/TNF-α, induced CD73 shedding was almost completely blocked (Fig. 7d, upper panel). This was not due to any alternation of MMP-9 activity (Fig. 7d, lower panel), but caused by mevastatin-blocked lipid rafts synthesis (Fig. 7e) and the formation CD73-ARA1 complex (Fig. 7e). At last, there is a potential contradiction needed to be clarified. One may ask if CD73 is absolutely essential for the association of ARA1 with lipoid rafts why the middle panel in Figure 7a showed the existence of ARA1 in lipid rafts fractions when there is almost no detectable CD73. Our concern is that CD73 is indispensable for introducing ARA1 to lipid rafts, and the formation of CD73-ARA1 complex in lipid rafts facilitated CD73 shedding. The whole procedure might be simplified as three steps: (i) following LPS/TNF-α treatment, CD73 was located to lipid rafts and gained high mobility. (2) CD73 contacted with ARA1 to form a CD73-ARA1 complex and introduced ARA1 to lipid rafts. (3) CD73 was catalyzed by MMP-9, while, ARA1 was remained in lipid rafts. CD73 is necessary for introducing ARA1 to lipid rafts; however, it is not critical for keeping ARA1 in lipid rafts fractions. Our following study may investigate for how long ARA1 can be maintained in lipid rafts without the existence of CD73.
To further evaluate the function of lipid rafts in mediating CD73 shuttling, CD73-ARA1 interaction and CD73 shedding, a FRET assay was adopted. The results shown in Fig. 9a indicated that when lipid raft synthesis was disturbed by mevastatin, FRET-induced red fluorescence was significantly decreased although CD73 and ARA1 were still expressed in the membrane. This evidence supported the idea that lipid rafts synthesis is indispensable for mediating CD73-ARA1 interaction. It needs to be aware that when fluorochromes-labeled antibodies were used in FRET assay, sometimes we cannot accurately estimate how close the targets are. Since FRET occurred at the ends of antibodies and there might be still some distance between the proteins of interest. Our immunoprecipitation result confirmed the association of CD73 and ARA1, which supported FRET assay firstly. Secondly, Fig. 9c showed that if LPS/TNF-α-induced CD73 shedding was blocked by a MMP-9 inhibitor FRET generated intensive red fluorescence which was hugely decreased by additively added mevastatin.
In summary, we demonstrated that multiple steps were involved in LPS/TNF-α-induced CD73 shedding, including elevated MMP-9 activity, induced ARA1 expression, and CD73-ARA1 complex formation in rafts. All these steps could potentially be targets for manipulating RPE’s immunoregulatory function. There are still some questions left which need to be clarified later. Interestingly, not only adenosine but AMP is also a functional ARA1 ligand. AMP and adenosine activate different ARA1 signaling, and induce different outcomes of immune response [51]. There must be a very interesting relationship between AMP, CD73, adenosine, and ARA1.
Electronic supplementary material
(DOCX 77 kb)
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 81570833).
Institutional approval was obtained, and institutional guidelines regarding animal experimentation were followed.
ᅟ
Conflicts of interest
Wei Zhang declares that he has no conflict of interest.
Shumin Zhou declares that she has no conflict of interest.
Guoping Liu declares that he has no conflict of interest.
Fanqiang Kong declares that he has no conflict of interest.
Song Chen declares that he has no conflict of interest.
References
- 1.Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: important modulators of purinergic signalling cascade. Biochim Biophys Acta. 2008;1783:673–694. doi: 10.1016/j.bbamcr.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 2.Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. 2014;509:310–317. doi: 10.1038/nature13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zarek PE, Huang C, Lutz ER, Kowalski J, Horton MR, Linden J. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood. 2008;111:251–259. doi: 10.1182/blood-2007-03-081646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. 2011;11:201–212. doi: 10.1038/nri2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smyth L.A. Ratnasothy K, Tsang JY, Boardman D, Warley A, Lechler R, Lombardi G. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur J Immunol. 2013;43(9):2430–2440. doi: 10.1002/eji.201242909. [DOI] [PubMed] [Google Scholar]
- 6.Takedachi M, Qu D, Ebisuno Y, Oohara H, Joachims ML, McGee ST, Maeda E, McEver RP, Tanaka T, Miyasaka M, Murakami S, Krahn T, Blackburn MR, Thompson LF. CD73-generated adenosine restricts lymphocyte migration into draining lymph nodes. J Immunol. 2008;180(9):6288–6296. doi: 10.4049/jimmunol.180.9.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coffey F, Lee SY, Buus TB, Lauritsen JP, Wong GW, Joachims ML, Thompson LF, Zúñiga-Pflücker JC, Kappes DJ, Wiest DL. The TCR ligand-inducible expression of CD73 marks γδ lineage commitment and a metastable intermediate in effector specification. J Exp Med. 2014;211(2):329–343. doi: 10.1084/jem.20131540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen S, Zhou S, Zang K, Kong F, Liang D, Yan H. CD73 expression in RPE cells is associated with the suppression of conventional CD4 cell proliferation. Exp Eye Res. 2014;127:26–36. doi: 10.1016/j.exer.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 9.Dong K, Gao ZW, Zhang HZ. The role of adenosinergic pathway in human autoimmune diseases. Immunol Res. 2016;64:1133–1141. doi: 10.1007/s12026-016-8870-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Regateiro FS, Cobbold SP, Waldmann H. CD73 and adenosine generation in the creation of regulatory microenvironments. Clin Exp Immunol. 2013;171:1–7. doi: 10.1111/j.1365-2249.2012.04623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chrobak P, Charlebois R, Rejtar P, El Bikai R, Allard B, Stagg J. CD73 plays a protective role in collagen-induced arthritis. J Immunol. 2015;194:2487–2492. doi: 10.4049/jimmunol.1401416. [DOI] [PubMed] [Google Scholar]
- 12.Botta Gordon-Smith S, Ursu S, Eaton S, Moncrieffe H, Wedderburn LR. Correlation of low CD73 expression on synovial lymphocytes with reduced adenosine generation and higher disease severity in juvenile idiopathic arthritis. Arthritis Rheumatol. 2015;67:545–554. doi: 10.1002/art.38959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Wang L, Chen X, Li L, Li Y, Ping Y, Huang L, Yue D, Zhang Z, Wang F, Li F, Yang L, Huang J, Yang S, Li H, Zhao X, Dong W, Yan Y, Zhao S, Huang B, Zhang B, Zhang Y. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology. 2017;6:e1320011. doi: 10.1080/2162402X.2017.1320011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whiteside TL. Targeting adenosine in cancer immunotherapy: a review of recent progress. Expert Rev Anticancer Ther. 2017;17:527–535. doi: 10.1080/14737140.2017.1316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. 2017;276:121–144. doi: 10.1111/imr.12528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, Huang Q, Liu J, Takeda K, Teng MW, Sachsenmeier K, Smyth MJ. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell. 2016;30:391–403. doi: 10.1016/j.ccell.2016.06.025. [DOI] [PubMed] [Google Scholar]
- 17.Doyle JW, Dowgiert RK, Buzney SM. Retinoic acid metabolism in cultured retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1995;36:708–717. [PubMed] [Google Scholar]
- 18.Chen M, Muckersie E, Robertson M, Fraczek M, Forrester JV, Xu H. Characterization of a spontaneous mouse retinal pigment epithelial cell line B6-RPE07. Invest Ophthalmol Vis Sci. 2008;49:3699–3706. doi: 10.1167/iovs.07-1522. [DOI] [PubMed] [Google Scholar]
- 19.Santerre RF, Allen NE, Hobbs JN, Jr, Rao RN, Schmidt RJ. Expression of prokaryotic genes for hygromycin B and G418 resistance as dominant-selection markers in mouse L cells. Gene. 1984;30:147–156. doi: 10.1016/0378-1119(84)90115-X. [DOI] [PubMed] [Google Scholar]
- 20.Macdonald JL, Pike LJ. A simplified method for the preparation of detergent-free lipid rafts. J Lipid Res. 2005;46:1061–1067. doi: 10.1194/jlr.D400041-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Aplin FP, Luu CD, Vessey KA, Guymer RH, Shepherd RK, Fletcher EL. ATP-induced photoreceptor death in a feline model of retinal degeneration. Invest Ophthalmol Vis Sci. 2014;55(12):8319–8329. doi: 10.1167/iovs.14-15732. [DOI] [PubMed] [Google Scholar]
- 22.Lu W, Hu H, Sévigny J, Gabelt BT, Kaufman PL, Johnson EC, Morrison JC, Zode GS, Sheffield VC, Zhang X, Laties AM, Mitchell CH. Rat, mouse, and primate models of chronic glaucoma show sustained elevation of extracellular ATP and altered purinergic signaling in the posterior eye. Invest Ophthalmol Vis Sci. 2015;56(5):3075–3083. doi: 10.1167/iovs.14-15891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakamoto K, Endo K, Suzuki T, Fujimura K, Kurauchi Y, Mori A, Nakahara T, Ishii K. P2X7 receptor antagonists protect against N-methyl-D-aspartic acid-induced neuronal injury in the rat retina. Eur J Pharmacol. 2015;756:52–58. doi: 10.1016/j.ejphar.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 24.Bar-Yehuda S, Luger D, Ochaion A, Cohen S, Patokaa R, Zozulya G, Silver PB, de Morales JM, Caspi RR, Fishman P. Inhibition of experimental auto-immune uveitis by the A3 adenosine receptor agonist CF101. Int J Mol Med. 2011;28(5):727–731. doi: 10.3892/ijmm.2011.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Avni I, Garzozi HJ, Barequet IS, Segev F, Varssano D, Sartani G, Chetrit N, Bakshi E, Zadok D, Tomkins O, Litvin G, Jacobson KA, Fishman S, Harpaz Z, Farbstein M, Yehuda SB, Silverman MH, Kerns WD, Bristol DR, Cohn I, Fishman P. Treatment of dry eye syndrome with orally administered CF101: data from a phase 2 clinical trial. Ophthalmology. 2010;117(7):1287–1293. doi: 10.1016/j.ophtha.2009.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobson KA, Civan MM. Ocular purine receptors as drug targets in the eye. J Ocul Pharmacol Ther. 2016;32(8):534–547. doi: 10.1089/jop.2016.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arab S, Kheshtchin N, Ajami M, Ashurpoor M, Safvati A, Namdar A, Mirzaei R, Mousavi Niri N, Jadidi-Niaragh F, Ghahremani MH, Hadjati J. Increased efficacy of a dendritic cell-based therapeutic cancer vaccine with adenosine receptor antagonist and CD73 inhibitor. Tumour Biol. 2017;39(3):1010428317695021. doi: 10.1177/1010428317695021. [DOI] [PubMed] [Google Scholar]
- 28.Allard B, Turcotte M, Stagg J. 2014.Targeting CD73 and downstream adenosine receptor signaling in triple-negative breast cancer. Expert Opin Ther Targets. 2014;18:863–881. doi: 10.1517/14728222.2014.915315. [DOI] [PubMed] [Google Scholar]
- 29.Beavis PA, Stagg J, Darcy PK, Smyth MJ. CD73: a potent suppressor of antitumor immune responses. Trends Immunol. 2012;33(5):231–237. doi: 10.1016/j.it.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 30.Stagg J, Beavis PA, Divisekera U, Liu MC, Möller A, Darcy PK, Smyth MJ. CD73-deficient mice are resistant to carcinogenesis. Cancer Res. 2012;72(9):2190–2196. doi: 10.1158/0008-5472.CAN-12-0420. [DOI] [PubMed] [Google Scholar]
- 31.Fausther M, Lavoie EG, Goree JR, Dranoff JA. An Elf2-like transcription factor acts as repressor of the mouse ecto-5′-nucleotidase gene expression in hepatic myofibroblasts. Purinergic Signal. 2017;13(4):417–428. doi: 10.1007/s11302-017-9570-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kao DJ, Saeedi BJ, Kitzenberg D, Burney KM, Dobrinskikh E, Battista KD, Vázquez-Torres A, Colgan SP, Kominsky DJ (2017) Intestinal epithelial Ecto-5'-Nucleotidase (CD73) regulates intestinal colonization and infection by nontyphoidal salmonella. Infect Immun 85(10) [DOI] [PMC free article] [PubMed]
- 33.Kling L, Benck U, Breedijk A, Leikeim L, Heitzmann M, Porubsky S, Krämer BK, Yard BA, Kälsch AI. Changes in CD73, CD39 and CD26 expression on T-lymphocytes of ANCA-associated vasculitis patients suggest impairment in adenosine generation and turn-over. Sci Rep. 2017;7(1):11683. doi: 10.1038/s41598-017-12011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monguió-Tortajada M, Roura S, Gálvez-Montón C, Franquesa M, Bayes-Genis A, Borràs FE. Mesenchymal stem cells induce expression of CD73 in human monocytes in vitro and in a swine model of myocardial infarction in vivo. Front Immunol. 2017;8:1577. doi: 10.3389/fimmu.2017.01577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Airas L, Niemelä J, Salmi M, Puurunen T, Smith DJ, Jalkanen S. Differential regulation and function of CD73, a glycosyl-phosphatidylinositol-linked 70-kD adhesion molecule, on lymphocytes and endothelial cells. J Cell Biol. 1997;136(2):421–431. doi: 10.1083/jcb.136.2.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kothekar D, Bandivdekar A, Dasgupta D. Increased activity of goat liver plasma membrane alkaline phosphatase upon release by phosphatidylinositol-specific phospholipase C. Indian J Biochem Biophys. 2014;51(4):263–270. [PubMed] [Google Scholar]
- 37.Yadav M, Raghupathy R, Saikam V, Dara S, Singh PP, Sawant SD, Mayor S, Vishwakarma RA. Synthesis of non-hydrolysable mimics of glycosylphosphatidylinositol (GPI) anchors. Org Biomol Chem. 2014;12(7):1163–1172. doi: 10.1039/c3ob42116c. [DOI] [PubMed] [Google Scholar]
- 38.Maksimow M, Kyhälä L, Nieminen A, Kylänpää L, Aalto K, Elima K, Mentula P, Lehti M, Puolakkainen P, Yegutkin GG, Jalkanen S, Repo H, Salmi M. Early prediction of persistent organ failure by soluble CD73 in patients with acute pancreatitis*. Crit Care Med. 2014;42(12):2556–2564. doi: 10.1097/CCM.0000000000000550. [DOI] [PubMed] [Google Scholar]
- 39.Morello S, Capone M, Sorrentino C, Giannarelli D, Madonna G, Mallardo D, Grimaldi AM, Pinto A, Ascierto PA. Soluble CD73 as biomarker in patients with metastatic melanoma patients treated with nivolumab. J Transl Med. 2017;15(1):244. doi: 10.1186/s12967-017-1348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen Q, Bourdais G, Pan H, Robatzek S, Tang D. Arabidopsis glycosylphosphatidylinositol-anchored protein LLG1 associates with and modulates FLS2 to regulate innate immunity. Proc Natl Acad Sci U S A. 2017;1142:5749–5754. doi: 10.1073/pnas.1614468114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tiengwe C, Bush PJ, Bangs JD. Controlling transferrin receptor trafficking with GPI-valence in bloodstream stage African trypanosomes. PLoS Pathog. 2017;13(5):e1006366. doi: 10.1371/journal.ppat.1006366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Augusto E, Matos M, Sévigny J, El-Tayeb A, Bynoe MS, Müller CE, Cunha RA, Chen JF. Ecto-5′-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J Neurosci. 2013;33(28):11390–11399. doi: 10.1523/JNEUROSCI.5817-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hart ML, Grenz A, Gorzolla IC, Schittenhelm J, Dalton JH, Eltzschig HK. Hypoxia-inducible factor-1α-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5′-nucleotidase (CD73) and the A2B adenosine receptor. J Immunol. 2011;186(7):4367–4374. doi: 10.4049/jimmunol.0903617. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Helms JB, Zurzolo C. Lipids as targeting signals: lipid rafts and intracelular trafficking. Traffic. 2004;5:247–254. doi: 10.1111/j.1600-0854.2004.0181.x. [DOI] [PubMed] [Google Scholar]
- 45.Cinek T, Horejsi V. The nature of large noncovalent complexes containing glycosyl-phosphatidylinositol-anchored membrane glycoproteins and protein tyrosine kinases. J Immunol. 1992;149:2262–2270. [PubMed] [Google Scholar]
- 46.Sargiacomo M, Sudol M, Tang Z, Lisanti MP. Signal transducing molecules and glycosyl-phosphatidylinositol-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J Cell Biol. 1993;122:789–807. doi: 10.1083/jcb.122.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zurzolo C, van't Hof W, van Meer G, Rodriguez-Boulan E. VIP21/caveolin, glycosphingolipid clusters and the sorting of glycosylphosphatidylinositol-anchored proteins in epithelial cells. EMBO J. 1994;13:42–53. doi: 10.1002/j.1460-2075.1994.tb06233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolczyk D, Zaremba-Czogalla M, Hryniewicz-Jankowska A, Tabola R, Grabowski K, Sikorski AF, Augoff K. TNF-α promotes breast cancer cell migration and enhances the concentration of membrane-associated proteases in lipid rafts. Cell Oncol (Dordr) 2016;39(4):353–363. doi: 10.1007/s13402-016-0280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tewari R, Choudhury SR, Mehta VS, Sen E (2012) TNFα regulates the localization of CD40 in lipid rafts of glioma cells. Mol Biol Rep 39(9):8695–8699 [DOI] [PubMed]
- 50.Schoeniger A, Fuhrmann H, Schumann J. LPS- or Pseudomonas aeruginosa-mediated activation of the macrophage TLR4 signaling cascade depends on membrane lipid composition. PeerJ. 2016;4:e1663. doi: 10.7717/peerj.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rittiner JE, Korboukh I, Hull-Ryde EA, Jin J, Janzen WP, Frye SV, Zylka MJ. AMP is an adenosine A1 receptor agonist. J Biol Chem. 2012;287(8):5301–5309. doi: 10.1074/jbc.M111.291666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 77 kb)