

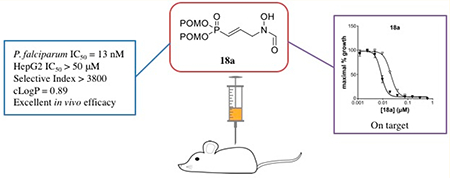
Abstract
Severe malaria due to Plasmodium falciparum remains a significant global health threat. DXR, the second enzyme in the MEP pathway, plays an important role to synthesize building blocks for isoprenoids. This enzyme is a promising drug target for malaria due to its essentiality as well as its absence in humans. In this study, we designed and synthesized a series of α,β-unsaturated analogues of fosmidomycin, a natural product that inhibits DXR in P. falciparum. All compounds were evaluated as inhibitors of P. falciparum. The most promising compound, 18a, displays on-target, potent inhibition against the growth of P. falciparum (IC50 = 13 nM) without significant inhibition of HepG2 cells (IC50 > 50 μM). 18a was also tested in a luciferase-based Plasmodium berghei mouse model of malaria and showed exceptional in vivo efficacy. Together, the data support MEPicide 18a as a novel, potent, and promising drug candidate for the treatment of malaria.
Graphical Abstract
INTRODUCTION
Malaria is a severe, life-threatening infectious disease with high mortality and morbidity rates.1 In 2016, there was a substantially high incidence rate of malaria with 216 million new malaria cases as well as 0.4 million deaths, 64% of which are children under 5 years of age.1 Malaria is caused by a group of Plasmodium parasites, with Plasmodium falciparum causing the majority of deaths and severe infections.2 Parasites are transmitted to humans via the bites of female Anopheles mosquitoes.3 After growing and replicating initially in human liver cells, the parasites reach the blood and cause malarial symptoms such as fever, headache, chills, or even death.4 Artemisinin-based combination therapy (ACT) is currently the best treatment for malaria and is typically highly effective.3 Resistance to artemisinin, however, has already spread in the Greater Mekong subregion.5 Thus, there is a pressing need for new therapeutics for malaria with novel modes of action that could provide alternate chemotherapies to combat sensitive and drug-resistant parasites.
P. falciparum uses the methyl erythritol phosphate (MEP) pathway for the biosynthesis of isopentenyl pyrophosphate (IPP) and its isomer dimethylallyl pyrophosphate (DMAPP), the C5 precursors of isoprenoids (Figure 1).6,7 Humans, however, use an alternate acetate/mevalonate pathway exclusively to synthesize these C5 isoprene building blocks.8 Blocking the MEP pathway terminates the biosynthesis of such important metabolites and results in cell death of P. falciparum.3,9 Thus, many enzymes in the MEP pathway could become promising drug targets to design MEPicides that work against malaria through a new mechanism of action and one not found in humans.10
Figure 1.

Methyl erythritol phosphate (MEP) pathway of isoprenoid biosynthesis.
DXR, 1-deoxy-D-xylulose 5-phosphate reductoisomerase, is the second enzyme in the MEP pathway and catalyzes the first committed step.11 DXR converts DOXP to MEP with the assistance of cofactor NADPH as well as a divalent metal cation (Figure 1).12 DXR is essential for the viability of several pathogens and is validated as a promising drug target for developing antitubercular agents13 and antimalarials.14 Fosmidomycin (Figure 2, 1a), isolated from Streptomyces lavendulae,15 is a potent inhibitor of P. falciparum DXR (IC50 = 0.034 μM).16 FR-900098 (Figure 2, 1b), the N-acetyl analogue of fosmidomycin isolated from Streptomyces rubellomurinus,15 is roughly equipotent to fosmidomycin (P. falciparum DXR IC50 = 0.024 μM).16 While these two natural products have submicromolar inhibition of P. falciparum growth (IC50 = 0.09–0.35 μM),7 their use as a single drug therapy is limited by low bioavailability, short serum half-life, and malaria recrudescence.17 Despite these disadvantages, fosmidomycin is a remarkably safe drug as proven by several clinical trials and a promising candidate for treating uncomplicated P. falciparum malaria in combination therapies.18,19 Thus, we selected fosmidomycin as the parent structure from which to design analogues that would effectively inhibit Pf DXR, have improved pharmacokinetic properties and lead to promising drug candidates against malaria.
Figure 2.

Fosmidomycin and selected previously reported analogues.
We and others have previously evaluated the structure—activity relationships (SAR) of fosmidomycin and FR-900098 as inhibitors of several DXR homologues as well as various microbial pathogens.20–29 Fosmidomycin binds to DXR competitively with substrate DOXP and uncompetitively with cofactor NADPH.30 SAR studies on fosmidomycin analogues reveal that the retrohydroxamate or hydroxamate moiety should be retained to mimic the crucial interaction of fosmidomycin with the divalent metal cation.21,24,25,27–29,31 Similarly, the phosphonate moiety should be retained as it forms numerous hydrogen bonds with neighboring amino acid residues.32–34 A three-carbon linker between the two moieties is also found to be crucial for DXR inhibition.24 As we reported earlier, the unsaturated FR-900098 analogue (Figure 2, 2) gained a 2-fold increase in potency against Mycobacterium tuberculosis (Mtb) DXR (IC50 = 1.07 μM) compared with parent compound FR900098.24 A prodrug strategy was applied to this structure, and the corresponding pivaloyloxymethyl (POM) phosphonate was synthesized (Figure 2, 3). Compound 3 displays an Mtb MIC99 value of 9.4 μg/mL (22 μM), thus gaining the needed lipophilicity to penetrate the Mtb cell wall.24 This compound likely regenerates 2 inside the bacteria, and this acid inhibits DXR.24,27 Prodrug 3 also shows potent inhibition against P. falciparum growth with an IC50 value of 18.3 nM,35 nearly as potent as artemisinin (P. falciparum IC50 = 10.4 nM),35 a current first-line antimalarial drug. As expected, prodrug 3 displays potent in vivo antimalarial activity.35
Since it was found that the NADPH-binding pocket of DXR is druggable,36 and because this pocket is adjacent to the cavity where the retrohydroxamate moiety of fosmidomycin binds,37 we previously synthesized analogues with extended aromatic groups on the N-alkoxy group of FR-900098 to develop improved DXR inhibitors as antimicrobials (Figure 2, 4, 5).27,28 These compounds are designed to act as bisubstrate inhibitors that could bind to both the DOXP and NADPH binding sites.27,28 The binding mode of compound 4 was then determined using classical Lineweaver—Burke double reciprocal plots. These experiments showed that compound 4 is competitive with DOXP and NADPH, confirming bisubstrate binding behavior.27 The POM prodrug of 4 was also synthesized (Figure 2, 5), showing effective Mtb growth inhibition (MIC99 = 18.75 μg/mL or 33.2 μM).27 The bisubstrate strategy increases the overall lipophilicity of the analogues, which is likely beneficial for penetration into several pathogens.
The purpose of the current work is to synthesize a series of α,β-unsaturated N-acyl (Figure 3A) and N-alkoxy (Figure 3B) fosmidomycin analogues and evaluate them as antimalarial agents that work via DXR inhibition. For the N-acyl analogues, we studied the influence of electronic effects on DXR inhibition via introduction of CF3 and OCH3 groups compared with the CH3 group of compound 2. The α,β-unsaturated N-formyl analogue was also made to assess the effect of size and lipophilicity compared with the methyl group in 2. To achieve the bisubstrate inhibitors that compete with both DOXP and NADPH, several aromatic groups (with a linker of 1 carbon atom) were selected as N-alkoxy analogues based on previous results.27,28 POM prodrugs of the initial hits that showed effective DXR inhibition, and which were synthetically accessible, were synthesized. These prodrug compounds were then evaluated as inhibitors of P. falciparum.
Figure 3.

α,β-Unsaturated N-acyl and N-alkoxy fosmidomycin analogues.
RESULTS AND DISCUSSION
Synthesis.
N-Formyl analogue 12a and N-acyl analogues 12b and 12c were prepared in seven steps from commercially available starting materials shown in Scheme 1. First, allyl phosphonate 6 was synthesized from triethylphosphite and allyl bromide via a Michaelis—Arbuzov reaction.38 Subsequent addition of bromine to compound 6 resulted in production of dibromide 7.39 O-Benzylhydroxylamine hydrochloride was neutralized in situ and protected using di-tert-butyldicarbonate to yield Boc-protected 8,40 which was then reacted with compound 7 and two equivalents of NaH to prepare compound 9.24 The first equivalent of NaH deprotonated 8 and generated a nucleophile to attack the primary bromide of compound 7. The second equivalent of NaH was used to eliminate the β-bromide to furnish α,β-unsaturated phosphonate 9. Under acidic conditions, compound 9 was hydrolyzed and generated the deprotected amine in situ that acted as a building block to be acylated with an acyl chloride or anhydride to synthesize N-acyl intermediates 10b,c. For compound 10a, the electrophilic reagent N-formylimidazole was prepared using formic acid and 1,1′-carbonyldiimidazole.41 Removal of the benzyl group using BCl3 yielded retrohydroxamic acids 11a–c.27 Treatment of these acids with TMSBr, followed by either NaOH or NH3, gave monosodium salts 12a,b or diammonium salt 12c, respectively.21,27 In the case of 11c, synthesis of the sodium salt resulted in an unstable compound, leading us to make the ammonium salt.
Scheme 1. Synthesis of N-Acyl Analogues 11a–c, 12a–ca.

aReagents and conditions: (a) allyl bromide, 60 °C, 2 days (85%); (b) Br2, CH2Cl2, 0 °C to rt, 2 h (88%); (c) Boc2O, TEA, H2O, THF, rt, 2.5 h (90%); (d) NaH, NaI, THF, 0 °C to rt, 20 h (77%); (e) (i) AcCl, MeOH, rt, 30 min, (ii) Na2CO3, RCOCl or (RCO)2O, 0 °C to rt, 30 min to 24 h (10a: Na2CO3, HCOOH, 1,1′-carbonyldiimidazole, 0 °C, 30 min) (50–73%); (f) BCl3, CH2Cl2, −70 °C, 30 min to 3 h (19–85%); (g) (i) TMSBr, CH2Cl2, 0 °C to rt, 24 h, (ii) NaOH, H2O, rt, 1 h (quant); (h) (i) TMSBr, CH2Cl2, 0 °C to rt, 24 h, (ii) NH4OH, H2O, rt, 1 h (quant).
Scheme 2 shows the four-step synthesis used to prepare the N-alkoxy analogues, starting from intermediate 9. Acetyl chloride was used to synthesize compound 13 following the method described above. Subsequent debenzylation of 13 by BCl3 generated 14, the diethyl ester of FR-900098.24 This compound acted as a core intermediate from which to synthesize the series of the N-alkoxy analogues. The synthesis of 15d–g was initially attempted using Williamson ether synthesis with either NaH or sodium tert-butoxide in polar solvents such as THF and DMF.27 Unfortunately, these reactions failed because of the instability of 14 in the presence of such harsh bases. Weaker bases, such as Na2CO3 or Et3N, were then applied to the reaction, and yet an overwhelming amount of side products were generated concomitantly, possibly due to the high polarity of the solvents. The reaction conditions were optimized using Na2CO3 with CH2Cl2 as a relatively nonpolar solvent. The reaction was carried out in a sealed tube and heated at 60 °C for 48 h to yield 15d–g, which were then converted to monosodium salts 16d–g in a manner similar to that used to prepare 12a–c.
Scheme 2. Synthesis of N-Hydroxyl Analogues 15d–g, 16d–ga.

aReagents and conditions: (a) (i) AcCl, MeOH, rt, 30 min, (ii) Na2CO3, CH3COCl, 0 °C to rt, 24 h (85%); (b) BCl3, CH2Cl2, −70 °C, 30 min to 3 h (77%); (c) R′CH2Br or R′CH(CH3)Br, Na2CO3, NaI, CH2Cl2, 60 °C, 2 days (25–45%); (d) (i) TMSBr, CH2Cl2, 0 °C to rt, 24 h, (ii) NaOH, H2O, rt, 1 h (51–95%).
The prodrugs of selected analogues were made (Scheme 3). To obtain the diPOM esters for N-acyl analogues (17a–c), compounds 10a–c were treated with TMSBr and then transformed to the esters using POM chloride and Et3N.27 Because of the low yield of 17b, only 17a and 17c were carried to the next reaction. Removal of the benzyl group using BCl3 gave the diPOM prodrugs 18a and 18c. Similarly, the diPOM esters of N-alkoxy analogues 19e–g were prepared from 16e–g via treatment with POM chloride and Et3N.
Scheme 3. Synthesis of POM Prodrugs 18a,c and 19e–ga.

aReagents and conditions: (a) (i) TMSBr, CH2Cl2, 0 °C to rt, 24 h, (ii) NaOH, H2O, rt, 1 h (1–30%); (b) chloromethylpivalate, TEA, NaI, DMF, 60 °C, 24 h (8–15%); (c) BCl3, CH2Cl2, −70 °C, 30 min to 3 h (34–55%).
Because of the low yields of N-acyl analogue 18a (overall yield 2.1%) and 17b (overall yield 0.42%), an optimized synthesis for N-acyl prodrugs was developed (Scheme 4). This route to synthesize diPOM esters was brought forward because the stability of compound 6 enabled a longer reaction time. By this method, compound 20 was obtained in higher yield (52%) compared with previous esterification reactions, and the synthetic attrition was greatly reduced. Bromine addition of the resulting product 20 yielded dibromide 21. Because of the instability of the POM group to acidic conditions, use of Boc-protected O-benzylhydroxylamine was inadvisible due to the acidic conditions that would be required to cleave the Boc group in the future steps. Thus, free amine 23 was prepared to react with monobromide 22, the eliminated product from 21 and NaH, to yield 24. This conversion enabled a variety of diPOM N-acyl analogues. Compound 24 was converted to acylated compounds 17a and 17b, with the latter compound obtained in a 17-fold increase in yield (overall yield 7.0%) versus the previous synthetic route (overall yield 0.42%). The final N-acyl prodrugs 18a (overall yield 3.9%) and 18b were subsequently obtained via a debenzylation reaction as in Scheme 3.
Scheme 4. Optimized Synthesis of N-Acyl Analogue Prodrugs 18a,ba.

aReagents and conditions: (a) (i) TMSBr, CH2Cl2, 0 °C to rt, 24 h, (ii) CH3OH, rt, 1 h, (iii) chloromethylpivalate, TEA, NaI, DMF, 60 °C, 24 h (52%); (b) Br2, CH2Cl2, 0 °C to rt, 2 h (78%); (c) NaH, THF, rt, 24h (70%); (d) NaOH, Et2O, H2O, rt, 30 min (97%); (e) TEA, THF, reflux, 3 h (34%); (f) TEA, (CF3CO)2O, 0 °C, 30 min or TEA, HCOOH, 1,1′-carbonyldiimidazole, 0 °C, 30 min (73–82%); (g) BCl3, CH2Cl2, −70 °C, 30 min to 3 h (34–55%).
Evaluation of 12a–c, 16d–g as DXR Inhibitors.
The phosphonic acid salts were evaluated as inhibitors of DXR from P. falciparum, and the results are shown in Table 1. Initially, the percent remaining enzyme activity was measured by treating the enzyme with each compound at a single concentration of 100 μM. This data shows the intrinsic activity of the compounds. Half-maximal inhibitory concentrations (IC50 values) were determined for compounds showing greater than 75% inhibition of DXR.
Table 1.
Inhibition of P. falciparum DXR by Phosphonic Acid Salts
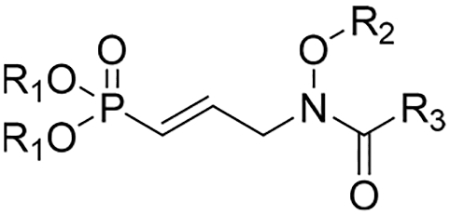
| compd | R1 | R2 | R3 | Pf DXR IC50 [μM]a |
|---|---|---|---|---|
| 1a | 0.064 | |||
| 1b | 0.023 | |||
| 2 | 0.206 | |||
| 12a | Na/H | H | H | 0.092 |
| 12b | Na/H | H | CF3 | (37) |
| 12c | NH4 | H | OCH3 | 14.45 |
| 16d | Na/H | CH(CH3)Ph | CH3 | 2.54 |
| 16e | Na/H | CH2(4-iprPh) | CH3 | 2.11 |
| 16f | Na/H | CH2(2-naphthyl) | CH3 | (25) |
| 16g | Na/H | CH2(4-biphenyl) | CH3 | 4.53 |
Values in parentheses are percent remaining enzyme activity at 100 μM. Pf = P. falciparum; IC50 = half-maximal inhibitory concentration
Of the new compounds, the most potent P. falciparum DXR inhibitor is 12a, with an IC50 value of 92 nM, slightly more potent than parent unsaturated compound 2. Within the N-alkoxy series of bisubstrate inhibitors 16d–g, compound 16e (4-iprPh) displays the most potent inhibition of the enzyme with an IC50 value of 2.11 μM. Interestingly, the N-alkoxy substituent of 16e was also found among the more active substituents in the saturated series.27 Compounds 16d and 16g with phenethyl and biphenyl substituents, respectively, were also active against the enzyme, displaying low μM inhibition.
Compounds 12b,c explore the influence of electronic effects in DXR inhibition, comparing an electron-withdrawing group (CF3, 12b) and electron-donating group (OCH3, 12c) with parent formyl analogue 12a and acetyl analogue 2. Compounds 12b and 12c display only weak and moderate inhibition of P. falciparum DXR, respectively. This result shows that electronic effects on the N-acyl group do not sway DXR inhibition as neither the CF3 group (12b) or OCH3 group (12c) improved the activity compared with the CH3 group of 2, which is a potent inhibitor of DXR.
In Vitro Effects on Pathogen Growth by 12a–c, 16d–g, 18a–c, 19e–g.
POM prodrugs of selected analogues were synthesized in an effort to improve their cellular activity (and possibly bioavailability). All target compounds were tested for growth inhibition against P. falciparum following reported procedures (Table 2).35 This data indicates the inhibitory concentration of compound required to decrease growth of P. falciparum by 50% (Pf IC50).
Table 2.
Growth Inhibition of the Analogues against P. falciparuma
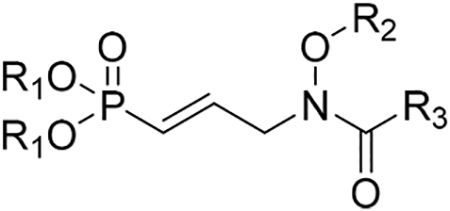
| compd | R1 | R2 | R3 | Pf DXR IC50 [μM] |
|---|---|---|---|---|
| 1a | 1.087 ± 0.064 | |||
| 1b | 0.511 ± 0.0535 | |||
| 2 | Na/H | H | CH3 | 0.202 ± 0.01535 |
| 3 | POM | H | CH3 | 0.018 ± 0.00235 |
| 12a | Na/H | H | H | 0.019 ± 0.002 |
| 12b | Na/H | H | CF3 | 113.3 ± 22.4 |
| 12c | NH4 | H | OCH3 | 28.6 ± 5.0 |
| 16d | Na/H | CH(CH3)Ph | CH3 | 1.2 ± 0.1 |
| 16e | Na/H | CH2(4-iprPh) | CH3 | 1.1 ± 0.0 |
| 16f | Na/H | CH2(2-naphthyl) | CH3 | 26.1 ± 1.1 |
| 16g | Na/H | CH2(4-biphenyl) | CH3 | 2.3 ± 0.4 |
| 18a | POM | H | H | 0.013 ± 0.002 |
| 18b | POM | H | CF3 | 14.3 ± 2.5 |
| 18c | POM | H | OCH3 | 23.7 ± 6.6 |
| 19e | POM | CH2(4-iprPh) | CH3 | 55.2 ± 1.9 |
| 19f | POM | CH2(2-naphthyl) | CH3 | 50.5 ± 1.1 |
| 19g | POM | CH2(4-biphenyl) | CH3 | 44.1 ± 1.1 |
Pf = P. falciparum; IC50 = concentration giving 50% inhibition of Pf growth
Because of the penetrable cell membrane of eukaryotic parasite P. falciparum, as well as the significant remodeling of host cell membranes by malaria parasites, we expect substantial cellular uptake into P. falciparum.42 In Table 2, the polar phosphonic acid salts show significant activity against P. falciparum parasites. Compound 12a is the most active compound of the phosphonate salts, with an activity surpassing that of parent compound (and clinically evaluated candidate) fosmidomycin (1a). The data also shows that the inhibition of P. falciparum growth corresponds well to the activities of these compounds against the enzyme target P. falciparum DXR. Of the salts, compounds 12a and 16e were the most active DXR inhibitors. These compounds are also the most active inhibitors of P. falciparum growth among the salts.
The cellular activity of the POM prodrugs is also shown in Table 2. As was the case with the phosphonic acid salts, several of the POM prodrugs are highly active against P. falciparum. Of the N-acyl series, compound 12a was the most potent P. falciparum DXR inhibitor. Its prodrug, compound 18a, is the most potent prodrug inhibitor of P. falciparum (IC50 = 13 nM) from the POM series. In the N-alkoxy series of compounds, compound 16e was the most potent DXR inhibitor and also shows the highest potency (IC50 = 1.1 μM) against P. falciparum parasites. Interestingly, addition of the prodrug did not improve the activity of this compound.
As is evident from the data in Table 2, several compounds show potent antimalarial activity. Much of our work focuses on analogues of fosmidomycin, which is itself a reasonably potent inhibitor of P. falciparum growth (1a, IC50 = 1.087 μM). Modification of fosmidomycin with the sole change of added α,β-unsaturation yields compound 12a as a highly potent P. falciparum inhibitor with an IC50 value of 19 nM. Its prodrug 18a also potently inhibits P. falciparum with an IC50 value of 13 nM. This value is comparable to the inhibitory activity of current first-line antimalarial drug artemisinin (P. falciparum IC50 = 10.4 nM).35
The cLogP, Cytotoxicity, and Selective Indices of 12a and 18a.
Because of the remarkable activities of compounds 12a and 18a, additional studies were pursued. The computed cLogP,43 inhibition of HepG2, and selectivity indices (ratio of the antimicrobial activity to the human cell toxicity) for compounds 12a and 18a are shown in Table 3. As expected, compound 12a has a low cLogP value of −5.7. The prodrug strategy significantly increased the lipophilicity of the compound, yielding 18a with a cLogP of 0.89 (an increase of over 6 orders of magnitude). Neither the phosphonic acid salt 12a nor the prodrug 18a show toxicity against HepG2 cell lines, with IC50 values >50 μM. Thus, these compounds have excellent selectivity indices of 2632 and 3846 for 12a and 18a, respectively, against P. falciparum. These compounds show promise as safe drug candidates for malaria.
Table 3.
Computed cLogP, Cytotoxicity, and Selectivity Indexa
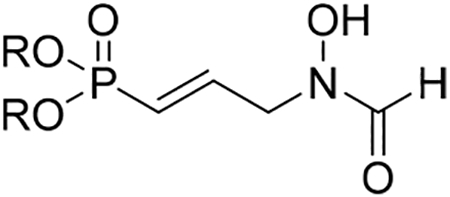
| compd | R | cLogP | HepG2 IC50 [μM] |
SIPf | metabolic stability MLM (t1/2) (min) |
plasma stability (min) |
|---|---|---|---|---|---|---|
| 12a | Na/H | −5.7 | >50 | 2632 | >60 | >120 |
| 18a | POM | 0.89 | >50 | 3846 | <5 | <5 |
cLogP calculated by DataWarrior;44 SIPf = selectivity index for P. falciparum (HepG2 IC50/P. falciparum IC50); MLM = mouse liver microsomes
Compound 18a Is Rapidly Converted to 12a in Vitro and in Vivo.
Compound 18a was designed to be a prodrug for 12a. To determine the rate of hydrolysis by plasma and hepatic esterases, compounds 12a and 18a were incubated in mouse liver microsomes (MLM) and in mouse plasma (Table 3). Compound 12a is stable in plasma (t1/2 > 120 min) and in mouse liver microsomes (t1/2 > 60 min). In contrast, POM prodrug 18a is very rapidly converted to compound 12a in plasma and microsomes (t1/2 < 5 min for both).
Compounds 12a and 18a Inhibit Isoprenoid Synthesis in P. falciparum.
Because of their significant activities against P. falciparum growth, we examined compounds 12a and 18a in further assays to determine their intracellular mechanism of action. First, we asked if parasites treated with these inhibitors could be rescued by MEP pathway product IPP supplementation. If the inhibitors target the MEP pathway, parasite growth should be restored if exogenous IPP were added to the growth media.35 As is shown in Figure 4, addition of exogenous IPP effectively rescues growth of P. falciparum treated with 12a or 18a. This pattern is similar to the restoration effect observed in fosmidomycin (1a)-treated parasites supplied with IPP.35 In addition, P. falciparum growth inhibited by the N-alkoxy analogue 16e is also restored by IPP supplementation (Supporting Information, Figure S3). This data hints that these α,β-unsaturated fosmidomycin analogues might act on target to inhibit parasitic growth by blocking the MEP pathway, the targeted intracellular pathway.
Figure 4.

Isoprenoid precursors rescue MEPicide-treated P. falciparum. P. falciparum strain 3D7 was treated with inhibitors at a range of concentrations and growth was quantified by PicoGreen (Life Technologies) after 72 h, as previously described.35 IPP, the product of the MEP pathway, rescues growth of drug-treated parasites (open shapes) hinting the compounds might be inhibitors of the MEP pathway in P. falciparum. Shown are representative graphs from three independent experiments. Data from remaining experiments is shown in Supporting Information, Figure S2.
Similarly, the proposed DXR inhibitor 18a should deplete MEP pathway intermediates beyond DXR from the treated P. falciparum. Thus, we employed a mass spectrometry-based method to quantify the MEP metabolites from untreated P. falciparum versus parasites treated with this compound (Figure 5).45 These metabolites include the DXR substrate (1-deoxy-D-xylulose 5-phosphate, DOXP), the DXR product (2-C-methylerythritol 4-phosphate, MEP), and the downstream metabolites (4-diphosphocytidyl-2-C-methylerythritol (CDP-ME) and 2-C-methyl-D-erythritol 2,4-cyclopyrophosphate (MEcPP)). Such metabolite levels were measured by LC-MS/MS after the 3D7 parasites were treated ± 18a after 10 h. The DOXP levels did not show significant difference between the untreated and treated parasites, while the metabolites downstream of DXR displayed reduced levels in treated parasites. This metabolic profiling data suggests that 18a inhibits DXR, the first committed enzyme in the MEP pathway from P. falciparum.
Figure 5.

Treating P. falciparum with 18a results in diminished concentrations of MEP pathway metabolites. Intracellular concentrations of MEP pathway metabolites were measured, comparing untreated (UNT) parasites and those treated with 18a at 5× the IC50 value of 13 nM. The cultures were saponin-lysed after treatment for 10 h and subsequently analyzed by LC-MS/MS. Shown are the metabolite levels from three independent experiments.
To further elucidate the mode of action of these analogues, compounds 12a and 18a were evaluated for efficacy against a unique mutated P. falciparum strain by using reported procedures.46 Because of a mutation in the metabolic regulator HAD1 (PF3D7_1033400), this P. falciparum strain produces high levels of DOXP, the substrate for DXR. Increased DOXP levels make it more difficult to inhibit DXR. For example, using this mutant, the (competitive) inhibition of DXR by fosmidomycin (1a) is impeded, resulting in fosmidomycin (1a)-resistant parasites (had1 parasites).35 As shown in Figure 6, had1 parasites (had1; open shapes, black line) were 3.4-fold and 2.5-fold more resistant to 12a and 18a, respectively, when compared with wild-type P. falciparum (3D7; closed shapes, gray line). Notably, the sensitivity of 12a and 18a were restored if the mutant strain was supplied with a wild-type copy of HAD1 (had1 + HAD1-GFP; closed shapes, black line). Similar results were observed with N-alkoxy analogue 16e, where had1 parasites were more resistant to these compounds than wild-type parasites (Supporting Information, Figure S2). These data corroborate earlier findings that these α,β-unsaturated fosmidomycin analogues inhibit P. falciparum growth via inhibition of the DXR enzyme in the MEP pathway.35
Figure 6.

P. falciparum had1 mutant strains resistant to fosmidomycin (1a) are also resistant to MEPicides. Dose-dependent growth inhibition was determined for P. falciparum strains treated with inhibitors as previously described.35 The had1 mutation results in higher levels of the DXR substrate DOXP, and mutants are resistant to DXR inhibition as indicated by a shift in the IC50 curve (had1; open shapes, black line) when compared to WT P. falciparum (3D7; closed shapes, gray line). Sensitivity was restored if a WT copy of had1 was supplied in the mutant strain (had1 + HAD1-GFP; closed shapes, black line). Data are representative of at least three independent biological replicates performed in duplicate.
In Vivo Evaluation of 18a in a Mouse Model of Malaria Infection.
Compound 18a stands out among these analogues and appears to be a promising antimalarial agent due to its potent inhibition of P. falciparum growth. Thus, compound 18a was further evaluated for in vivo efficacy in a Plasmodium berghei-infected mouse model of malaria using reported procedures.35 Groups of mice were infected with luciferase-based blood-stage P. berghei ANKA by intraperitoneal (ip) injection. After being infected for 2 days, the mice were treated daily for 5 days with vehicle, 20 mg/kg chloroquine, 20 mg/kg 18a, or 50 mg/kg 18a via ip injection. Seven days after infection, intensity of the luciferin signal was measured, correlating to the parasitemia burden. As shown in Figure 7, parasitemia was greatly reduced in mice treated with the control drug chloroquine (20 mg/kg), lowering the luciferin signal intensity to 2.13 × 103. Interestingly, mice treated with 18a at the same dose showed a similar result as chloroquine, with a 3-log drop in luciferin signal intensity (2.87 × 103) when compared to the vehicle (2.62 × 106). When a higher dose of 18a was administered, the average luciferin signal intensity of the P. berghei-infected mice is 2.34 × 103, not significantly different from the result with the lower dose. Additionally, 18a was well tolerated in mice at these dosages, as no adverse effects were observed, corroborating our results with the HepG2 cells.
Figure 7.

Compound 18a is effective in an in vivo mouse model of efficacy. Swiss Webster mice in groups of 2–5 were infected with 103 P. berghei parasites expressing luciferase. From day 2 to day 7, mice were dosed daily with vehicle, 20 mg/kg chloroquine, 20 mg/kg 18a, or 50 mg/kg 18a. Mice were imaged using an IVIS imager at 7 days postinfection (A) and parasitemia was quantified (B). All doses were administered ip.
On the basis of our in vitro stability studies, we anticipated that 18a would be rapidly converted to 12a in the in vivo study. We designed a compound exposure study to determine plasma concentration of 12a under similar conditions to the efficacy study. Compound 18a was dosed at 20 mg/kg ip in Swiss Webster mice (n = 3). and plasma samples were removed at select time points over the course of 8 h (Figure 8). Over the course of 8 h, 12a was observed at high concentrations, with a concentration of 485 ng/mL (2.8 μM) at 8 h which is approximately 200-fold above the Pf IC50 of 18a in vitro.
Figure 8.

Compound 18a is rapidly converted to 12a in vivo. Swiss Webster mice were dosed with 18a at 20 mg/kg ip. Plasma samples were collected over 8 h and analyzed for concentration of 12a by LC-MS/MS.
CONCLUSIONS
With a growing number of drug-resistant cases of malaria, there is an urgent demand for new antimalarial agents with novel modes of action. DXR, an essential enzyme in the causative pathogens of malaria, while completely absent in humans, appears to be a promising drug target for the treatment of this disease. We aim to develop compounds that selectively inhibit this crucial enzyme to eliminate P. falciparum while avoiding toxicity in humans. Thus, several α,β-unsaturated analogues of fosmidomycin were designed and synthesized, including the N-alkoxy and N-acyl series. Our lead compound, 18a, stands out as a superior antimalarial agent. The excellent safety profile, transparent mode of action, and encouraging in vivo efficacy of 18a in eliminating Plasmodium spp. parasites establish 18a as a promising drug candidate for the treatment of malaria. Encouraging activities of these N-alkoxy and N-acyl fosmidomycin analogues suggests a new direction for the development of more prospective drug candidates in the fight against this important human pathogen.
EXPERIMENTAL SECTION
General.
1H and 13C NMR spectra were recorded in CDCl3, CD3OD, or D2O on Agilent spectrometer at 400 and 101 MHz, respectively, with TMS, H2O, or solvent signal as internal standard. Chemical shifts are given in parts per million (ppm). Spin multiplicities are given with the following abbreviations: s (singlet), br s (broad singlet), d (doublet), dd (doublet of doublets), ddd (doublet of doublets of doublets), t (triplet), dt (doublet of triplets), ddt (doublet of doublet of triplets), q (quadruplet), qt (quintuplet), and m (multiplet). Mass spectra were measured in the ESI mode on an HPLC-MS (Agilent 1100) or in the EI mode on an GC-MS (Shimadzu GCMS-QP2010S). Thin layer chromatography (TLC) was performed on Baker-flex Silica Gel IB2-F silica plates, and flash column chromatography was carried out using SiliCycle SiliaFlash P60 silica gel (40–63 μm). All reagents were purchase from commercial suppliers and used without further purification. Anhydrous solvents were purified by MBRAUN MB-SPS solvent purification system before use. All air sensitive reactions were carried out under nitrogen atmosphere. The purity of synthesized compounds (>95%) was determined by 1H/13C NMR in combination with HPLC-MS (Agilent 1100). Column: Thermo Fisher Scientific Hypersil GOLD aQ C-18 3 μm particle (250 mm × 4.6 mm). Mobile phase (containing 0.1% formic acid as the additive): linear gradient of acetonitrile (50%–100%) in water at a flow rate of 0.8 mL/min over 12.5 min, followed by 100% acetonitrile that was maintained for another 12.5 min. The UV detection wavelength was 210 and 254 nm. High-resolution mass spectroscopy spectra (HRMS) were recorded in positive or negative ESI mode on a Waters Q-TOF Ultima mass spectrometer (UIUC Mass Spectrometry Laboratory) or in positive FAB mode on a VG Analytical VG70SE magnetic sector mass spectrometer (JHU Mass Spectrometry Facility).
General Procedure for Synthesis of Amides 10a–c and 13.
To a solution of MeOH (10.1 equiv) in dry CH2Cl2 (1 M) under N2 was added acetyl chloride (10 equiv) dropwise at room temperature, and the mixture was stirred for 10 min. The reaction mixture was then added a solution of 9 (1 equiv) in dry CH2Cl2 (1 M) and stirred at room temperature for 30 min. After the completion of deprotection, dry Na2CO3 (12 equiv) was added at 0 °C and the mixture was stirred at the same temperature for 10 min. The reaction mixture at 0 °C was added dropwise RCOCl, (CF3CO)2O, or N-formylimidazole* (2 equiv). The mixture was then warmed up to room temperature and stirred for 30 min to 24 h, quenched with saturated NaHCO3 (aq), and extracted with CH2Cl2 (3×). The combined organic layers were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude concentrate was then purified by column chromatography on silica gel using EtOAc and CH2Cl2 (with a ratio from 1/10 to 2/1) to give the pure title compound.
General Procedure for Synthesis of 11a–c, 14, and 18a–c.24
To a solution of 10, 13, or 17 (1 equiv) in dry CH2Cl2 (0.1 M) under N2 was added boron trichloride (1 M in CH2Cl2, 4 equiv) at −78 °C dropwise. The reaction mixture was stirred at −78 °C for 30 min to 3 h, quenched with saturated NaHCO3 (aq), and extracted with EtOAc (5×). The combined organic layers were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude residue was then purified by column chromatography on silica gel using EtOAc and MeOH (EtOAc and CH2Cl2 for 18a–c) to give the pure title compound.
General Procedure for Synthesis of 12a,b and 16d–g.
To a solution of 11a,b or 15d–g (1 equiv) in dry CH2Cl2 (0.1 M) under N2 was added TMSBr (10 equiv) dropwise at 0 °C. The reaction mixture was warmed to room temperature, stirred overnight, and then concentrated under reduced pressure. The mixture was dissolved in CH2Cl2, evaporated under reduced pressure, and dried under vacuum. The crude residue was then stirred in 0.5 M NaOH (1 equiv) in H2O at room temperature for 1 h, washed with Et2O three times, and lyophilized to give the title compounds.
General Procedure for Synthesis of 17a–c.
To a solution of 10a–c (1 equiv) in dry CH2Cl2 (0.1 M) under N2 was added TMSBr (10 equiv) dropwise at 0 °C. The reaction mixture was warmed to room temperature, stirred overnight, and then concentrated under reduced pressure. The mixture was dissolved in CH2Cl2, evaporated under reduced pressure, and dried under vacuum. The crude residue was then stirred in 0.5 M NaOH (2 equiv) in H2O at room temperature for 1 h, washed with Et2O (3×), and lyophilized to give disodium salts as white solids. The crude solid was then dissolved in dry DMF (0.1M), and TEA (6 equiv), chloromethylpivalate (6 equiv), and NaI (0.1 equiv) were added. The reaction mixture was stirred at 60 °C for 24 h, quenched with H2O, and extracted with Et2O (3×). The combined organic layers were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was then purified by column chromatography on silica gel using hexanes and EtOAc or CH2Cl2 and EtOAc to give the pure title compound.
Diethyl [(1E)-3-[N-(Benzyloxy)formamido]prop-1-en-1-yl]-phosphonate (10a).
Light-yellow oil (594 mg, 73%). 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 7.41–7.29 (m, 5H), 6.72–6.57 (m, 1H), 5.82 (ddd, J = 12.4, 10.0, 6.3 Hz, 1H), 4.84 (s, 2H), 4.30–4.20 (m, 2H), 4.13–3.98 (m, 4H), 1.32–1.27 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 163.3, 144.40 (d, J = 5.7 Hz), 134.1, 129.4, 129.2, 128.8, 120.8 (d, J = 188.7 Hz), 78.3, 61.9 (d, J = 5.6 Hz), 47.2 (d, J = 26.7 Hz), 16.3 (d, J = 6.3 Hz). LC-MS (ESI+): 328.2 m/z [M + H]+, 655.2 m/z [2M + H]+.
Diethyl [(1E)-3-(N-Hydroxyformamido)prop-1-en-1-yl]-phosphonate (11a).
Light-yellow oil (106 mg, 55%). 1H NMR (400 MHz, CDCl3) δ 10.06 (s, 1H), 8.32 (s, 1H), 6.75–6.55 (m, 1H), 5.99–5.74 (m, 1H), 4.32–4.25 (m, 2H), 4.09–3.92 (m, 4H), 1.25 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 172.5, 162.78, 146.3 (d, J = 5.5 Hz), 118.7 (d, J = 189.1 Hz), 62.3 (d, J = 5.7 Hz), 48.
Sodium Hydrogen [(1E)-3-(N-Hydroxyformamido)prop-1-en-1-yl]phosphonate (12a).
Light-yellow solids (29 mg, quantitative yield). 1H NMR (400 MHz, CD3OD) δ 8.32 and 7.99 (s, 1H), 6.52–6.30 (m, 1H), 6.11–5.93 (m, 1H), 4.32–4.17 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 162.6, 137.5 (d, J = 5.0 Hz), 127.3 (d, J = 178.0 Hz), 48.7 (d, J = 23.6 Hz). LC-MS (ESI−): 361.0 m/z [2M-2Na + H]−, 542.2 m/z [3M-3Na + 2H]−. HRMS (ESI−) calculated for C4H7NNaO5P 202.9960, found 180.0065 [M − Na]−.
({[(1E)-3-[N-(Benzyloxy)formamido]prop-1-en-1-yl]({[(2,2-dimethylpropanoyl)oxy]methoxy})phosphoryl}oxy)methyl 2,2-Dimethylpropanoate (17a).
Light-yellow oil (38 mg, 9%). 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 1H), 7.43–7.30 (m, 5H), 6.72 (ddt, J = 22.4, 17.2, 5.1 Hz, 1H), 5.96–5.82 (m, 1H), 5.66 (dd, J = 13.1, 0.8 Hz, 4H), 4.84 (s, 2H), 4.30–4.20 (m, 2H), 1.21 (s, 18H). 13C NMR (101 MHz, CDCl3) δ 176.7, 163.2, 146.0 (d, J = 6.0 Hz), 129.9, 129.5, 129.3, 128.8, 119.4 (d, J = 193.0 Hz), 81.5 (d, J = 5.4 Hz), 78.3, 47.0 (d, J = 25.8 Hz), 38.7, 26.8. LC-MS (ESI+): 500.2 m/z [M + H]+, 999.2 m/z [2M + H]+.
Optimized Synthesis of ({[(1E)-3-[N-(Benzyloxy)formamido]prop-1-en-1-yl]({[(2,2-dimethylpropanoyl)oxy]methoxy})phosphoryl}oxy)methyl 2,2-Dimethylpropanoate (17a).
To a suspension of 1,1′-carbonyldiimidazole (688 mg, 4.3 mmol, 2 equiv) in CH2Cl2 (3 mL) was added formic acid (0.16 mL, 4.3 mmol, 2 equiv) dropwise at room temperature. The mixture was stirred at room temperature for 5 min, and then transferred dropwise to a solution of 24 (1.0 g, 2.1 mmol, 1 equiv) and TEA (0.85 mL, 6.4 mmol, 3 equiv) in CH2Cl2 (15 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 30 min, quenched with saturated aqueous NaHCO3 (30 mL), and extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Chromatographic separation on silica gel (hexanes/EtOAc = 2/1 to 1/3) gave the title compound as a light-yellow oil (862 mg, 82%). 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 1H), 7.43–7.30 (m, 5H), 6.72 (ddt, J = 22.4, 17.2, 5.1 Hz, 1H), 5.96–5.82 (m, 1H), 5.66 (dd, J = 13.1, 0.8 Hz, 4H), 4.84 (s, 2H), 4.30–4.20 (m, 2H), 1.21 (s, 18H). 13C NMR (101 MHz, CDCl3) δ 176.7, 163.2, 146.0 (d, J = 6.0 Hz), 129.9, 129.5, 129.3, 128.8, 119.4 (d, J = 193.0 Hz), 81.5 (d, J = 5.4 Hz), 78.3, 47.0 (d, J = 25.8 Hz), 38.7, 26.8. LC-MS (ESI+): 500.2 m/z [M + H]+, 999.2 m/z [2M + H]+.
[({[(2,2-Dimethylpropanoyl)oxy]methoxy}[(1E)-3-(N-hydroxyformamido)prop-1-en-1-yl]phosphoryl)oxy]methyl 2,2-Dimethylpropanoate (18a).
Light-yellow oil (103 mg, 55%). 1H NMR (400 MHz, CDCl3) δ 9.41 (s, 1H), 8.42 and 7.94 (s, 1H), 6.87–6.69 (m, 1H), 6.07–5.88 (m, 1H), 5.71–5.59 (m, 4H), 4.37–4.32 (m, 2H), 1.21 (s, 18H). 13C NMR (101 MHz, CDCl3) δ 177.1, 163.0, 157.3, 147.3 (d, J = 6.1 Hz), 118.6 (d, J = 191.8 Hz), 81.6 (d, J = 4.9 Hz), 48.8 (d, J = 26.1 Hz), 38.7, 26.8. LC-MS (ESI+): 410.2 m/z [M + H]+, 819.2 m/z [2M + H]+. HRMS (ESI+) calculated for C16H28NO9P 409.1502, found 432.1388 [M + Na]+.
({[(1E)-3-[(Benzyloxy)amino]prop-1-en-1-yl]({[(2,2-dimethylpropanoyl)oxy]methoxy})phosphoryl}oxy)methyl 2,2-Dimethylpropanoate (24).
To a solution of 22 (2.8 g, 6.5 mmol, 1 equiv) in dry THF (50 mL) was added 23 (980 mg, 8 mmol, 1.2 equiv) and TEA (1.73 mL, 13 mmol, 2 equiv). The reaction mixture was stirred at reflux for 3 h and then concentrated under reduced pressure. Chromatographic separation on silica gel (hexanes/EtOAc = 2/1 to 1/2) gave the title compound as a colorless oil (1.0 g, 34%). 1H NMR (400 MHz, CDCl3) δ 7.41–7.26 (m, 5H), 6.87 (ddt, 1H),6.02–5.85 (m, 1H), 5.70–5.62 (m, 4H), 4.68 (s, 2H), 3.67–3.63 (m, 2H), 1.21 (s, 18H). 13C NMR (101 MHz, CDCl3) δ 176.8, 150.1 (d, J = 5.3 Hz), 137.5, 128.4, 128.4, 128.0, 117.8 (d, J = 192.1 Hz), 81.5 (d, J = 5.5 Hz), 76.4, 54.0 (d, J = 24.2 Hz), 38.7, 26.8. LC-MS (ESI+): 472.2 m/z [M + H]+, 943.2 m/z [2M + H]+.
P. falciparum DXR Enzyme Inhibition Assay.
P. falciparum DXR activity was assayed at 37 °C by spectrophotometrically monitoring the enzyme catalyzed oxidation of NADPH upon addition of 1-deoxy-D-xylulose 5-phosphate (DOXP; Echelon Biosciences, Salt Lake City, UT) to the assay mixture, as described previously.47,48 Briefly, the assay system contained 100 mM Tris pH 7.8, 25 mM MgCl2, 0.86 μM Pf DXR, and 150 μM NADPH. The reaction was initiated by adding 144 μM DOXP to the complete assay mixture. One unit of P. falciparum DXR activity is defined as the amount of enzyme that catalyzes the oxidation of 1 μM NADPH per min. The oxidation of NADPH was monitored at 340 nm using an Agilent 8453 UV—visible spectrophotometer equipped with a temperature regulated cuvette holder. All assays were performed in duplicate.
P. falciparum Growth Inhibition Assays.46
Asynchronous P. falciparum cultures (strain 3D7, MR4/ATCC) were diluted to 1% parasitemia and treated with inhibitors at concentrations ranging from 1.2 nM to 492.4 μM. Growth inhibition assays were performed in opaque 96-well plates at 100 μL culture volume. After 3 days, parasite growth was quantified by measuring DNA content using PicoGreen (Life Technologies) as described.46 Fluorescence was measured on a FLUOstar Omega microplate reader (BMG Labtech) at 485 nm excitation and 528 nm emission. Half-maximal inhibitory concentration (IC50) values were calculated by nonlinear regression analysis using GraphPad Prism software. For isopentenyl pyrophosphate (IPP) (Echelon) rescue experiments, 125 μM IPP was added to the appropriate wells for the duration of the experiment. Experiments were performed in triplicate.
In Vivo Studies in a Mouse Model of Malaria Infection.
All animal procedures were conducted in compliance with the New York University Institutional Animal Care and Use Committee under Protocol No. 160720. Female Swiss Webster mice, weighing 25–30 g, were infected via ip injection with 103 transgenic P. berghei expressing luciferase (PMID: 16051702). Two days later, mice groups of five mice were injected ip with vehicle (2% methylcellulose, 0.5% Tween 80) or treatments (18a at 20 and 50 mg/kg). As control, two infected mice were treated with chloroquine (ip 20 mg/kg). All mice were injected daily for 5 days. One day after 5 days of treatment (day 7 after infection), the mice were anesthetized by inhalation of isofluorane (controlled flow of 2.5% isofluorane in air was administered through a nose cone via a gas anesthesia system). Mice were injected ip with 150 mg/kg of D-luciferin potassium salt (Goldbio) dissolved in PBS. Mice were imaged 5–10 min after injection of luciferin with an IVIS 100 (Xenogen, Alameda, CA). Data acquisition and analysis were performed with the software LivingImage (Xenogen).
Supplementary Material
ACKNOWLEDGMENTS
This work was generously supported by the George Washington University (GWU) Department of Chemistry, the GWU University Facilitating Fund, the NIH (AI 123433 to C.S.D. and AI 123808 to A.R.O.J.), and the Division of Intramural Research, NIAID, NIH. We thank Ana Rodriguez at the Anti-Infectives Screening core at NYU School of Medicine for testing the in vivo efficacy of 18e. We also thank Phil Mortimer (JHU) and Furong Sun (UIUC) for assistance with HRMS analysis. In vivo PK analysis was performed by Charles River Laboratories.
ABBREVIATIONS USED
- ACT
artemisinin-based combination therapy
- CDP-ME
4-diphosphocytidyl-2-C-methylerythritol
- DMAPP
dimethylallyl pyrophosphate
- DOXP
1-deoxy-D-xylulose 5-phosphate
- DXR
1-deoxy-D-xylulose 5-phosphate reductoisomerase
- IC50
drug concentration yielding 50 percent inhibition of activity
- ip
intraperitoneal
- IPP
isopentenyl pyrophosphate
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- MEcPP
2-C-methyl-D-erythritol 2,4-cyclopyrophosphate
- MEP
methyl erythritol phosphate
- MIC
minimum inhibitory concentration
- MLM
mouse liver microsomes
- Mtb
Mycobacterium tuberculosis
- NADPH
nicotinamide adenine dinucleotide phosphate (reduced form)
- POM
pivaloyloxymethyl
- Pf
Plasmodium falciparum
- PK
pharmacokinetic
- SAR
structure—activity relationship
- SI
selectivity index
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01026.
General experimental information, synthetic preparation of intermediates and target compounds, expression/purification/kinetic characterization of Pf DXR, parasitic growth and inhibition, HepG2 cytotoxicity, MEP pathway metabolite measurement, in vitro and in vivo PK, and in vivo efficacy, data for diethyl analogues against P. falciparum, IPP rescue for compounds 1a and 16e, and had1 resistance to compounds 1a and 16e (PDF)
Molecular formula strings (XLSX)
The authors declare no competing financial interest.
REFERENCES
- (1).World Malaria Report 2016; World Health Organization: Geneva, 2016; http://www.who.int/malaria/publications/world-malaria-report-2016/report/en/. [Google Scholar]
- (2).Rich SM; Leendertz FH; Xu G; LeBreton M; Djoko CF; Aminake MN; Takang EE; Diffo JLD; Pike BL; Rosenthal BM; Formenty P; Boesch C; Ayala FJ; Wolfe ND The origin of malignant malaria. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 14902–14907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ralph SA; van Dooren GG; Waller RF; Crawford MJ; Fraunholz MJ; Foth BJ; Tonkin CJ; Roos DS; McFadden GI Metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat. Rev. Microbiol 2004, 2, 203–216. [DOI] [PubMed] [Google Scholar]
- (4).Gerald N; Mahajan B; Kumar S Mitosis in the human malaria parasite Plasmodium falciparum. Eukaryotic Cell 2011, 10, 474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ashley EA; Dhorda M; Fairhurst RM; Amaratunga C; Lim P; Suon S; Sreng S; Anderson JM; Mao S; Sam B; Sopha C; Chuor CM; Nguon C; Sovannaroth S; Pukrittayakamee S; Jittamala P; Chotivanich K; Chutasmit K; Suchatsoonthorn C; Runcharoen R; Hien TT; Thuy-Nhien NT; Thanh NV; Phu NH; Htut Y; Han KT; Aye KH; Mokuolu OA; Olaosebikan RR; Folaranmi OO; Mayxay M; Khanthavong M; Hongvanthong B; Newton PN; Onyamboko MA; Fanello CI; Tshefu AK; Mishra N; Valecha N; Phyo AP; Nosten F; Yi P; Tripura R; Borrmann S; Bashraheil M; Peshu J; Faiz MA; Ghose A; Hossain MA; Samad R; Rahman MR; Hasan MM; Islam A; Miotto O; Amato R; MacInnis B; Stalker J; Kwiatkowski DP; Bozdech Z; Jeeyapant A; Cheah PY; Sakulthaew T; Chalk J; Intharabut B; Silamut K; Lee SJ; Vihokhern B; Kunasol C; Imwong M; Tarning J; Taylor WJ; Yeung S; Woodrow CJ; Flegg JA; Das D; Smith J; Venkatesan M; Plowe CV; Stepniewska K; Guerin PJ; Dondorp AM; Day NP; White NJ Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med 2014, 371, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Eoh H; Brennan PJ; Crick DC The Mycobacterium tuberculosis MEP (2C-methyl-d-erythritol 4-phosphate) pathway as a new drug target. Tuberculosis (Oxford, U. K.) 2009, 89, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jomaa H; Wiesner J; Sanderbrand S; Altincicek B; Weidemeyer C; Hintz M; Türbachova I; Eberl M; Zeidler J; Lichtenthaler HK; Soldati D; Beck E Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 1999, 285, 1573–1576. [DOI] [PubMed] [Google Scholar]
- (8).Goldstein JL; Brown MS Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [DOI] [PubMed] [Google Scholar]
- (9).Yeh E; DeRisi JL Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 2011, 9, e1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Masini T; Hirsch AKH Development of inhibitors of the 2C-methyl-D-erythritol 4-phosphate (MEP) pathway enzymes as potential anti-infective agents. J. Med. Chem 2014, 57, 9740–9763. [DOI] [PubMed] [Google Scholar]
- (11).Rohmer M The discovery of a mevalonate-independent pathway for isoprenoid biosynthesis in bacteria, algae and higher plants. Nat. Prod. Rep 1999, 16, 565–574. [DOI] [PubMed] [Google Scholar]
- (12).Argyrou A; Blanchard JS Kinetic and chemical mechanism of Mycobacterium tuberculosis 1-deoxy-d-xylulose-5-phosphate isomeroreductase. Biochemistry 2004, 43, 4375–4384. [DOI] [PubMed] [Google Scholar]
- (13).Brown AC; Parish T Dxr is essential in Mycobacterium tuberculosisand fosmidomycin resistance is due to a lack of uptake. BMC Microbiol. 2008, 8, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Odom AR; Van Voorhis WC Functional genetic analysis of the Plasmodium falciparum deoxyxylulose 5-phosphate reductoisomerase gene. Mol. Biochem. Parasitol 2010, 170, 108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Okuhara M; Kuroda Y; Goto T; Okamoto M; Terano H; Kohsaka M; Aoki H; Imanaka H Studies on new phosphonic acid antibiotics. III. Isolation and characterization of FR-31564, FR-32863 and FR-33289. J. Antibiot 1980, 33, 24–28. [DOI] [PubMed] [Google Scholar]
- (16).Armstrong CM; Meyers DJ; Imlay LS; Freel Meyers C; Odom AR Resistance to the antimicrobial agent fosmidomycin and an FR900098 prodrug through mutations in the deoxyxylulose phosphate reductoisomerase gene (dxr). Antimicrob. Agents Chemother. 2015, 59, 5511–5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wiesner J; Henschker D; Hutchinson DB; Beck E; Jomaa H In vitro and in vivo synergy of fosmidomycin, a novel antimalarial drug, with clindamycin. Antimicrob. Agents Chemother. 2002, 46, 2889–2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Borrmann S; Adegnika AA; Matsiegui P-B; Issifou S; Schindler A; Mawili-Mboumba DP; Baranek T; Wiesner J; Jomaa H; Kremsner PG Fosmidomycin-clindamycin for Plasmodium falciparum infections in African children. J. Infect. Dis 2004, 189, 901–908. [DOI] [PubMed] [Google Scholar]
- (19).Borrmann S; Lundgren I; Oyakhirome S; Impouma B; Matsiegui P-B; Adegnika AA; Issifou S; Kun JFJ; Hutchinson D; Wiesner J; Jomaa H; Kremsner PG Fosmidomycin plus clindamycin for treatment of pediatric patients aged 1 to 14 years with Plasmodium falciparum malaria. Antimicrob. Agents Chemother. 2006, 50, 2713–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bjorkelid C; Bergfors T; Unge T; Mowbray SL; Jones TA Structural studies on Mycobacterium tuberculosis DXR in complex with the antibiotic FR-900098. Acta Crystallogr., Sect. D: Biol. Crystallogr 2012, 68, 134–143. [DOI] [PubMed] [Google Scholar]
- (21).Chofor R; Sooriyaarachchi S; Risseeuw MDP; Bergfors T; Pouyez J; Johny C; Haymond A; Everaert A; Dowd CS; Maes L; Coenye T; Alex A; Couch RD; Jones TA; Wouters J; Mowbray SL; Van Calenbergh S Synthesis and bioactivity of β-substituted fosmidomycin analogues targeting 1-deoxy-d-xylulose-5-phosphate reductoisomerase. J. Med. Chem 2015, 58, 2988–3001. [DOI] [PubMed] [Google Scholar]
- (22).DeSieno MA; van der Donk WA; Zhao H Characterization and application of the Fe(ii) and [small alpha]-ketoglutarate dependent hydroxylase FrbJ. Chem. Commun. (Cambridge, U. K.) 2011, 47, 10025–10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Devreux V; Wiesner J; Jomaa H; Van der Eycken J; Van Calenbergh S Synthesis and evaluation of α,β-unsaturated α-aryl-substituted fosmidomycin analogues as DXR inhibitors. Bioorg. Med. Chem. Lett 2007, 17, 4920–4923. [DOI] [PubMed] [Google Scholar]
- (24).Jackson ER; San Jose G; Brothers RC; Edelstein EK; Sheldon Z; Haymond A; Johny C; Boshoff HI; Couch RD; Dowd CS The effect of chain length and unsaturation on Mtb Dxr inhibition and antitubercular killing activity of FR900098 analogs. Bioorg. Med. Chem. Lett 2014, 24, 649–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Jansson AM; Więckowska A; Björkelid C; Yahiaoui S; Sooriyaarachchi S; Lindh M; Bergfors T; Dharavath S; Desroses M; Suresh S; Andaloussi M; Nikhil R; Sreevalli S; Srinivasa BR; Larhed M; Jones TA; Karlén A; Mowbray SL DXR Inhibition by potent mono- and disubstituted fosmidomycin analogues. J. Med. Chem 2013, 56, 6190–6199. [DOI] [PubMed] [Google Scholar]
- (26).Kunfermann A; Lienau C; Illarionov B; Held J; Gräwert T; Behrendt CT; Werner P; Hähn S; Eisenreich W; Riederer U; Mordmüller B; Bacher A; Fischer M; Groll M; Kurz T IspC as target for antiinfective drug discovery: synthesis, enantiomeric separation, and structural biology of fosmidomycin thia isosters. J. Med. Chem 2013, 56, 8151–8162. [DOI] [PubMed] [Google Scholar]
- (27).San Jose G; Jackson ER; Haymond A; Johny C; Edwards RL; Wang X; Brothers RC; Edelstein EK; Odom AR; Boshoff HI; Couch RD; Dowd CS Structure–activity relationships of the MEPicides: N-acyl and O-linked analogs of FR900098 as inhibitors of Dxr from Mycobacterium tuberculosis and Yersinia pestis. ACS Infect. Dis 2016, 2, 923–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).San Jose G; Jackson ER; Uh E; Johny C; Haymond A; Lundberg L; Pinkham C; Kehn-Hall K; Boshoff HI; Couch RD; Dowd CS Design of potential bisubstrate inhibitors against Mycobacterium tuberculosis (Mtb) 1-deoxy-d-xylulose 5-phosphate reductoisomerase (Dxr)-evidence of a novel binding mode. MedChemComm 2013, 4, 1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Uh E; Jackson ER; San Jose G; Maddox M; Lee RE; Lee RE; Boshoff HI; Dowd CS Antibacterial and antitubercular activity of fosmidomycin, FR900098, and their lipophilic analogs. Bioorg. Med. Chem. Lett 2011, 21, 6973–6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Koppisch AT; Fox DT; Blagg BSJ; Poulter CDE coli MEP synthase: steady-state kinetic analysis and substrate binding. Biochemistry 2002, 41, 236–243. [DOI] [PubMed] [Google Scholar]
- (31).Jackson ER; Dowd CS Inhibition of 1-deoxy-D-Xylulose-5-phosphate reductoisomerase (Dxr): a review of the synthesis and biological evaluation of recent inhibitors. Curr. Top. Med. Chem 2012, 12, 706–728. [DOI] [PubMed] [Google Scholar]
- (32).Chofor R; Risseeuw DM; Pouyez J; Johny C; Wouters J; Dowd SC; Couch DR; Van Calenbergh S Synthetic fosmidomycin analogues with altered chelating moieties do not inhibit 1-deoxy-D-xylulose 5-phosphate reductoisomerase or Plasmodium falciparum growth in vitro. Molecules 2014, 19, 2571–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hemmi K; Takeno H; Hashimoto M; Kamiya T Studies on phosphonic acid antibiotics. IV. Synthesis and antibacterial activity of analogs of 3-(N-acetyl-N-hydroxyamino)-propylphosphonic acid (FR-900098). Chem. Pharm. Bull 1982, 30, 111–118. [DOI] [PubMed] [Google Scholar]
- (34).Woo Y-H; Fernandes RPM; Proteau PJ Evaluation of fosmidomycin analogs as inhibitors of the Synechocystis sp. PCC6803 1-deoxy-d-xylulose 5-phosphate reductoisomerase. Bioorg. Med. Chem 2006, 14, 2375–2385. [DOI] [PubMed] [Google Scholar]
- (35).Edwards RL; Brothers RC; Wang X; Maron MI; Ziniel PD; Tsang PS; Kraft TE; Hruz PW; Williamson KC; Dowd CS; Odom John AR MEPicides: potent antimalarial prodrugs targeting isoprenoid biosynthesis. Sci. Rep 2017, 7, 8400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Masini T; Kroezen BS; Hirsch AKH Druggability of the enzymes of the non-mevalonate-pathway. Drug Discovery Today 2013, 18, 1256–1262. [DOI] [PubMed] [Google Scholar]
- (37).Henriksson LM; Unge T; Carlsson J; Åqvist J; Mowbray SL; Jones TA Structures of Mycobacterium tuberculosis 1-deoxy-D-xylulose-5-phosphate reductoisomerase provide new insights into catalysis. J. Biol. Chem 2007, 282, 19905–19916. [DOI] [PubMed] [Google Scholar]
- (38).Fourgeaud P; Midrier C; Vors J-P; Volle J-N; Pirat J-L; Virieux D Oxaphospholene and oxaphosphinene heterocycles via RCM using unsymmetrical phosphonates or functional phosphinates. Tetrahedron 2010, 66, 758–764. [Google Scholar]
- (39).Laureyn I; Stevens CV; Soroka M; Malysa P Synthesis of γ-amino-α,β-unsaturated phosphonates via a substitution-elimination sequence of dibromophosphonates. ARKIVOC 2003, No. 4, 102–115. [Google Scholar]
- (40).Kadi N; Oves-Costales D; Barona-Gomez F; Challis GL A new family of ATP-dependent oligomerization-macrocyclization biocatalysts. Nat. Chem. Biol 2007, 3, 652–656. [DOI] [PubMed] [Google Scholar]
- (41).Haemers T; Wiesner J; Poecke SV; Goeman J; Henschker D; Beck E; Jomaa H; Calenbergh SV Synthesis of α-substituted fosmidomycin analogues as highly potent Plasmodium falciparum growth inhibitors. Bioorg. Med. Chem. Lett 2006, 16, 1888–1891. [DOI] [PubMed] [Google Scholar]
- (42).Guggisberg AM; Amthor RE; Odom AR Isoprenoid biosynthesis in Plasmodium falciparum. Eukaryotic Cell 2014, 13, 1348–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).I. CLOGP; Pomona College and BioByte: Claremont, CA, 1988–2011. [Google Scholar]
- (44).DataWarrior, version 4.6.1; openmolecules.org, 2017; http://www.openmolecules.org/index.html.
- (45).Zhang Z; Jakkaraju S; Blain J; Gogol K; Zhao L; Hartley RC; Karlsson CA; Staker BL; Edwards TE; Stewart LJ; Myler PJ; Clare M; Begley DW; Horn JR; Hagen TJ Cytidine derivatives as IspF inhibitors of Burkolderia pseudomallei. Bioorg. Med. Chem. Lett 2013, 23, 6860–6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Guggisberg AM; Park J; Edwards RL; Kelly ML; Hodge DM; Tolia NH; Odom AR A sugar phosphatase regulates the methylerythritol phosphate (MEP) pathway in malaria parasites. Nat. Commun 2014, 5, 4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Haymond A; Johny C; Dowdy T; Schweibenz B; Villarroel K; Young R; Mantooth CJ; Patel T; Bases J; Jose GS; Jackson ER; Dowd CS; Couch RD Kinetic characterization and allosteric inhibition of the Yersinia pestis 1-deoxy-D-xylulose 5-phosphate reductoisomerase (MEP synthase). PLoS One 2014, 9, e106243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Jawaid S; Seidle H; Zhou W; Abdirahman H; Abadeer M; Hix JH; van Hoek ML; Couch RD Kinetic characterization and phosphoregulation of the Francisella tularensis 1-deoxy-D-xylulose 5-phosphate reductoisomerase (MEP synthase). PLoS One 2009, 4, e8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.