

ABSTRACT
Kainate receptors (KARs) regulate neuronal excitability and network function. Most KARs contain the subunit GluK2 (also known as GRIK2), and the properties of these receptors are determined in part by ADAR2 (also known as ADARB1)-mediated mRNA editing of GluK2, which changes a genomically encoded glutamine residue into an arginine residue (Q/R editing). Suppression of synaptic activity reduces ADAR2-dependent Q/R editing of GluK2 with a consequential increase in GluK2-containing KAR surface expression. However, the mechanism underlying this reduction in GluK2 editing has not been addressed. Here, we show that induction of KAR upscaling, a phenomenon in which surface expression of receptors is increased in response to a chronic decrease in synaptic activity, results in proteasomal degradation of ADAR2, which reduces GluK2 Q/R editing. Because KARs incorporating unedited GluK2(Q) assemble and exit the ER more efficiently, this leads to an upscaling of KAR surface expression. Consistent with this, we demonstrate that partial ADAR2 knockdown phenocopies and occludes KAR upscaling. Moreover, we show that although the AMPA receptor (AMPAR) subunit GluA2 (also known as GRIA2) also undergoes ADAR2-dependent Q/R editing, this process does not mediate AMPAR upscaling. These data demonstrate that activity-dependent regulation of ADAR2 proteostasis and GluK2 Q/R editing are key determinants of KAR, but not AMPAR, trafficking and upscaling.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Synapse, Kainate receptor, AMPA receptor, GluK2, GluA2, ADAR2, mRNA editing, Homeostatic plasticity, Scaling
Highlighted Article: Synaptic suppression promotes proteasomal degradation of the mRNA-editing enzyme ADAR2. Decreased ADAR2 levels reduce Q/R editing of the kainate receptor subunit GluK2 leading to enhanced surface expression and homeostatic upscaling of GluK2-containing KARs.
INTRODUCTION
Kainate receptors (KARs) are glutamate receptors comprising tetrameric assemblies of combinations of five receptor subunits, GluK1–GluK5 (also known as GRIK1–GRIK5), with GluK2 and GluK5 being the most abundant subunit combination (Kumar et al., 2011; Petralia et al., 1994). KARs can be located pre-, post- and/or extra-synaptically, where they contribute to neurotransmitter release, postsynaptic depolarisation and the regulation of neuronal and network excitability. The variety of possible subunit combinations, together with co-assembly with Neto auxiliary subunits (Griffith and Swanson, 2015), creates a wide range of possible KAR subtypes (Evans et al., 2017a).
Additional KAR diversity arises from RNA editing (Egebjerg et al., 1994; Howe, 1996) mediated by the nuclear enzyme ADAR2 (also known as ADARB1), which edits pre-mRNAs encoding GluK2 and GluK1, as well as that for the AMPA receptor (AMPAR) subunit GluA2 (also known as GRIA2) and other non-coding RNAs (Nishikura, 2016; Sommer et al., 1991). ADAR2-mediated editing of a glutamine residue into an arginine residue (Q/R editing) in the pore-lining region of GluK2 alters a genomically encoded glutamine residue into an arginine residue, changing receptor assembly efficiency, forward trafficking, Ca2+ permeability and biophysical properties of the KARs (Egebjerg et al., 1994; Howe, 1996). More specifically, edited GluK2(R) has markedly reduced tetramerisation, leading to its accumulation in the ER (Ball et al., 2010). Furthermore, GluK2(R)-containing KARs that do assemble, exit the ER and reach the plasma membrane are Ca2+ impermeable and have a channel conductance of less than 1% of the non-edited GluK2(Q)-containing KARs (Swanson et al., 1996).
ADAR2 levels are very low during embryogenesis but increase in the first postnatal week (Behm et al., 2017), and ∼80% of GluK2, ∼40% of GluK1 and ∼99% of GluA2 subunits are edited in the mature brain (Bernard et al., 1999; Filippini et al., 2016; Paschen et al., 1997). ADAR2-knockout mice die at the early postnatal stage, but can be rescued by expressing the edited form of GluA2, demonstrating that unedited AMPARs are fatally excitotoxic (Higuchi et al., 2000). In contrast, mice specifically deficient in GluK2 Q/R editing are viable but are seizure prone and adults retain an immature form of NMDA receptor (NMDAR)-independent long-term potentiation (LTP) (Vissel et al., 2001). Thus, although not critical for survival, GluK2 editing plays important roles in network function and LTP. These observations have become particularly intriguing in the light of recent data from our laboratory showing that KARs can induce a novel form of LTP (KAR-LTPAMPAR) even in mature rats (Petrovic et al., 2017).
As well as directly inducing synaptic plasticity of AMPARs, KARs themselves undergo long-term depression (LTD) (Chamberlain et al., 2012) and LTP (González-González et al., 2012; Martin et al., 2008; Martin and Henley, 2004). Furthermore, it has recently been shown that KARs also undergo homeostatic plasticity (scaling) (Evans et al., 2017b) which, for AMPARs and NMDARs, is a crucial regulator of neuronal network and brain function because it constrains neuronal firing to within a tunable physiological range (Mu et al., 2003; Turrigiano, 2011; Turrigiano et al., 1998). This homeostatic control prevents runaway excitation thereby maintaining synaptic stability, critical to network formation, development and stability. Furthermore, defects in this process have been implicated in neurological diseases including epilepsy and schizophrenia (Wondolowski and Dickman, 2013).
We recently reported that, as for AMPARs, KAR upscaling can be induced by suppression of synaptic activity with tetrodotoxin (TTX) for 24 h and is accompanied by a decrease in GluK2 Q/R editing (Evans et al., 2017a). Upscaling is a form of homeostatic plasticity in which surface expression of glutamate receptors is increased in order to counteract a chronic loss in synaptic activity. However, the molecular and cellular mechanisms that reduce Q/R editing and whether this change in editing is a direct cause of, and sufficient to mediate, KAR upscaling are not known.
Here, we show that the widely used and well-established ‘classical’ protocol for inducing homeostatic plasticity of AMPARs through prolonged suppression of network activity with TTX in neuronal cultures (Turrigiano, 2011; Turrigiano et al., 1998) leads to KAR upscaling by promoting the proteosomal degradation of ADAR2. Decreased levels of ADAR2 reduce GluK2 pre-mRNA Q/R editing and lead to enhanced surface expression of GluK2-containing KARs. Importantly, this upscaling mechanism is specific to KARs, as TTX did not change the editing status of GluA2. Taken together, these data demonstrate a selective role of mRNA editing by ADAR2 in homeostatic upscaling of KARs, and identify alterations in ADAR2 stability as a novel mechanism for inducing plasticity.
RESULTS
Suppression of synaptic activity decreases ADAR2 levels
Our previous results demonstrated that a 24 h incubation of rat primary cultured hippocampal neurons with TTX leads to reduced Q/R editing of GluK2 and KAR upscaling (Evans et al., 2017a), but did not address the underlying mechanisms. We therefore examined levels of the enzyme ADAR2, which mediates Q/R editing of GluK2. Chronic blockade of action potentials with TTX for 24 h decreased ADAR2 levels by ∼50%, with no effect on ADAR1 levels (Fig. 1A–D). Longer periods of TTX treatment did not decrease ADAR2 levels any further (Fig. 1E,F), suggesting that a basal level of ADAR2 is retained even under long-term suppression of synaptic activity. Importantly, TTX treatment did not alter total levels of either GluK2 or GluK5 KAR subunits (Fig. S1A–C).
Fig. 1.

Chronic suppression of network activity decreases ADAR2 levels. (A) Representative western blots of total ADAR2 and GAPDH levels in hippocampal neurons with or without 24 h of TTX treatment to suppress synaptic activity. (B) Quantification of results from A with total levels of ADAR2 normalised to levels of GAPDH from five independent experiments. ****P<0.0001 (unpaired t-test). (C) Representative western blots of total ADAR1 and GAPDH levels in hippocampal neurons with or without 24 h TTX treatment. (D) Quantification of results from C with total levels of ADAR1 normalised to levels of GAPDH from eight independent experiments. Both bands were quantified. ns, not significant (P>0.05; unpaired t-test). (E) Representative western blots showing total ADAR2 and GAPDH levels with increasing durations of TTX treatment. (F) Quantification of results from E with total levels of ADAR2 normalised to levels of GAPDH from six independent experiments. **P<0.01, ***P<0.001 (one-way ANOVA with Dunnett's multiple comparison test). (G) Representative western blot of nuclear ADAR2 levels in hippocampal neurons with or without a 24 h TTX treatment. Cell fractionation was performed to determine the ADAR2 levels in the nucleus. Lamin B was used as a nuclear marker and RhoGDI as cytosol marker. (H) Quantification of results from G, with the nuclear ADAR2 immunoblots levels normalised to levels of Lamin B from three independent experiments. **P<0.01 (unpaired t-test). (I) Representative images of hippocampal neurons with or without 24 h TTX treatment labelled with nuclear DAPI stain (blue), and anti-ADAR2 (red) and anti-fibrillarin (nucleolar marker; cyan) antibodies. Bottom panels show magnified images as indicated and the red arrows indicate cells expressing ADAR2. Scale bar: 10 µm. (J) Quantification of results from I for the total value of ADAR2 intensity per nucleus. N=3 independent dissections and n=60 cells for control and 62 cells for TTX treatment. ****P<0.0001 (Wilcoxon matched-pairs signed rank test). (K) Analysis of the percentage of cells expressing ADAR2 (I) with or without TTX. N=3 independent dissections and n=15 fields of view. *P<0.05 (unpaired t-test). (L) Representative western blots of total ADAR2 and GAPDH levels in hippocampal neurons after 24 h TTX or 24 h AP5 or both treatments, to suppress synaptic activity. (M) Quantification of results from L with total levels of total ADAR2 normalised to levels of GAPDH from four independent experiments. ****P<0.0001 (two-way ANOVA with Tukey's multiple comparisons test).
As expected, the decrease in ADAR2 following a 24 h TTX treatment occurs in the nucleus (Fig. 1G,H), where ADAR2 binds to the pre-mRNA substrates prior to mRNA splicing and maturation (Herb et al., 1996). Both the abundance of ADAR2 within cells and the proportion of cells expressing ADAR2 were decreased (Fig. 1I–K) by TTX treatment. We therefore hypothesised that this activity-dependent modulation of ADAR2 could underpin the previously reported upscaling of GluK2-containing KARs in response to suppression of synaptic activity (Evans et al., 2017b).
Importantly, in addition to TTX, chronic block of NMDARs with the antagonist AP5 also significantly decreased ADAR2 levels, with no additional decrease when cells were treated with both TTX and AP5 for 24 h (Fig. 1L,M). These data demonstrate that different methods of suppressing synaptic and network activity can modulate ADAR2 levels in the same manner.
GluK2 Q/R editing in KARs is more sensitive to changes in ADAR2 levels than GluA2 Q/R editing in AMPARs
Following exposure of neurons to TTX for 24 h, KARs undergo robust upscaling and decreased GluK2 Q/R editing. Moreover, partial knockdown of ADAR2 to levels similar to those observed following 24 h TTX treatment caused upscaling of KARs (Evans et al., 2017b). We now show that, while AMPARs also undergo TTX-mediated upscaling (Turrigiano, 2011), the Q/R editing status of GluA2 is not changed by TTX (Fig. S1D–F) indicating that AMPAR scaling occurs via a different mechanism. To further investigate this, we tested whether mRNA editing of the AMPAR subunit GluA2 was also affected by depletion of ADAR2.
We used the same shRNA construct that reduced ADAR2 levels to ∼50% of the control (shRNA‘Partial’) to reduce the amount of ADAR2 per cell and the percentage of cells expressing ADAR2 to levels similar to those seen upon TTX treatment (Evans et al., 2017b). In addition, we also validated a different ADAR2 shRNA (shRNA‘Complete’) that ablated essentially all ADAR2 (Fig. S2).
We then compared how the extent of ADAR2 loss affects GluK2 and GluA2 editing. Knockdown of ADAR2 by shRNA‘Complete’ reduced GluK2 Q/R editing by over 60%, whereas, as reported previously (Evans et al., 2017b), shRNA‘Partial’ only reduced GluK2 Q/R editing by ∼20%, (Fig. 2A,B). Consistent with this, DNA sequencing chromatographs from cells treated with shRNA‘Complete’ show a dramatic change in the base read of the editing site to the unedited CAG (Q) rather than edited CGG (R), while neurons treated with shRNA‘Partial’ show a mixture of both CGG and CAG (Fig. 2C). Interestingly, shRNA‘Partial’ knockdown of ADAR2 had no effect on the Q/R editing of the AMPAR subunit GluA2 while shRNA‘Complete’ only reduced GluA2 editing by ∼30% (Fig. 2D–F).
Fig. 2.

Complete and partial ADAR2 knockdown differentially alter GluK2 and GluA2 Q/R editing. (A) RT-PCR and BbvI digestion analysis of GluK2 Q/R editing from hippocampal neurons infected with scrambled, complete or partial shRNAs for ADAR2 knockdown (KD). (B) Quantification of results from A for four independent experiments. *P<0.05, **P<0.01, ****P<0.0001 (one-way ANOVA with Tukey's multiple comparisons test). (C) Sanger sequencing chromatographs of the GluK2 PCR products from hippocampal neurons infected with scrambled, complete or partial shRNAs for ADAR2 KD, showing dual A and G peaks at the editing site, indicated by the green arrows. The green peak represents an A (unedited) base read and black represents a G (edited) base read. (D) RT-PCR and BbvI digestion analysis of GluA2 Q/R editing from hippocampal neurons infected with either scrambled, complete or partial shRNAs for ADAR2 knockdown (KD). (E) Quantification of results from D for four independent experiments. ****P<0.0001 (one-way ANOVA with Tukey's multiple comparisons test). (F) Sanger sequencing chromatographs of the GluA2 PCR products from hippocampal neurons infected with either scrambled, complete or partial shRNAs for ADAR2 KD. Green arrows indicate the editing site. Only samples treated for complete ADAR2 knockdown show a dual A and G peak at the editing site. The green peak represents an A (unedited) base read and black represents a G (edited) base read.
Partial ADAR2 knockdown mimics and occludes TTX-evoked KAR upscaling
Fig. 2 shows that shRNA‘Partial’ reduces ADAR2 levels to a similar extent to that seen following TTX treatment and results in a similar shift in the extent of GluK2 editing, while not affecting editing of the AMPAR subunit GluA2. We therefore wondered whether shRNA‘Partial’ ADAR2 knockdown alone is sufficient to upscale KARs. Indeed, shRNA‘Partial’ in the absence of TTX significantly increased GluK2 surface expression with no effect on EGFR surface expression (Fig. 3A–C). Total levels of both GluK2 and EGFR remained unaltered (Fig. S3A,B). Moreover, the effects of shRNA‘Partial’ on GluK2 upscaling were reversed upon rescue of the levels of ADAR2 (Fig. S3C–G).
Fig. 3.

Partial ADAR2 knockdown mimics TTX-evoked KAR upscaling and also occludes any further TTX-mediated upscaling. (A) Representative western blot of total and surface levels of GluK2, EGFR and GAPDH in cells infected with scrambled or partial shRNAs for ADAR2 knockdown (KD) in the presence or absence of TTX. EGFR was used as a negative control, while GAPDH was used as a control to show that only surface proteins were labelled with biotin. (B) Quantification results from A showing surface levels of GluK2 from 10 independent experiments. Surface levels were normalised to their total levels. *P<0.05, **P<0.01, ***P<0.001 (two-way ANOVA with Tukey's multiple comparisons test). (C) Quantification of results from A showing surface levels of EGFR from 10 independent experiments. Surface levels were normalised to their total levels. ns, not significant (P>0.05; two-way ANOVA with Tukey's multiple comparisons test). (D) RT-PCR and BbvI digestion analysis of GluK2 Q/R editing from hippocampal neurons infected with either scrambled or partial shRNAs for ADAR2 KD with or without TTX. (E) Quantification of results from D for five independent experiments. **P<0.01, ***P<0.001 (one-way ANOVA with Tukey's multiple comparisons test).
Since in our knockdown–rescue experiments there was an ‘over rescue’ of ADAR2 (Fig. S3F,G), we also tested whether Q/R editing of GluK2 was correspondingly increased compared to the scrambled control condition. Interestingly, the levels of GluK2 Q/R editing were restored to basal level (∼80%) (Fig. S3E–G) despite the overexpression of ADAR2 in the rescue condition. These results suggest that a proportion of GluK2 is resistant to Q/R editing even when excess ADAR2 is present.
Surprisingly, complete ablation of ADAR2 with shRNA‘Complete’ had no effect on GluK2 surface expression (Fig. S4A,B), suggesting the effect of physiologically relevant partial loss of ADAR2 differs from that of the complete knockdown. It is likely that compensatory mechanisms exist to restore cellular homeostasis when ADAR2 is completely ablated. Furthermore, it is also important to note that complete knockdown of ADAR2 leads to further reductions in GluK2, as well as GluA2, editing, making these results difficult to interpret.
Application of both TTX and shRNA‘Partial’ was not additive (Fig. 3A,B) and did not further decrease GluK2 Q/R editing compared to that seen with each individual treatment alone (Fig. 3D,E). The fact that partial ADAR2 knockdown is sufficient to upscale KARs and that the effects of TTX are occluded by shRNA‘Partial’ provide further support for the proposal that TTX-induced GluK2 upscaling is primarily mediated by a reduction in ADAR2 levels.
TTX promotes proteasomal degradation of ADAR2
We next explored the mechanisms underlying ADAR2 loss during scaling. As shown in Fig. 4A, TTX does not alter ADAR2 mRNA levels, indicating that transcriptional changes are not involved, so we investigated possible mechanisms for activity-dependent ADAR2 degradation.
Fig. 4.

TTX promotes proteasomal degradation of ADAR2. (A) RT-qPCR analysis of mRNA levels of ADAR2 post TTX treatment showing no changes in the ADAR2 mature mRNA transcripts from seven independent experiments. ns, not significant (P>0.05; unpaired t-test). (B) Representative western blots of total ADAR2, total ubiquitin and GAPDH levels in neurons treated with either DMSO or 1 µM BTZ for 20 h either in the presence or absence of 24 h TTX. (C) Quantification of levels of total ADAR2 normalised to levels of GAPDH from immunoblots for six independent experiments. **P<0.01; ****P<0.0001; ns, not significant, P>0.05 (two-way ANOVA with Tukey's multiple comparisons test). (D) Quantification of the level of total ubiquitin-conjugated products normalised to the level of GAPDH for six independent experiments. ****P<0.0001 (two-way ANOVA with Tukey's multiple comparisons test). (E) Representative western blots of ADAR2 in the nucleus and cytoplasm in the presence of DMSO or BTZ with or without TTX treatment. Lamin B was used as a nuclear marker and RhoGDI as cytosol marker. (F) Quantification of the level of nuclear ADAR2 from seven independent experiments. Nuclear ADAR2 levels were normalised to the levels of Lamin B. *P<0.05; **P<0.01; ns, not significant, P>0.05 (two-way ANOVA with Tukey's multiple comparisons test). (G) Quantification of cytosolic ADAR2 from seven independent experiments. Cytosolic ADAR2 was normalised to RhoGDI level. *P<0.05, **P<0.01, ****P<0.0001 (two-way ANOVA with Tukey's multiple comparisons test). (H) Representative western blots of total and surface levels of GluK2, EGFR and GAPDH in DMSO- and BTZ (20 h, 1 µM)-treated cells in the presence or absence of 24 h TTX. EGFR was used as a negative control while GAPDH was used as a control to show only surface proteins were labelled with biotin. (I) Quantification of surface levels of GluK2 from five independent experiments. Surface levels were normalised to their total levels. *P<0.05; ns, not significant, P>0.05 (two-way ANOVA with Tukey's multiple comparisons test). (J) Quantification of surface levels of EGFR from five independent experiments. Surface levels were normalised to their total levels. ns, not significant, P>0.05 (two-way ANOVA with Tukey's multiple comparisons test).
The nuclear protein Pin1 retains ADAR2 in the nucleus to prevent its export to the cytosol where it is ubiquitylated and degraded (Marcucci et al., 2011). It has also been reported that Pin1-mediated stabilisation is an important regulator of ADAR2 editing activity during development in cortical neurons (Behm et al., 2017). We therefore wondered whether destabilisation of the Pin1–ADAR2 interaction would cause TTX-mediated ADAR2 loss. However, this is not the case since Pin1 levels were unchanged following TTX treatment (Fig. S5A,B).
ADAR2 phosphorylation at threonine 32 (T32) has also been reported to be crucial for the ADAR2–Pin1 interaction (Marcucci et al., 2011) so we made phosphorylation-null (phosphonull) (T32A) and phosphomimetic (T32D) ADAR2 mutants. ADAR2(T32D) binds very strongly to Pin1 in GFP-trap assays compared to wild type (WT) and ADAR2(T32A) (Fig. S5C,D). We therefore tested whether the phosphonull or phosphomimetic ADAR2 mutants were more sensitive to TTX treatment.
We first knocked down endogenous ADAR2 and replaced it with HA-tagged WT ADAR2 (Fig. S5E,F). More than 80% of the cells expressed this ADAR2 knockdown-rescue protein, similar to the percentage of scrambled-treated neurons that express endogenous ADAR2. We then investigated the stability of the phosphonull or phosphomimetic ADAR2 mutants in response to TTX treatment. Similar to WT ADAR2, levels of both mutants were significantly decreased by TTX treatment (Fig. S5G,H). Since both phosphonull and phosphomimetic mutants of ADAR2, which decrease or enhance binding to Pin1 respectively, were equally susceptible to the TTX-mediated loss these experiments suggest that alterations in the Pin1–ADAR2 interaction do not underpin ADAR2 loss during TTX-mediated upscaling.
The effects of TTX on ADAR2 stability were determined using the proteasomal inhibitor Bortezomib (BTZ) (Chen et al., 2011). BTZ prevented the TTX-evoked decrease in ADAR2 (Fig. 4B,C) and resulted in the accumulation of ubiquitylated proteins (Fig. 4B,D). We performed nuclear and cytoplasmic fractionation experiments to determine whether ADAR2 is exported from the nucleus for degradation in the cytosol. BTZ prevented the TTX-evoked decrease in ADAR2 in both the nuclear and cytosolic fractions, and actually led to a significant accumulation of ADAR2 in the cytosol (Fig. 4E–G). These data suggest that ADAR2 may be (1) exported to the cytosol for ubiquitination, (2) ubiquitylated in the nucleus and exported to the cytosol, or (3) subject to a combination of both processes. Thus, while the exact mechanisms remain to be determined, these experiments show that suppression of synaptic activity induces ADAR2 ubiquitylation and proteasomal degradation.
Since BTZ prevents the loss of ADAR2 upon TTX treatment, we next tested whether BTZ also blocks KAR upscaling. Indeed, surface biotinylation experiments showed that BTZ prevents TTX-induced increases in surface-localised GluK2 (Fig. 4H,I) with no effect on EGFR (Fig. 4H,J). These results support the hypothesis that proteasomal degradation of ADAR2 following TTX treatment is both necessary and sufficient for KAR upscaling.
DISCUSSION
Unlike other mRNA-editing sites in KAR subunits, GluK2 Q/R editing has been shown to affect KAR trafficking to the plasma membrane (Ball et al., 2010). ADAR2 also regulates GluA2 mRNA editing and, as for GluK2, this affects the trafficking and surface expression of GluA2-containing AMPARs (Greger et al., 2003). Indeed, ADAR2-knockout mice are deficient in both GluK2 and GluA2 editing (Higuchi et al., 2000). Moreover, GluA2 Q/R editing is modulated by changes in ADAR2 levels that occur during ischaemia (Peng et al., 2006) and in response to excitotoxic levels of glutamate (Mahajan et al., 2011).
We have previously reported that 24 h of TTX treatment causes a robust upscaling of KARs with a concomitant decrease in GluK2 Q/R editing. We also showed that partial knockdown of ADAR2 to levels similar to those observed following 24 h of TTX treatment upscaled KARs (Evans et al., 2017b). In this report, we build on those findings to show that, while AMPARs also undergo TTX-mediated upscaling (Turrigiano, 2011), the Q/R editing status of GluA2 is not changed by TTX (Fig. S1D–F) indicating that AMPAR scaling occurs via a different mechanism.
These data demonstrate that the editing levels of GluK2 are selectively sensitive to changes in ADAR2 that occur as a result of TTX treatment and support a model whereby synaptic suppression-evoked loss of ADAR2 during homeostatic scaling directly promotes surface expression of GluK2-containing KARs through a reduction in GluK2 editing. This is consistent with Q/R editing of GluA2 being preferentially maintained to prevent neurotoxicity associated with Ca2+-permeable AMPARs and suggests that mechanisms other than ADAR2 can regulate GluA2 editing. How this is achieved is currently unclear but it is notable that ADAR1 remains unchanged during TTX treatment. Although it has been reported that ADAR1 primarily, but not exclusively, mediates arginine to glycine (R/G) mRNA editing (Wong et al., 2001), we speculate that under conditions where levels of ADAR2 are diminished, ADAR1 may compensate to maintain Q/R editing of GluA2 but not GluK2. It has also been suggested that additional regulatory steps during GluA2 pre-mRNA maturation contribute to ensuring its editing levels are maintained (Penn et al., 2013).
Taken together, our data demonstrate that suppression of synaptic activity reduces ADAR2, leading to decreased KAR editing which, in turn, directly mediates KAR upscaling via increased KAR assembly and ER exit of unedited GluK2(Q) compared to edited GluK2(R) (Fig. 5). Moreover, the fact that application of both TTX and shRNA‘Partial’ were not additive and that partial ADAR2 knockdown is sufficient to upscale KARs provide further support for the proposal that TTX-induced GluK2 upscaling is primarily mediated by a reduction in ADAR2 levels.
Fig. 5.
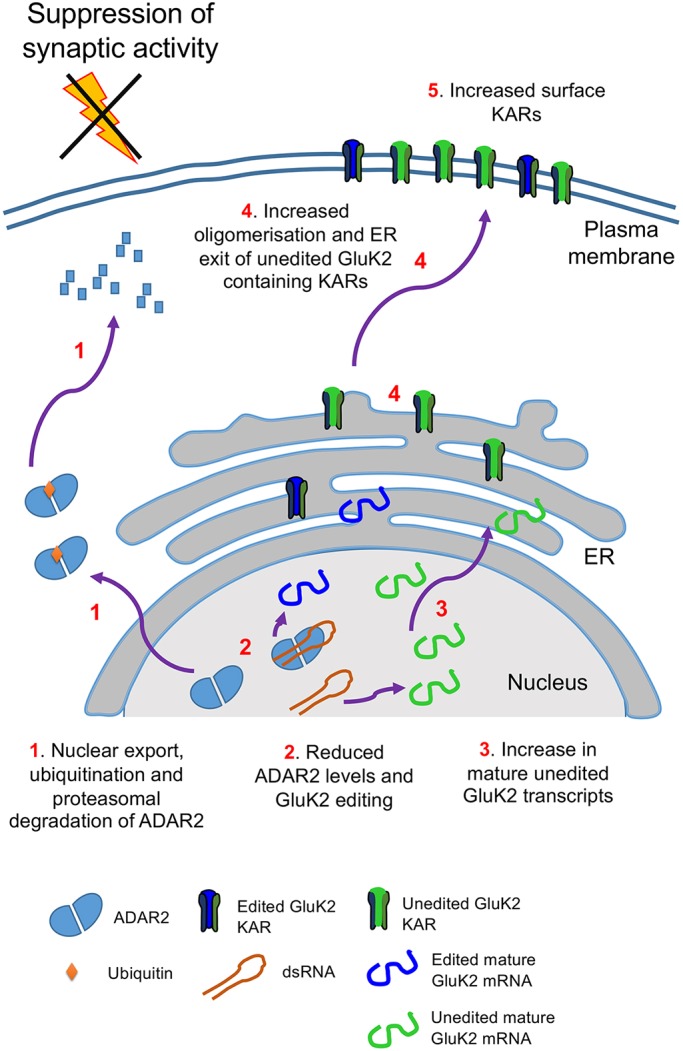
Schematic of ADAR2-mediated Q/R editing regulating GluK2 containing KARs homeostatic upscaling. Under basal conditions unedited GluK2 transcripts are edited at their Q/R site by ADAR2 resulting in ∼80% of mature GluK2 transcripts being edited. The resultant edited and unedited GluK2 subunits oligomerise in the ER and traffic to the surface. Under conditions of synaptic activity suppression with TTX treatment, ADAR2 undergoes proteasomal degradation in the cytosol (1). This results in less ADAR2 editing of GluK2 pre-mRNA transcripts (2) and increased levels of unedited mature GluK2 transcripts (3). The subsequent increase in the proportion of unedited GluK2(Q) allows enhanced oligomerisation and ER exit (4) to increase surface expression of GluK2-containing KARs on the surface (5).
These results show that regulation of ADAR2 stability and changes in GluK2 editing underpin a novel and specific mechanism to tune the surface expression of KARs. Thus, changes in levels of ADAR2 and ADAR2-mediated GluK2 Q/R editing levels provides a flexible and rapidly tunable system to control the KAR forward trafficking and scaling that is not present for AMPARs.
Given that KARs play many roles in controlling neuronal network activity (Contractor et al., 2011; Evans et al., 2017a), that they have recently been identified as inducers of AMPAR plasticity (Petrovic et al., 2017) and that their dysfunction has been implicated in a number of neurological disorders (Crépel and Mulle, 2015; Lerma and Marques, 2013), it is likely that ADAR2-mediated control of KAR surface expression plays a wide role in neuronal function and dysfunction.
MATERIALS AND METHODS
Primary neuronal cultures
Primary rat hippocampal neurons were dissected from embryonic day (E)18 Han Wistar rat pups as previously described (Rocca et al., 2017). Pregnant E18 Han Wistar rats were anaesthetised using isoflurane with pure oxygen flow and humanely killed using cervical dislocation under Home Office Schedule 1 regulations. The animal was ordered in-house from University of Bristol Animal Services Unit. Briefly, neurons were dissected from E18 Wistar rats followed by trypsin dissociation and culturing for up to 2 weeks. For the first 24 h, cells were grown in plating medium [neurobasal medium (Gibco) supplemented with 10% horse serum (Sigma), B27 (1×, Gibco), penicillin-streptomycin (P/S, 100 units penicillin and 0.1 mg/ml streptomycin; ThermoScientific) and 5 mM Glutamax (Gibco)]. After 24 h, plating medium was replaced with feeding medium (neurobasal medium containing 2 mM Glutamax and lacking horse serum and P/S). For biochemistry experiments, cells were plated at a density of 500,000 cells per 35 mm well and 250,000 cells per coverslip for imaging experiments.
ADAR2 cloning
ADAR2 was cloned from rat neuronal cDNA, and ADAR2 shRNA knockdown and knockdown-rescue viruses were generated as previously described (Rocca et al., 2017). ADAR2 was cloned from rat neuronal cDNA into the KpnI and XbaI sites of the vector pcDNA3 with a HA tag at its N-terminus. Phosphomutants of ADAR2 were generated by site-directed mutagenesis. Pin1 was cloned from rat neuronal cDNA into the EcoRI and BamHI sites of the vector pEGFP-N1.
Lentivirus generation
For ADAR2 knockdown experiments, shRNA sequences targeting ADAR2 cloned into a modified pXLG3-GFP vector (Rocca et al., 2017) under the control of a H1 promoter. The ADAR2 target sequences were: complete shRNA 5′-AAGAACGCCCTGATGCAGCTG-3′; partial shRNA, 5′-AACAAGAAGCTTGCCAAGGCC-3′.
For rescue experiments, shRNA-insensitive HA-ADAR2 was cloned into a modified pXLG3-GFP vector under the control of an SFFV promoter.
The viruses were produced in HEK293T cells as reported previously (Rocca et al., 2017), harvested and added to hippocampal neurons at 9–10 days in vitro (DIV) for 5 days and lysed accordingly. For 24 h treatment experiments, cells were treated with on the fourth day after virus addition and harvested accordingly on the fifth day after the completion of the time course.
Scaling, developmental and TTX time course and BTZ treatment
For scaling experiments, cells were treated with 1 µM TTX (Tocris) for 24 h and were either lysed directly in 1× sample buffer [4× sample buffer (0.24 M Tris-HCl, 8% SDS, 40% glycerol, 10% β-mercaptoethanol and 0.009% Bromophenol Blue) diluted in water] and heated for 10 min at 95°C or were used for either surface biotinylation or fractionation experiments (see below). For the TTX time course, cells were harvested directly into 1× sample buffer. In experiments inhibiting proteosomal degradation, cells were treated with Bortezomib (BTZ; Cell Signalling) dissolved in DMSO for 20 h at 1 µM concentration. The control cells were treated with an equal volume of DMSO.
Cell surface biotinylation and streptavidin pulldown
Cell surface biotinylation was performed essentially as previously described (Evans et al., 2017b). All steps were performed on ice with ice-cold buffers unless stated otherwise. Live hippocampal neurons post stated treatments were washed twice in phosphate-buffered saline (PBS). Surface proteins were labelled with membrane-impermeable Sulfo-NHS-SS biotin (0.3 mg/ml, Thermo Scientific) for 10 min on ice and washed 3× with PBS. 100 mM NH4Cl was added to quench free biotin-reactive groups and cells were extracted with lysis buffer [50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% triton, 0.1% SDS, protease inhibitors (Roche)], incubated on ice for 30 min and centrifuged (15,000 g at 4°C for 20 min) to remove cell debris. For streptavidin pulldown, each lysate was added to 30 µl of streptavidin beads (Sigma) and left on a wheel to rotate for 90 min at 4°C. The beads were then washed three times with wash buffer (lysis buffer without protease inhibitors) and proteins were eluted with 2× sample buffer and boiled for 10 min at 95°C. The samples were then resolved by SDS-PAGE and immunoblotted.
Subcellular fractionation
All the steps were performed on ice with ice-cold buffers unless stated otherwise. Following stated treatments, the cells were washed with PBS followed by addition of buffer 1 (150 mM NaCl, 50 mM HEPES pH 7.4, 25 µg/ml digitonin and protease inhibitors), incubated for 20 min, scraped, homogenised and centrifuged at 15,000 g for 30 min at 4°C. The supernatant consisted of the cytosolic proteins, while the pellet was resuspended in buffer 2 (buffer 1 with 1% Triton X-100), incubated for 20 min and again centrifuged at 15,000 g for 30 min at 4°C. The supernatant consisted of mitochondrial proteins, while the pellet was resuspended in buffer 3 (150 mM NaCl, 50 mM HEPES pH 7.4, 0.5% sodium deoxycholate, 0.1% SDS, protease inhibitors and 0.5% Triton X-100), incubated for 1 h and centrifuged at 15,000 g for 30 min at 4°C. The supernatant was discarded and the pellet, consisting of nuclear proteins, was resuspended in buffer 3. The cytosolic supernatant was concentrated using four volumes of acetone (kept at −20°C), incubated at −20°C for 1 h and spun for 20 min at 1500 g and resuspended in buffer 3. A BCA assay was then performed to determine protein concentration and allow equal loading.
BCA assay
The BCA assay was performed using a commercial kit (Pierce, Thermo Scientific) following the manufacturer's instructions. Following 30 min incubation at 37°C, the samples were read using a plate reader (Versamax Microplate reader, Molecular Devices) at a wavelength of 562 nm.
Antibodies used
A comprehensive list of the specific antibodies used, the suppliers, batch numbers, dilutions and references are provided in Table S1.
Western blotting
Proteins were separated by SDS-PAGE and transferred to PVDF membrane for western blotting. Membranes were blocked in 5% w/w non-fat milk powder in PBS-T. Primary antibodies used are mentioned in the antibodies section and HRP-conjugated secondary antibodies were used to incubate the membrane for visualisation by enhanced chemiluminescence. Western blots were imaged and quantified using Image Studio Lite Version 5.2 (LI-COR) or developed on X-ray film in a dark room using developer and fixer solutions. The blots were then scanned and quantified using FIJI (ImageJ studio). Surface levels were normalised to their respective total levels. Nuclear protein levels were normalised to LaminB and cytosolic protein levels to RhoGDI. Treated samples were normalised to their control samples.
RNA extraction, RT-PCR, BbvI digestion and RT-qPCR
RNA samples were extracted from DIV14/15 hippocampal neurons following the stated treatments by using an RNeasy mini kit (Qiagen) according to the manufacturer's instructions. 1 µg of RNA was used per condition and reverse transcribed to cDNA using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific) following the manufacturer's instructions. The following primers [spanning the M2 region of GluK2 (Bernard et al., 1999) and GluA2] were used, giving PCR products of 452 bp and 252 bp: GluK2 F, 5′-GGTATAACCCACACCCTTGCAACC-3′, GluK2 R, 5′-TGACTCCATTAAGAAAGCATAATCCGA-3′; GluA2 F, 5′-GTGTTTGCCTACATTGGGGTC-3′, GluA2 R, 5′-TCCTCCTACACGGCTAACTTA-3′.
5 µl of cDNA was used to set up PCRs (50 µl total, 35 cycles, 20 s denaturing at 95°C, 10 s annealing at 60°C and 15 s elongation at 70°C).
To determine the level of RNA editing, BbvI (New England Biolabs) digestion was used as previously (Bernard et al., 1999). A total 20 µl digestion was set up using 10 µl of PCR product at 37°C for 2 h. All of the digested product was run on 4% agarose gels, and the ethidium bromide-stained bands were imaged using UV transilluminator and quantified using FIJI (NIH ImageJ). To determine the level of editing in GluK2, the following formula was used: {intensity of 376 bp band (edited)/intensity of [376 bp band (edited)+269 bp band (unedited)]}×100. The band at 76 bp allowed to determine equal loading. For GluA2, {intensity of 158 bp band (edited)/intensity of [158 bp band (edited)+94 bp band (unedited)]}×100.
Purified PCR products were also sent for sequencing to Eurofins Genomics at 4 ng/µl along with the above GluK2 and GluA2 F primers, to obtain sequence chromatographs.
For quantitative PCR (RT-qPCR), 2 µl of the cDNA samples per condition were mixed with PowerUp SYBR Green Master Mix (Life Technologies) and forward and reverse primers targeting ADAR2 and GAPDH, and amplified quantitatively using a real-time PCR System (MiniOpticon, BioRAD) for 40 cycles and Ct values were recorded. Each reaction was performed in triplicate and average Ct was measured per condition. ADAR2 Ct values were normalised to GAPDH Ct values, and ADAR2 mRNA fold difference value for TTX-treated conditions was normalised against the value for the untreated control. The melting curve of the primers were also determined to ensure the specificity of the primers and lack of primer dimer formation. The primers used were: ADAR2 F, 5′-TCCCGCCTGTGTAAGCAC-3′ and ADAR2 R, 5′-TGGGCTTGGTGATCTTGG-3′; GAPDH F, 5′-AATCCCATCACCATCTTCCA-3′ and GAPDH R, 5′-GTCCACCACCCTGTTGCTGTAG-3′.
HEK293T cells and GFP trap
The human embryonic kidney (HEK) 293T cell lines were obtained from The European Collection of Cell Cultures (ECACC) in 2017. Stocks were maintained in 1% DMSO and stored in liquid nitrogen. Each batch of thawed cells was routinely tested for microplasma infection.
GFP-trap protocols were as previously published (Guo et al., 2017). HEK293T cells were transfected the next day using Lipofectamine™ 3000 and 2.5 µg of each construct. At 48 h post transfection, cells were washed with PBS and lysed and harvested in lysis buffer [20 mM Tris-HCl pH7.4, 137 mM NaCl, 2 mM sodium pyrophosphate, 2 mM EDTA, 1% triton X-100, 0.1% SDS, 25 mM β-glycerophosphate, 10% glycerol, protease inhibitors (Roche), phosphatase inhibitor cocktail 2 (1:100, Sigma)]. The lysates were left to incubate for 30 min on ice and centrifuged at 1500 g for 20 min at 4°C to remove any cell debris. The supernatant was then added to 5 µl of GFP-trap beads (Chromotek), incubated on a wheel at 4°C for 90 min and washed three times with wash buffer (lysis buffer without protease or phosphatase inhibitors). The samples were then lysed in 2× sample buffer, heated at 95°C for 10 min and separated using SDS-PAGE.
Fixed immunostaining, imaging and analysis
Immunostaining was performed as previously described (Glebov et al., 2015). For fixed immunostaining, cells post TTX treatment or lentiviral treatment as indicated were fixed with 4% formaldehyde for 10 min, washed three times with PBS, treated with 100 mM glycine to quench any remaining formaldehyde and washed three times with PBS. The cells were permeabilised and blocked with 3% BSA in PBS and 0.1% Triton-X for 30 min. The cells were then incubated for 1 h with primary antibodies (Table S1) in 3% BSA at room temperature, washed three times for 5 min each with PBS and incubated for 45 min with the indicated secondary antibodies (Jackson Immunoresearch Antibodies, 1:400) in 3% BSA at room temperature. Three 5-min washes were performed with PBS and the cells were mounted using DAPI containing fluoromount.
A Leica SP5-II confocal laser-scanning microscope attached to a Leica DMI 6000 inverted epifluorescence microscope was used to image the coverslips. The confocal images were captured under a 63× objective, with a 1024×1024 pixel resolution and a 1× optical zoom. A frame average of 2 was taken with a Z-stack of 6–8 Z-planes with 0.5 µm interval.
FIJI (NIH Image J) was used to compress the Z-stacks and analyse the mean intensity per nucleus using the DAPI channel to draw regions of interest. To calculate the percentage of cells expressing ADAR2, all the cells expressing ADAR2 were manually counted per image taken.
Methodology and statistical analysis
All samples were included in the analysis. For purposes of randomisation, a set of experiments was performed in a six-well dish and each time different wells were assigned into different experimental group. Samples were also loaded in a different order each time on SDS-PAGE gels. During imaging, the DAPI channel was used to determine regions of interest to avoid bias. No blinding was used during data analysis.
The mean values were calculated for all data and all error bars show standard deviation. All statistical analysis was performed using GraphPad (Prism) software version 7.0 as stated. Each independent experiment refers to independent dissections. For imaging, N=number of independent dissections and n=number of cells. An unpaired t-test (two-tailed) was performed when comparing changes between two different groups. An F-test was performed to ensure variations within compared groups were not significantly different. One-way analysis of variance (ANOVA) was performed to compare mean changes within more than two groups and two-way ANOVA was used to compare mean differences between multiple groups with two independent variables. A Dunnett's multiple post test was performed to determine any significant changes when compared to the control group while Tukey's multiple post comparisons were performed to compare multiple groups at a time. Where possible, a D'Agostino and Pearson normality test was performed to determine normal distribution of the samples. Everywhere else, individual data points were plotted to show the spread of samples. P-values of less than 0.05 were considered statistically significant.
At least three repeats from three independent dissections were chosen as a minimum criterion for sample size to minimise animal use while ensuring enough replicates to obtain statistical significance.
Supplementary Material
Acknowledgements
We thank Dr Yasuko Nakamura for excellent technical and logistical support.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
S.G. performed all of the experiments. A.J.E. and K.A.W. provided specialised constructs, advice and assisted in some experiments. J.M.H. supervised the research. All authors contributed to writing the manuscript.
Author contributions
Conceptualization: S.G., A.E., K.W., J.H.; Methodology: A.E., K.W., S.G., A.E., K.W.; Investigation: S.G.; Data curation: S.G.; Writing - original draft: J.H.; Writing - review & editing: S.G., A.E., K.W., J.H.; Project administration: K.W., J.H.; Funding acquisition: J.H.
Funding
We are grateful to the Medical Research Council (MRC), Biotechnology and Biological Sciences Research Council (BBSRC) and Wellcome Trust for financial support. S.G. and A.J.E. were supported by Wellcome Trust Dynamic Cell Biology PhD studentships. Deposited in PMC for release after 6 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.222273.supplemental
References
- Ball S. M., Atlason P. T., Shittu-Balogun O. O. and Molnár E. (2010). Assembly and intracellular distribution of kainate receptors is determined by RNA editing and subunit composition. J. Neurochem. 114, 1805-1818. 10.1111/j.1471-4159.2010.06895.x [DOI] [PubMed] [Google Scholar]
- Behm M., Wahlstedt H., Widmark A., Eriksson M. and Öhman M. (2017). Accumulation of nuclear ADAR2 regulates adenosine-to-inosine RNA editing during neuronal development. J. Cell Sci. 130, 745-753. 10.1242/jcs.200055 [DOI] [PubMed] [Google Scholar]
- Bernard A., Ferhat L., Dessi F., Charton G., Represa A., Ben-Ari Y. and Khrestchatisky M. (1999). Q/R editing of the rat GluR5 and GluR6 kainate receptors in vivo and in vitro: evidence for independent developmental, pathological and cellular regulation. Eur. J. Neurosci. 11, 604-616. 10.1046/j.1460-9568.1999.00479.x [DOI] [PubMed] [Google Scholar]
- Chamberlain S. E. L., González-González I. M., Wilkinson K. A., Konopacki F. A., Kantamneni S., Henley J. M. and Mellor J. R. (2012). SUMOylation and phosphorylation of GluK2 regulate kainate receptor trafficking and synaptic plasticity. Nat. Neurosci. 15, 845-852. 10.1038/nn.3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D., Frezza M., Schmitt S., Kanwar J. and Dou Q. P. (2011). Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr. Cancer Drug Targets 11, 239-253. 10.2174/156800911794519752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A., Mulle C. and Swanson G. T. (2011). Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci. 34, 154-163. 10.1016/j.tins.2010.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crépel V. and Mulle C. (2015). Physiopathology of kainate receptors in epilepsy. Curr. Opin. Pharmacol. 20, 83-88. 10.1016/j.coph.2014.11.012 [DOI] [PubMed] [Google Scholar]
- Egebjerg J., Kukekov V. and Heinemann S. F. (1994). Intron sequence directs RNA editing of the glutamate receptor subunit GluR2 coding sequence. Proc. Natl. Acad. Sci. USA 91, 10270-10274. 10.1073/pnas.91.22.10270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans A. J., Gurung S., Henley J. M., Nakamura Y. and Wilkinson K. A. (2017a). Exciting times: new advances towards understanding the regulation and roles of kainate receptors. Neurochem. Res., https://doi.org/10.1007/s11064-017-2450-2 10.1007/s11064-017-2450-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans A. J., Gurung S., Wilkinson K. A., Stephens D. J. and Henley J. M. (2017b). Assembly, secretory pathway trafficking, and surface delivery of kainate receptors is regulated by neuronal activity. Cell Rep. 19, 2613-2626. 10.1016/j.celrep.2017.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippini A., Bonini D., La Via L. and Barbon A. (2016). The good and the bad of glutamate receptor RNA editing. Mol. Neurobiol. 54, 6795-6805. 10.1007/s12035-016-0201-z [DOI] [PubMed] [Google Scholar]
- Glebov O. O., Tigaret C. M., Mellor J. R. and Henley J. M. (2015). Clathrin-independent trafficking of AMPA receptors. J. Neurosci. 35, 4830-4836. 10.1523/JNEUROSCI.3571-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-González I. M., Konopacki F. A., Rocca D. L., Doherty A. J., Jaafari N., Wilkinson K. A. and Henley J. M. (2012). Kainate receptor trafficking. WIRES Membr. Trasnsp. Signal. 1, 31-44. 10.1002/wmts.23 [DOI] [Google Scholar]
- Greger I. H., Khatri L., Kong X. and Ziff E. B. (2003). AMPA receptor tetramerization is mediated by Q/R editing. Neuron 40, 763-774. 10.1016/S0896-6273(03)00668-8 [DOI] [PubMed] [Google Scholar]
- Griffith T. N. and Swanson G. T. (2015). Identification of critical functional determinants of kainate receptor modulation by auxiliary protein Neto2. J. Physiol. 593, 4815-4833. 10.1113/JP271103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C., Wilkinson K. A., Evans A. J., Rubin P. P. and Henley J. M. (2017). SENP3-mediated deSUMOylation of Drp1 facilitates interaction with Mff to promote cell death. Sci. Rep. 7, 43811 10.1038/srep43811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herb A., Higuchi M., Sprengel R. and Seeburg P. H. (1996). Q/R site editing in kainate receptor GluR5 and GluR6 pre-mRNAs requires distant intronic sequences. Proc. Natl. Acad. Sci. USA 93, 1875-1880. 10.1073/pnas.93.5.1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M., Maas S., Single F. N., Hartner J., Rozov A., Burnashev N., Feldmeyer D., Sprengel R. and Seeburg P. H. (2000). Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406, 78-81. 10.1038/35017558 [DOI] [PubMed] [Google Scholar]
- Howe J. R. (1996). Homomeric and heteromeric ion channels formed from the kainate-type subunits GluR6 and KA2 have very small, but different, unitary conductances. J. Neurophysiol. 76, 510-519. 10.1152/jn.1996.76.1.510 [DOI] [PubMed] [Google Scholar]
- Kumar J., Schuck P. and Mayer M. L. (2011). Structure and assembly mechanism for heteromeric kainate receptors. Neuron 71, 319-331. 10.1016/j.neuron.2011.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma J. and Marques J. M. (2013). Kainate receptors in health and disease. Neuron 80, 292-311. 10.1016/j.neuron.2013.09.045 [DOI] [PubMed] [Google Scholar]
- Mahajan S. S., Thai K. H., Chen K. and Ziff E. (2011). Exposure of neurons to excitotoxic levels of glutamate induces cleavage of the RNA editing enzyme, adenosine deaminase acting on RNA 2, and loss of GLUR2 editing. Neuroscience 189, 305-315. 10.1016/j.neuroscience.2011.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcucci R., Brindle J., Paro S., Casadio A., Hempel S., Morrice N., Bisso A., Keegan L. P., Del Sal G. and O'Connell M. A. (2011). Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J. 30, 4211-4222. 10.1038/emboj.2011.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S. and Henley J. M. (2004). Activity-dependent endocytic sorting of kainate receptors to recycling or degradation pathways. EMBO J. 23, 4749-4759. 10.1038/sj.emboj.7600483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S., Bouschet T., Jenkins E. L., Nishimune A. and Henley J. M. (2008). Bidirectional regulation of kainate receptor surface expression in hippocampal neurons. J. Biol. Chem. 283, 36435-36440. 10.1074/jbc.M806447200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Y., Otsuka T., Horton A. C., Scott D. B. and Ehlers M. D. (2003). Activity-dependent mRNA splicing controls ER export and synaptic delivery of NMDA receptors. Neuron 40, 581-594. 10.1016/S0896-6273(03)00676-7 [DOI] [PubMed] [Google Scholar]
- Nishikura K. (2016). A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 17, 83-96. 10.1038/nrm.2015.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschen W., Schmitt J., Gissel C. and Dux E. (1997). Developmental changes of RNA editing of glutamate receptor subunits GluR5 and GluR6: in vivo versus in vitro. Brain Res. Dev. Brain Res. 98, 271-280. 10.1016/S0165-3806(96)00193-9 [DOI] [PubMed] [Google Scholar]
- Peng P. L., Zhong X., Tu W., Soundarapandian M. M., Molner P., Zhu D., Lau L., Liu S., Liu F. and Lu Y. M. (2006). ADAR2-dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron 49, 719-733. 10.1016/j.neuron.2006.01.025 [DOI] [PubMed] [Google Scholar]
- Penn A. C., Balik A. and Greger I. H. (2013). Steric antisense inhibition of AMPA receptor Q/R editing reveals tight coupling to intronic editing sites and splicing. Nucleic Acids Res. 41, 1113-1123. 10.1093/nar/gks1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia R. S., Wang Y.-X. and Wenthold R. J. (1994). Histological and ultrastructural localization of the kainate receptor subunits, KA2 and GluR6/7, in the rat nervous system using selective antipeptide antibodies. J. Comp. Neurol. 349, 85-110. 10.1002/cne.903490107 [DOI] [PubMed] [Google Scholar]
- Petrovic M. M., Viana da Silva S., Clement J. P., Vyklicky L., Mulle C., González-González I. M. and Henley J. M. (2017). Metabotropic action of postsynaptic kainate receptors triggers hippocampal long-term potentiation. Nat. Neurosci. 20, 529-539. 10.1038/nn.4505 [DOI] [PubMed] [Google Scholar]
- Rocca D. L., Wilkinson K. A. and Henley J. M. (2017). SUMOylation of FOXP1 regulates transcriptional repression via CtBP1 to drive dendritic morphogenesis. Sci. Rep. 7, 877 10.1038/s41598-017-00707-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer B., Köhler M., Sprengel R. and Seeburg P. H. (1991). RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67, 11-19. 10.1016/0092-8674(91)90568-J [DOI] [PubMed] [Google Scholar]
- Swanson G. T., Feldmeyer D., Kaneda M. and Cull-Candy S. G. (1996). Effect of RNA editing and subunit co-assembly single-channel properties of recombinant kainate receptors. J. Physiol. 492, 129-142. 10.1113/jphysiol.1996.sp021295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. (2011). Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu. Rev. Neurosci. 34, 89-103. 10.1146/annurev-neuro-060909-153238 [DOI] [PubMed] [Google Scholar]
- Turrigiano G. G., Leslie K. R., Desai N. S., Rutherford L. C. and Nelson S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892-896. 10.1038/36103 [DOI] [PubMed] [Google Scholar]
- Vissel B., Royle G. A., Christie B. R., Schiffer H. H., Ghetti A., Tritto T., Perez-Otano I., Radcliffe R. A., Seamans J., Sejnowski T. et al. (2001). The role of RNA editing of kainate receptors in synaptic plasticity and seizures. Neuron 29, 217-227. 10.1016/S0896-6273(01)00192-1 [DOI] [PubMed] [Google Scholar]
- Wondolowski J. and Dickman D. (2013). Emerging links between homeostatic synaptic plasticity and neurological disease. Front. Cell Neurosci. 7, 223 10.3389/fncel.2013.00223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S. K., Sato S. and Lazinski D. W. (2001). Substrate recognition by ADAR1 and ADAR2. RNA 7, 846-858. 10.1017/S135583820101007X [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.