

Abstract
The extant literature supports the role of stress in enhancing the susceptibility of drug abuse progressing to a substance use disorder diagnosis. However, the molecular mediators by which stress enhances the progression from cocaine abuse to cocaine use disorder via the mesolimbic pathway remain elusive. In this mini-review article, we highlight three mechanisms by which glucocorticoids (GCs) and the dopaminergic system interact. First, GCs upregulate tyrosine hydroxylase (TH), the rate-limiting enzyme in dopamine (DA) synthesis. Second, GCs downregulate monoamine-oxidase (MAO), an enzyme responsible for DA removal. Lastly, GCs are hypothesized to decrease DA reuptake, subsequently increasing synaptic DA. Based on these interactions, we review preclinical literature highlighting how stress modulates the mesolimbic pathway, including the ventral tegmental area (VTA) and nucleus accumbens (NAcs), to alter cocaine abuse-related effects. Taken together, stress enhances cocaine’s abuse-related effects at multiple points along the VTA mesolimbic projection, and uniquely in the NAcs through a positive feedback type mechanism. Furthermore, we highlight future directions to elucidate the interaction between the prefrontal cortex (PFC) and key intermediaries including ΔFosB, cAMP response element binding protein (CREB) and cyclin-dependent kinase 5 (CDK5) to highlight possible mechanisms that underlie stress-induced acceleration of the progression to a cocaine use disorder diagnosis.
Keywords: stress, addiction, cocaine, VTA, NAc, drugs
Introduction
The Diagnostic and Statistical Manual of Mental Disorders (DSM)-V criteria for substance use disorders is defined as “recurrent use of alcohol and/or other drugs causes clinically and functionally significant impairment, such as health problems, disability and failure to meet major responsibilities at work, school, or home” (American Psychiatric Association, 2013). Substance use disorders may range from mild-to-severe and include a variety of substances such as opiates, nicotine, alcohol, cocaine and others, each of which has different mechanisms of action and protein targets. While cocaine exposure does not always progress to a cocaine use disorder diagnosis, a subset of individuals will progress to severe cocaine use disorder or what is referred to as cocaine “addiction” in the preclinical literature. Although epidemiological reports vary, cocaine use disorder is estimated to have an incidence of 0.1% worldwide (Shield et al., 2018). Although the factors that drive progression to substance use disorders are not fully defined, several lines of evidence suggest stress exacerbates susceptibility to the abuse-related effects of drugs (Piazza and Le Moal, 1998; Sinha, 2001; Cleck and Blendy, 2008). For example, neonatal stress selectively enhances the acquisition of cocaine self-administration in rats, but does not augment self-administration when the reinforcer is food (Kosten et al., 2000). Social housing stress in nonhuman primates enhances the reinforcing effects of cocaine in subordinate monkeys (Morgan et al., 2002); however, early life stress produced by maternal separation does not enhance the abuse-related effects of cocaine in nonhuman primates (Ewing Corcoran and Howell, 2010).
Moreover, cumulative adversity is significantly predictive of drug abuse in a dose-dependent manner (Sinha, 2008). In fact, the limbic-hypothalamic-pituitary-adrenal axis (LHPA) axis, responsible for governing the stress response, has substantial overlap with the mesolimbic “reward” pathway involved in reward circuitry (Koob, 2009). The mesolimbic pathway involves dopaminergic projections from the ventral tegmental area (VTA) to the nucleus accumbens (NAcs) and olfactory tubercle in the brain (Quintero, 2013). This pathway is hypothesized to have a critical role in the perception of pleasure and is conceptualized by Koob (2011) to have several key functions: associating meaning to reward-related cues, motivating goal-oriented behavior and general activation. In this mini-review article, we will focus on the impact of stress on cocaine abuse-related effects mediated through the mesolimbic dopamine (DA) “reward” pathway. Given the considerable evidence supporting an impact of stress on substance use disorder susceptibility and relapse, improved understanding of the mechanisms by which stress alters the abuse-related effects of drugs may provide insight into novel molecular targets for therapeutic interventions.
Underlying Mechanisms of Cocaine Abuse
Cocaine nonselectively binds to all three monoamine transporters (DA, norepinephrine, and serotonin) and prevents the reuptake of these monoamines into the presynaptic terminal thereby enhancing monoamine neurotransmission. Cocaine inhibition of the DA transporter is thought to be the primary mediator of the abuse-related effects of cocaine (Ritz et al., 1987; Volkow et al., 1997). Despite the DA transporter being the primary target for cocaine’s abuse-related effects, repeated cocaine exposure does not alter presynaptic DA transporter availability in either humans (Wang et al., 1997) or nonhuman primates (Czoty et al., 2007). However, repeated cocaine exposure has been shown to increase serotonin and norepinephrine transporter densities in nonhuman primates (Macey et al., 2003; Beveridge et al., 2005; Banks et al., 2008). Furthermore, repeated cocaine exposure downregulates both presynaptic and postsynaptic DA receptors in humans (Volkow et al., 1990, 1993), nonhuman primates (Nader et al., 2006) and rats (Laurier et al., 1994). These cocaine-induced decreases in DA receptors on both pre- and post-synaptic terminals, and the resulting reduced dopaminergic tone, are thought to contribute to the depressive-like symptoms of cocaine withdrawal and relapse of cocaine abuse (Volkow et al., 1993; Thomas et al., 2001).
In substance use disorders, relapse can be triggered by drug-related cues that function as discriminative stimuli to signal contingencies of drug availability and promote drug-taking behavior. For example, following drug-associated cue presentation, the amygdala signals to dopaminergic cell bodies in the VTA (Nestler and Carlezon, 2006; Cleck and Blendy, 2008). These VTA dopaminergic neurons then signal to the NAcs to release DA, which triggers increased gamma-aminobutyric acid (GABA)-ergic input to the thalamus (Koob, 1992; Nestler and Carlezon, 2006). This GABAergic thalamic input leads to hypoactivation of the prefrontal cortex (PFC), impairing judgment and reasoning (Volkow and Morales, 2015). Thus, a combination of increased DA output in the mesolimbic pathway and decreased PFC activation in cortical pathways appear to result in increased drug-taking behavior. Curiously, various types of stressors have been shown to promote drug-taking behavior in preclinical models of drug relapse (Mantsch et al., 2016; Dong et al., 2017), further highlighting the interconnection between stress and reward pathways in the brain.
Mechanisms of Stress Response
The LHPA influences a variety of functions including the digestive system, immune system, reproductive system, mood and energy expenditure (Vázquez, 1998). The LHPA undergoes self-regulation through feedback and modulates the extrahypothalamic stress neurocircuit (Koob and Kreek, 2007). In addition, the LHPA activates the brain reward circuit (Koob and Kreek, 2007), bridging the interdependent relationship of glucocorticoids (GCs) and the dopaminergic system.
The LHPA is activated following hypothalamic release of corticotropin-releasing hormone (CRH) and vasopressin through a hypophyseal portal system to the anterior pituitary (Aguilera, 2011). CRH may be triggered by either internal or external cues. Synergistically interacting with vasopressin, CRH induces adrenocorticotrophic hormone (ACTH) release by the anterior pituitary. ACTH then acts on the adrenal gland inducing GC secretion into the bloodstream. Cortisol, the primary GC in humans, binds to the GC receptor (GR) in the brain and other end organ tissues facilitating the stress response. The LHPA modulates the stress response through negative feedback on the axis, specifically through negative feedback on the anterior pituitary and hypothalamus that inhibits ACTH and CRH release, ultimately decreasing blood cortisol levels through reduced release.
The GR is a transcription factor, and following translocation to the nucleus, the GR can modulate 10%–20% of genes in the human genome (Oakley and Cidlowski, 2013). While unbound GR remains in the cytosol, in the presence of cortisol, bound GR translocates to the nucleus and interacts with GC response elements (GREs) to modulate transcription (Chrousos et al., 2009). Moreover, GR interacts with other transcription factors, including nuclear factor-κB (NF-κB; Russo et al., 2007) and activator protein-1 (AP-1), which have been implicated in the progression to severe substance use disorder (Hope, 1998; Chrousos et al., 2009).
Interactions Between Glucocorticoids and the Dopaminergic System
The interactions between LHPA-induced GC release and the dopaminergic system are pivotal to understanding interactions between stress and substance use disorders. Both stressors and drugs of abuse have been shown to activate the mesolimbic “reward” pathway. For example, both increase glutamate receptor activation of VTA dopaminergic neurons (Cleck and Blendy, 2008). In addition, the LHPA axis also enhances glutamatergic plasticity in the VTA (Stelly et al., 2016). Furthermore, Barrot et al. (2000) have shown that adrenalectomy leading to decreased GC levels resulted in decreased basal and cocaine-induced increase in NAcs shell DA levels. Figure 1 shows three potential mechanisms by which GCs are hypothesized to alter dopaminergic activity. First, GCs increase DA biosynthesis by enhancing tyrosine hydroxylase (TH) activity, the rate-limiting enzyme in DA synthesis (Daubner et al., 2011). This is illustrated by the observation that rats exposed to social isolation have increased TH levels in the NAcs shell (Trainor, 2011). A second mechanism by which GCs are hypothesized to alter dopaminergic activity is through GC-induced reductions in monoamine-oxidase (MAO) activity (Poletto et al., 2011). MAO is another method, in addition to monoamine reuptake by presynaptic transporters as described above, for terminating monoamine neurotransmission. Decreased MAO activity would increase synaptic DA levels and enhance dopaminergic neurotransmission. Lastly, GCs acting at GRs have been shown to regulate DA transporter expression under both basal and cocaine-stimulated conditions (Wheeler et al., 2017). These results are also consistent with reduced DA transporters in rats that underwent early life stress (Meaney et al., 2002). Overall, this literature supports a role of GC regulation of the mesolimbic DA pathway at multiple levels to alter both basal and cocaine-induced dopaminergic neurotransmission.
Figure 1.
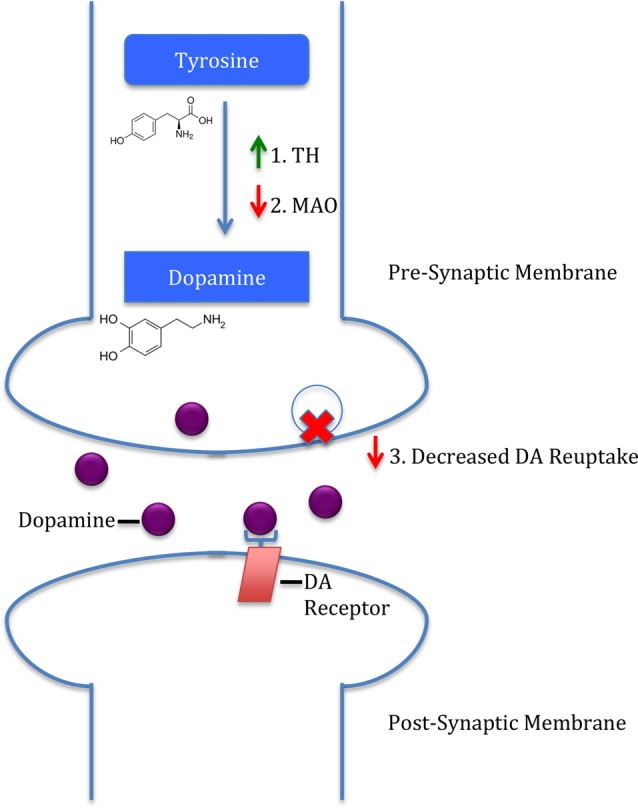
Three mechanisms by which glucocorticoids (GCs) induce dopamine (DA) release. First, GCs upregulate tyrosine hydroxylase (TH), the rate-limiting enzyme in DA synthesis. Second, GCs downregulate monoamine-oxidase (MAO), an enzyme responsible for DA removal. Lastly, GCs are hypothesized to decrease DA reuptake, subsequently increasing synaptic DA.
VTA
Increased Glutamatergic Plasticity
Both stress and drugs of abuse have been shown to increase glutamatergic plasticity in the VTA (Saal et al., 2003). Furthermore, exposure to stressful events enhances VTA glutamatergic plasticity that may further enhance the abuse-related effects of cocaine (Fitzgerald et al., 1996; Kauer and Malenka, 2007; Stelly et al., 2016). In a recent study by Stelly et al. (2016), rats first underwent a resident-intruder social defeat paradigm in conjunction with corticosterone injections, and then cocaine rewarding effects were assessed using a conditioned place preference (CPP) procedure. Repeated social defeat selectively enhanced long-term potentiation (LTP) of N-Methyl-D-aspartic acid receptors (NMDARs) in the VTA. This LTP manifested as enhanced VTA dopaminergic neuron firing in response to cocaine-associated cues during CPP only in the stressed group. This additional dopaminergic burst was interpreted as enhancing the conditioned stimulus-response relationship between drug-associated cues and the abused drug that may be involved in drug relapse (Stelly et al., 2016). These results suggest stress-induced glutamatergic plasticity of NMDAR and subsequent enhancement of cocaine abuse-related effects may be attenuated in the VTA by a GC antagonist. Deletion of nuclear receptor subfamily 3, group C, member 1 (nr3c1), a gene encoding a GR, blunted cocaine reinforcement in a drug self-administration procedure and VTA dopaminergic firing (Ambroggi et al., 2009; Barik et al., 2013). These results provide further evidence that GRs modulate VTA dopaminergic plasticity that directly impacts the abuse-related effects of cocaine.
Accumulating evidence suggests one molecular mechanism by which both stress and drugs of abuse impact glutamatergic plasticity in the mesolimbic pathway is through extracellular signal-regulated kinases (ERK). For example, stress exposure increased inositol 1,4,5-trisphosphate receptors (IP3R) sensitization that was mediated by protein kinase A (PKA), an upstream activator in ERK pathway (Vanhoutte et al., 1999; Stelly et al., 2016; Figure 2). Consistent with these previous results, social-defeat stress increased ERK signaling in the VTA (Yap et al., 2015). Moreover, ERK signaling appears to rely on the relative ratio of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) and NMDARs. For example, stress exposure increases the AMPA/NMDA ratio in the VTA (Saal et al., 2003; Dong et al., 2004). However, inhibition of ERK activation has produced equivocal results on the abuse-related effects of cocaine. Administration of SL327, a mitogen-activated protein kinase (MEK) inhibitor used to inhibit ERK, decreased both context and cocaine-induced CPP (Valjent et al., 2000, 2006; Pan et al., 2011). This trend may be indicative of neuroadaptive changes post ERK inhibition. In contrast, administration of U0126, another MEK inhibitor, directly into the VTA enhanced both context and cocaine cue-induced reinstatement in non-stressed rats (Lu et al., 2004, 2009). However, in rats undergoing social stress first, U0126 directly into the VTA attenuated stress-enhanced cocaine locomotor sensitization (Stelly et al., 2016). Taken together, the role of ERK activation in cocaine’s abuse-related effects seems fundamental to understanding downstream physiological and behavioral alterations initiated in the VTA.
Figure 2.
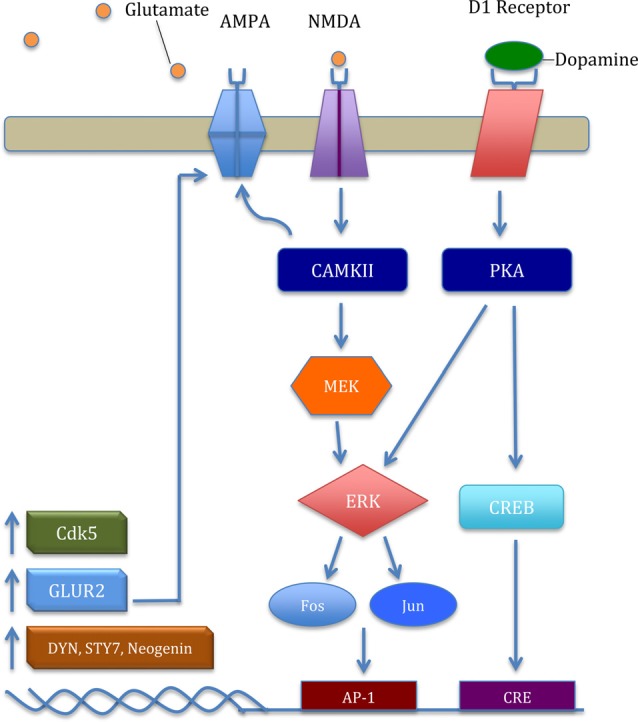
The general mechanism of the extracellular signal-regulated kinase (ERK) pathway and key downstream products important in cocaine addiction. Glutamate binding excites the N-Methyl-D-aspartic acid receptor (NMDAR) and upregulates intracellular calcium. Excitation provokes a signaling cascade, upregulating transcription factors Fos and Jun. Subsequently, increased ΔFosB acts on activator protein-1 (AP-1) and upregulates transcription and translation of cyclin-dependent kinase 5 (CDK5), GLUR2, dynorphin (Dyn), synaptotagmin VII (Syt7), and neogenin. CDK5 mediates localization and GLUR2-mediated plasticity in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) through phosphorylation of δ-Catenin (Poore et al., 2010). Increased GLUR2 upregulates the ERK pathway in a positive-feedback type manner through increased AMPAR in the nucleus accumbens (NAcs). Moreover, increased D1 receptor activation upregulates protein kinase A (PKA), phosphorylating the transcription factor cAMP response element binding protein (CREB), leading to further increase in CDK5 and GLUR2 protein levels.
CRF-R1 Modulation
Corticotropin-releasing factor acting at type 1 receptor (CRF-R1) has also emerged as one potential molecular mechanism linking stress and drug abuse. For example, intermittent social defeat stress elicits CRF release in the VTA (Holly et al., 2016). Furthermore, social defeat stress or intra-VTA CRF enhanced the abuse-related effects of cocaine in rats (Boyson et al., 2014; Leonard et al., 2017). Consistent with these previous findings, administration of a CRF antagonist before each social defeat stress attenuated both cocaine-induced locomotor sensitization and escalated cocaine self-administration in rats (Boyson et al., 2011). However, CRF antagonists also decrease escalated cocaine self-administration in non-stressed rats (Specio et al., 2008) suggesting the role of CRF on interactions between social stress and cocaine abuse-related effects have not been fully elucidated. Further complicating the role of CRF in cocaine reinforcement are results from nonhuman primates demonstrating a CRF antagonist does not attenuate cocaine self-administration (Mello et al., 2006). In congruence with this observation, the CRF antagonist verucerfont failed to attenuate alcohol craving in anxious alcoholic women, despite blocking HPA axis responsivity to dexamethasone (Schwandt et al., 2016). Overall, in contrast to the preclinical reports using rodents, nonhuman primate and clinical results do not provide compelling evidence for a significant role of CRF in altering the abuse-related effects of abused drugs in either stress or non-stressed research subjects.
Nucleus Accumbens NAcs
Increased LTP From D1 Activation
In addition to drug-induced changes in the VTA, chronic cocaine use and stress exposure can directly alter the NAcs (Wolf and Ferrario, 2010; Koya and Hope, 2011). Preclinical models show cocaine-induced morphological changes in dendritic spine density and greater AMPAR/NMDAR firing in the NAcs after administration alone (Wolf and Ferrario, 2010; Koya and Hope, 2011). Furthermore, chronic stress may alter relapse and self-administration via epigenetic modifications to histone dimethyltransferase G9a in the NAcs (Anderson et al., 2018). In addition to drug and stress induced changes in the NAcs, chronic stress exposure may further substance abuse via a feedback loop with the VTA. The D1 receptor is a Gs-protein coupled post-synaptic receptor that is linked to upregulation of FBJ murine osteosarcoma viral oncogene homolog B (ΔFosB), cAMP response element binding protein (CREB), and cyclin-dependent kinase 5 (CDK5; Catalano et al., 2009; Lebel et al., 2009; Zhang et al., 2002). Increased D1 receptor activation leads to upregulated glutamatergic receptors in the NAcs (Chao et al., 2002; Mangiavacchi and Wolf, 2004). In addition, increased D1 activation attenuates GABA-B, a metabotropic transmembrane receptor, inhibition due to changes in adenosine levels after cocaine exposure in the VTA (Bonci and Williams, 1996). Reduced inhibition by GABA-B can subsequently increase LTP (Nicola et al., 2000) and decrease long-term depression (LDP) leading to increased synaptic plasticity in the NAcs (Bonci and Williams, 1996; Nicola et al., 2000; Fourgeaud et al., 2004). NAcs inhibitory neurons can project back to the VTA, resulting in a possible feedback loop of increased neurogenic excitability and DA release (Omelchenko and Sesack, 2009; Xia et al., 2011). The increase in potentiation further excites DA cells, causing DA release (Gonon and Sundstrom, 1996; Gonon, 1997). This theory aligns with recent data suggesting increased DA release after CGP55845 administration, a GABA-B antagonist (Melchior et al., 2015). Subsequently, greater DA in the synapse reduces D1 DA receptor availability in the ventral striatum according to recent PET scans (Martinez et al., 2009). Additional research is needed to support a pattern of a positive feedback loop and greater VTA response to the drug. Furthermore, stress-induced cocaine seeking is initiated by GABA-B receptor-dependent CRF actions in the VTA (Blacktop et al., 2016). Although this modulation by stress is carried out in the VTA, effects of GABA-B and CRF interactions are exerted in the postsynaptic membrane in the NAcs. Additional evidence suggests brain-derived neurotrophic factor (BDNF) may mediate neuronal excitability through activation of tropomyosin receptor kinase B (TrkB) receptors in the NAcs (Berton et al., 2006). Lobo et al. (2010) found a loss of TrkB receptors, mimicked through upregulation of D2 neurons, lead to decreased cocaine reward; in contrast, upregulation of D1 excitability showed an increase in cocaine reward. In addition to BDNF’s mediating role, stress is implicated in facilitation of further synaptic adaptations in the NAcs. To this end, Chaudhury et al. (2013) demonstrated that repeated social defeat stress may induce VTA DA neuron phasic firing to the NAcs in mice. These data suggest that stress-induced phasic firing of the VTA may augment synaptic excitability in the NAcs of cocaine-addicted brains (Chaudhury et al., 2013).
The proposition that stress exerts effects through inhibition of positive feedback is not fully supported in the extant literature. For example, Sinha (2008), reported that chronic stress inhibits DA synthesis in the NAcs. However, it is well supported that GC concentrations directly correlate with extracellular DA release (Brake et al., 2004; Sinha, 2008). Although DA synthesis may be inhibited by chronic stress, cocaine sensitization has been repeatedly shown to increase by gene and protein regulators such as ΔFosB, CREB and CDK5 (Kelz et al., 1999; Bibb et al., 2001; McClung and Nestler, 2003; Mattson et al., 2005). Therefore, the combined data leads us to conclude that stress increases drug addiction susceptibility through increased sensitization in a positive feedback manner (Figure 3). Furthermore, the literature suggests that stress perpetuates drug dependence through allostasis by reinforcement in an analogous feedback manner (Koob and Le Moal, 2001; Ahmed et al., 2002). Taken together, the available findings collectively suggest that stress may mediate drug dependence at multiple levels, through positive feedback mechanisms.
Figure 3.
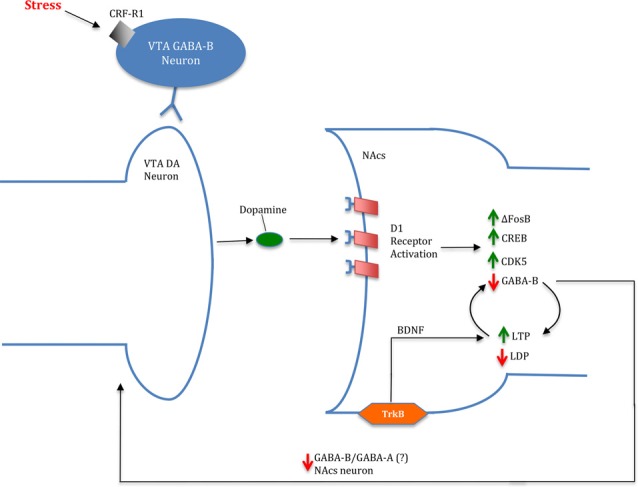
Image of a potential mechanism for stress- and cocaine-induced drug dependence via a feed-forward cycle in the NAcs. In the presence of stress, ventral tegmental area (VTA) DA release is upregulated resulting in increased D1 receptor activation. Cortisol is implicated in increasing DA release through corticotropin-releasing factor acting at type 1 receptor (CRF-R1) binding to gamma-aminobutyric acid (GABA)-B VTA neurons acting on VTA DA neurons. Increased DA levels promote D1 activation leading to an increase in ΔFosB, CREB and CDK5 levels in the NAcs. Moreover, D1 activation is linked to decreased GABA-B activation in the NAcs, resulting in greater long-term potentiation (LTP): long-term depression (LDP). Attenuation of GABA projections from NAcs to the VTA is suggested to further DA release; however, the particular projection (GABA-A/GABA-B) is currently unknown. Furthermore, brain-derived neurotrophic factor (BDNF) is implicated in contributing to LTP in the NAcs through activation of tropomyosin receptor kinase B (TrkB) receptors.
Conclusion and Future Directions
Although this mini-review article has focused on the effects of stress on the mesolimbic DA pathway, the effects of stress on other brain regions implicated in substance use disorders are important considerations beyond the capacity of this brief synopsis. For example, GRs are highly expressed in the PFC. GCs can act locally in the PFC to modulate cognitive impairments in working memory due to acute stress (Butts et al., 2011). Similar to GC effects on the mesolimbic DA pathway, corticosterone administered directly into the PFC can increase DA efflux (Butts et al., 2011). However, despite the relevant function of the PFC in substance use disorders (Volkow et al., 2016), relatively little research has been done to determine the extent to which molecular intermediaries such as ΔFosB, CREB, or CDK5 are involved in the PFC with regard to stress-induced enhancement of cocaine abuse-related effects.
Collectively, this mini-review article details three potential molecular mechanisms relating DA and GC interactions as they relate to stress-induced enhancement of cocaine abuse-related behaviors. In all three mechanisms, stress-induced GC release and subsequent activation of GRs primes the mesolimbic DA pathway. The overall net effect is enhanced abuse-related effects of cocaine and enhanced susceptibility of progressing to a cocaine use disorder diagnosis (Sinha, 2008). Thus, stress may serve as a positive feedback mechanism in the NAcs for enhancing the susceptibility to, or progression to, substance use disorder.
Author Contributions
DM and GN discussed ideas for the initial submission. DM led the literature search and discussed results with GN. GN advised on direction and additional resources. DM wrote the majority of the initial manuscript and GN provided revisions and reformatting of content. MB provided substantial editorial comments following the first stage of review including writing additional content and recommendations on revisions for existing content. All individuals have approved the final version and agree to be responsible for the content.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by the National Institutes of Health National Institute of Nursing Research (NR014886; GN).
References
- Aguilera G. (2011). HPA axis responsiveness to stress: implications for healthy aging. Exp. Gerontol. 46, 90–95. 10.1016/j.exger.2010.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S. H., Kenny P. J., Koob G. F., Markou A. (2002). Neurobiological evidence for hedonic allostasis associated with escalating cocaine use. Nat. Neurosci. 5, 625–626. 10.1038/nn872 [DOI] [PubMed] [Google Scholar]
- Ambroggi F., Turiault M., Milet A., Deroche-Gamonet V., Parnaudeau S., Balado E., et al. (2009). Stress and addiction: glucocorticoid receptor in dopaminoceptive neurons facilitates cocaine seeking. Nat. Neurosci. 12, 247–249. 10.1038/nn.2282 [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association (2013). Diagnostic and Statistical Manual of Mental Disorders (DSM-5)®. Arlington, VA: American Psychiatric Publishing. [Google Scholar]
- Anderson E. M., Larson E. B., Guzman D., Wissman A. M., Neve R. L., Nestler E. J., et al. (2018). Overexpression of the histone dimethyltransferase G9a in nucleus accumbens shell increases cocaine self-administration, stress-induced reinstatement, and anxiety. J. Neurosci. 38, 803–813. 10.1523/JNEUROSCI.1657-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks M. L., Czoty P. W., Gage H. D., Bounds M. C., Garg P. K., Garg S., et al. (2008). Effects of cocaine and MDMA self-administration on serotonin transporter availability in monkeys. Neuropsychopharmacology 33, 219–225. 10.1038/sj.npp.1301420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barik J., Marti F., Morel C., Fernandez S. P., Lanteri C., Godeheu G., et al. (2013). Chronic stress triggers social aversion via glucocorticoid receptor in dopaminoceptive neurons. Science 339, 332–335. 10.1126/science.1226767 [DOI] [PubMed] [Google Scholar]
- Barrot M., Marinelli M., Abrous D. N., Rougé-Pont F., Le Moal M., Piazza P. V. (2000). The dopaminergic hyper-responsiveness of the shell of the nucleus accumbens is hormone-dependent. Eur. J. Neurosci. 12, 973–979. 10.1046/j.1460-9568.2000.00996.x [DOI] [PubMed] [Google Scholar]
- Berton O., McClung C. A., DiLeone R. J., Krishnan V., Renthal W., Russo S. J., et al. (2006). Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 311, 864–868. 10.1126/science.1120972 [DOI] [PubMed] [Google Scholar]
- Beveridge T. J., Smith H. R., Nader M. A., Porrino L. J. (2005). Effects of chronic cocaine self-administration on norepinephrine transporters in the nonhuman primate brain. Psychopharmacology 180, 781–788. 10.1007/s00213-005-2162-1 [DOI] [PubMed] [Google Scholar]
- Bibb J. A., Chen J., Taylor J. R., Svenningsson P., Nishi A., Snyder G. L., et al. (2001). Effects of chronic exposure to cocaine are regulated by the neuronal protein Cdk5. Nature 410, 376–380. 10.1038/35066591 [DOI] [PubMed] [Google Scholar]
- Blacktop J. M., Vranjkovic O., Mayer M., Van Hoof M., Baker D. A., Mantsch J. R. (2016). Antagonism of GABA-B but not GABA-A receptors in the VTA prevents stress- and intra-VTA CRF-induced reinstatement of extinguished cocaine seeking in rats. Neuropharmacology 102, 197–206. 10.1016/j.neuropharm.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A., Williams J. T. (1996). A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron 16, 631–639. 10.1016/s0896-6273(00)80082-3 [DOI] [PubMed] [Google Scholar]
- Boyson C. O., Holly E. N., Shimamoto A., Albrechet-Souza L., Weiner L. A., DeBold J. F., et al. (2014). Social stress and CRF–dopamine interactions in the VTA: role in long-term escalation of cocaine self-administration. J. Neurosci. 34, 6659–6667. 10.1523/JNEUROSCI.3942-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyson C. O., Miguel T. T., Quadros I. M., Debold J. F., Miczek K. A. (2011). Prevention of social stress-escalated cocaine self-administration by CRF-R1 antagonist in the rat VTA. Psychopharmacology 218, 257–269. 10.1007/s00213-011-2266-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brake W. G., Zhang T. Y., Diorio J., Meaney M. J., Gratton A. (2004). Influence of early postnatal rearing conditions on mesocorticolimbic dopamine and behavioural responses to psychostimulants and stressors in adult rats. Eur. J. Neurosci. 19, 1863–1874. 10.1111/j.1460-9568.2004.03286.x [DOI] [PubMed] [Google Scholar]
- Butts K. A., Weinberg J., Young A. H., Phillips A. G. (2011). Glucocorticoid receptors in the prefrontal cortex regulate stress-evoked dopamine efflux and aspects of executive function. Proc. Natl. Acad. Sci. U S A 108, 18459–18464. 10.1073/pnas.1111746108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano S., Giordano C., Rizza P., Gu G., Barone I., Bonofiglio D., et al. (2009). Evidence that leptin through STAT and CREB signaling enhances cyclin D1 expression and promotes human endometrial cancer proliferation. J. Cell. Physiol. 218, 490–500. 10.1002/jcp.21622 [DOI] [PubMed] [Google Scholar]
- Chao S. Z., Ariano M. A., Peterson D. A., Wolf M. E. (2002). D1 dopamine receptor stimulation increases GluR1 surface expression in nucleus accumbens neurons. J. Neurochem. 83, 704–712. 10.1046/j.1471-4159.2002.01164.x [DOI] [PubMed] [Google Scholar]
- Chaudhury D., Walsh J. J., Friedman A. K., Juarez B., Ku S. M., Koo J. W., et al. (2013). Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature 493, 532–536. 10.1038/nature11713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrousos G. P., Kino T., Charmandari E. (2009). Evaluation of the hypothalamic-pituitary-adrenal axis function in childhood and adolescence. Neuroimmunomodulation 16, 272–283. 10.1159/000216185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleck J. N., Blendy J. A. (2008). Making a bad thing worse: adverse effects of stress on drug addiction. J. Clin. Invest. 118, 454–461. 10.1172/jci33946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czoty P. W., Gage H. D., Nader S. H., Reboussin B. A., Bounds M., Nader M. A. (2007). PET imaging of dopamine D2 receptor and transporter availability during acquisition of cocaine self-administration in rhesus monkeys. J. Addict. Med. 1, 33–39. 10.1097/adm.0b013e318045c038 [DOI] [PubMed] [Google Scholar]
- Daubner S. C., Le T., Wang S. (2011). Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 508, 1–12. 10.1016/j.abb.2010.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y., Saal D., Thomas M., Faust R., Bonci A., Robinson T., et al. (2004). Cocaine-induced potentiation of synaptic strength in dopamine neurons: behavioral correlates in GluRA (−/−) mice. Proc. Natl. Acad. Sci. U S A 101, 14282–14287. 10.1073/pnas.0401553101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y., Taylor J. R., Wolf M. E., Shaham Y. (2017). Circuit and synaptic plasticity mechanisms of drug relapse. J. Neurosci. 37, 10867–10876. 10.1523/JNEUROSCI.1821-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing Corcoran S. B., Howell L. L. (2010). Impact of early life stress on the reinforcing and behavioral-stimulant effects of psychostimulants in rhesus monkeys. Behav. Pharmacol. 21, 69–76. 10.1097/fbp.0b013e3283359f53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald L. W., Ortiz J., Hamedani A. G., Nestler E. J. (1996). Drugs of abuse and stress increase the expression of GluR1 and NMDAR1 glutamate receptor subunits in the rat ventral tegmental area: common adaptations among cross-sensitizing agents. J. Neurosci. 16, 274–282. 10.1523/JNEUROSCI.16-01-00274.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourgeaud L., Mato S., Bouchet D., Hémar A., Worley P. F., Manzoni O. J. (2004). A single in vivo exposure to cocaine abolishes endocannabinoid-mediated long-term depression in the nucleus accumbens. J. Neurosci. 24, 6939–6945. 10.1523/JNEUROSCI.0671-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonon F. (1997). Prolonged and extrasynaptic excitatory action of dopamine mediated by D1 receptors in the rat striatum in vivo. J. Neurosci. 17, 5972–5978. 10.1523/JNEUROSCI.17-15-05972.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonon F., Sundstrom L. (1996). Excitatory effects of dopamine released by impulse flow in the rat nucleus accumbens in vivo. Neuroscience 75, 13–18. 10.1016/0306-4522(96)00320-x [DOI] [PubMed] [Google Scholar]
- Holly E. N., Boyson C. O., Montagud-Romero S., Stein D. J., Gobrogge K. L., DeBold J. F., et al. (2016). Episodic social stress-escalated cocaine self-administration: role of phasic and tonic corticotropin releasing factor in the anterior and posterior ventral tegmental area. J. Neurosci. 36, 4093–4105. 10.1523/JNEUROSCI.2232-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope B. T. (1998). Cocaine and the AP-1 transcription factor complex. Ann. N Y Acad. Sci. 844, 1–6. 10.1111/j.1749-6632.1998.tb08216.x [DOI] [PubMed] [Google Scholar]
- Kauer J. A., Malenka R. C. (2007). Synaptic plasticity and addiction. Nat. Rev. Neurosci. 8, 844–858. 10.1038/nrn2234 [DOI] [PubMed] [Google Scholar]
- Kelz M. B., Chen J., Carlezon W. A., Jr., Whisler K., Gilden L., Beckmann A. M., et al. (1999). Expression of the transcription factor ΔFosB in the brain controls sensitivity to cocaine. Nature 401, 272–276. 10.1038/45790 [DOI] [PubMed] [Google Scholar]
- Koob G. F. (1992). Drugs of abuse: anatomy, pharmacology and function of reward pathways. Trends Pharmacol. Sci. 13, 177–184. 10.1016/0165-6147(92)90060-j [DOI] [PubMed] [Google Scholar]
- Koob G. F. (2009). Dynamics of neuronal circuits in addiction: reward, antireward, and emotional memory. Pharmacopsychiatry 42, S32–S41. 10.1055/s-0029-1216356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob G. F. (2011). Theoretical frameworks and mechanistic aspects of alcohol addiction: alcohol addiction as a reward deficit disorder. Curr. Top. Behav. Neurosci. 13, 3–30. 10.1007/7854_2011_129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob G. F., Le Moal M. (2001). Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24, 97–129. 10.1016/s0893-133x(00)00195-0 [DOI] [PubMed] [Google Scholar]
- Koob G., Kreek M. J. (2007). Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am. J. Psychiatry 164, 1149–1159. 10.1176/appi.ajp.2007.05030503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosten T. A., Miserendino M. J., Kehoe P. (2000). Enhanced acquisition of cocaine self-administration in adult rats with neonatal isolation stress experience. Brain Res. 875, 44–50. 10.1016/s0006-8993(00)02595-6 [DOI] [PubMed] [Google Scholar]
- Koya E., Hope B. T. (2011). Cocaine and synaptic alterations in the nucleus accumbens. Biol. Psychiatry 69, 1013–1014. 10.1016/j.biopsych.2011.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurier L. G., Corrigall W. A., George S. R. (1994). Dopamine receptor density, sensitivity and mRNA levels are altered following self-administration of cocaine in the rat. Brain Res. 634, 31–40. 10.1016/0006-8993(94)90255-0 [DOI] [PubMed] [Google Scholar]
- Lebel M., Patenaude C., Allyson J., Massicotte G., Cyr M. (2009). Dopamine D1 receptor activation induces tau phosphorylation via cdk5 and GSK3 signaling pathways. Neuropharmacology 57, 392–402. 10.1016/j.neuropharm.2009.06.041 [DOI] [PubMed] [Google Scholar]
- Leonard M. Z., DeBold J. F., Miczek K. A. (2017). Escalated cocaine “binges” in rats: enduring effects of social defeat stress or intra-VTA CRF. Psychopharmacology 234, 2823–2836. 10.1007/s00213-017-4677-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo M. K., Covington H. E., III., Chaudhury D., Friedman A. K., Sun H., Damez-Werno D., et al. (2010). Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science 330, 385–390. 10.1126/science.1188472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L., Dempsey J., Liu S. Y., Bossert J. M., Shaham Y. (2004). A single infusion of brain-derived neurotrophic factor into the ventral tegmental area induces long-lasting potentiation of cocaine seeking after withdrawal. J. Neurosci. 24, 1604–1611. 10.1523/JNEUROSCI.5124-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L., Wang X., Wu P., Xu C., Zhao M., Morales M., et al. (2009). Role of ventral tegmental area glial cell line-derived neurotrophic factor in incubation of cocaine craving. Biol. Psychiatry 66, 137–145. 10.1016/j.biopsych.2009.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macey D. J., Smith H. R., Nader M. A., Porrino L. J. (2003). Chronic cocaine self-administration upregulates the norepinephrine transporter and alters functional activity in the bed nucleus of the stria terminalis of the rhesus monkey. J. Neurosci. 23, 12–16. 10.1523/JNEUROSCI.23-01-00012.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiavacchi S., Wolf M. E. (2004). D1 dopamine receptor stimulation increases the rate of AMPA receptor insertion onto the surface of cultured nucleus accumbens neurons through a pathway dependent on protein kinase A. J. Neurochem. 88, 1261–1271. 10.1046/j.1471-4159.2003.02248.x [DOI] [PubMed] [Google Scholar]
- Mantsch J. R., Baker D. A., Funk D., Lê A. D., Shaham Y. (2016). Stress-induced reinstatement of drug seeking: 20 years of progress. Neuropsychopharmacology 41, 335–356. 10.1038/npp.2015.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez D., Slifstein M., Narendran R., Foltin R. W., Broft A., Hwang D. R., et al. (2009). Dopamine D1 receptors in cocaine dependence measured with PET and the choice to self-administer cocaine. Neuropsychopharmacology 34, 1774–1782. 10.1038/npp.2008.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson B. J., Bossert J. M., Simmons D. E., Nozaki N., Nagarkar D., Kreuter J. D., et al. (2005). Cocaine-induced CREB phosphorylation in nucleus accumbens of cocaine-sensitized rats is enabled by enhanced activation of extracellular signal-related kinase, but not protein kinase A. J. Neurochem. 95, 1481–1494. 10.1111/j.1471-4159.2005.03500.x [DOI] [PubMed] [Google Scholar]
- McClung C. A., Nestler E. J. (2003). Regulation of gene expression and cocaine reward by CREB and ΔFosB. Nat. Neurosci. 6, 1208–1215. 10.1038/nn1143 [DOI] [PubMed] [Google Scholar]
- Meaney M. J., Brake W., Gratton A. (2002). Environmental regulation of the development of mesolimbic dopamine systems: a neurobiological mechanism for vulnerability to drug abuse? Psychoneuroendocrinology 27, 127–138. 10.1016/s0306-4530(01)00040-3 [DOI] [PubMed] [Google Scholar]
- Melchior J. R., Ferris M. J., Stuber G. D., Riddle D. R., Jones S. R. (2015). Optogenetic versus electrical stimulation of dopamine terminals in the nucleus accumbens reveals local modulation of presynaptic release. J. Neurochem. 134, 833–844. 10.1111/jnc.13177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello N. K., Negus S. S., Rice K. C., Mendelson J. H. (2006). Effects of the CRF1 antagonist antalarmin on cocaine self-administration and discrimination in rhesus monkeys. Pharmacol. Biochem. Behav. 85, 744–751. 10.1016/j.pbb.2006.11.008 [DOI] [PubMed] [Google Scholar]
- Morgan D., Grant K. A., Gage H. D., Mach R. H., Kaplan J. R., Prioleau O., et al. (2002). Social dominance in monkeys: dopamine D2 receptors and cocaine self-administration. Nat. Neurosci. 5, 169–174. 10.1038/nn798 [DOI] [PubMed] [Google Scholar]
- Nader M. A., Morgan D., Gage H. D., Nader S. H., Calhoun T. L., Buchheimer N., et al. (2006). PET imaging of dopamine D2 receptors during chronic cocaine self-administration in monkeys. Nat. Neurosci. 9, 1050–1056. 10.1038/nn1737 [DOI] [PubMed] [Google Scholar]
- Nestler E. J., Carlezon W. A. (2006). The mesolimbic dopamine reward circuit in depression. Biol. Psychiatry 59, 1151–1159. 10.1016/j.biopsych.2005.09.018 [DOI] [PubMed] [Google Scholar]
- Nicola S. M., Surmeier D. J., Malenka R. C. (2000). Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu. Rev. Neurosci. 23, 185–215. 10.1146/annurev.neuro.23.1.185 [DOI] [PubMed] [Google Scholar]
- Oakley R. H., Cidlowski J. A. (2013). The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 132, 1033–1044. 10.1016/j.jaci.2013.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omelchenko N., Sesack S. R. (2009). Ultrastructural analysis of local collaterals of rat ventral tegmental area neurons: GABA phenotype and synapses onto dopamine and GABA cells. Synapse 63, 895–906. 10.1002/syn.20668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B., Zhong P., Sun D., Liu Q. S. (2011). Extracellular signal-regulated kinase signaling in the ventral tegmental area mediates cocaine-induced synaptic plasticity and rewarding effects. J. Neurosci. 31, 11244–11255. 10.1523/JNEUROSCI.1040-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza P. V., Le Moal M. (1998). The role of stress in drug self-administration. Trends Pharmacol. Sci. 19, 67–74. 10.1016/s0165-6147(97)01115-2 [DOI] [PubMed] [Google Scholar]
- Poletto R., Cheng H. W., Meisel R. L., Richert B. T., Marchant-Forde J. N. (2011). Gene expression of serotonin and dopamine receptors and monoamine oxidase-A in the brain of dominant and subordinate pubertal domestic pigs (Sus scrofa) fed a β-adrenoreceptor agonist. Brain Res. 1381, 11–20. 10.1016/j.brainres.2010.11.035 [DOI] [PubMed] [Google Scholar]
- Poore C. P., Sundaram J. R., Pareek T. K., Fu A., Amin N., Mohamed N. E., et al. (2010). Cdk5-mediated phosphorylation of δ-catenin regulates its localization and GluR2-mediated synaptic activity. J. Neurosci. 30, 8457–8467. 10.1523/JNEUROSCI.6062-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero G. C. (2013). Role of nucleus accumbens glutamatergic plasticity in drug addiction. Neuropsychiatr. Dis. Treat. 9, 1499–1512. 10.2147/ndt.s45963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz M. C., Lamb R. J., Goldberg S. R., Kuhar M. J. (1987). Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science 237, 1219–1223. 10.1126/science.2820058 [DOI] [PubMed] [Google Scholar]
- Russo S. J., Rental W., Kumar A. (2007). NFkB Signaling Regulates Cocaine-Induced Behavioral and Cellular Plasticity. San Diego, CA: Society for Neuroscience. [Google Scholar]
- Saal D., Dong Y., Bonci A., Malenka R. C. (2003). Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37, 577–582. 10.1016/s0896-6273(03)00021-7 [DOI] [PubMed] [Google Scholar]
- Schwandt M. L., Cortes C. R., Kwako L. E., George D. T., Momenan R., Sinha R., et al. (2016). The CRF1 antagonist verucerfont in anxious alcohol -dependent women: translation of neuroendocrine, but not of anti-craving effects. Neuropsychopharmacology 41, 2818–2829. 10.1038/npp.2016.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shield K. D., Imtiaz S., Probst C., Rehm J. (2018). “The epidemiology and public health burden of addictive disorders,” in Integrating Psychological and Pharmacological Treatments for Addictive Disorders: An Evidence-Based Guide, eds MacKillop J., Kenna G. A., Leggio L., Ray L. A. (New York, NY: Routledge/Taylor & Francis Group; ), 3–31. [Google Scholar]
- Sinha R. (2001). How does stress increase risk of drug abuse and relapse? Psychopharmacology 158, 343–359. 10.1007/s002130100917 [DOI] [PubMed] [Google Scholar]
- Sinha R. (2008). Chronic stress, drug use and vulnerability to addiction. Ann. N Y Acad. Sci. 1141, 105–130. 10.1196/annals.1441.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specio S. E., Wee S., O’Dell L. E., Boutrel B., Zorrilla E. P., Koob G. F. (2008). CRF1 receptor antagonists attenuate escalated cocaine self-administration in rats. Psychopharmacology 196, 473–482. 10.1007/s00213-007-0983-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelly C. E., Pomrenze M. B., Cook J. B., Morikawa H. (2016). Repeated social defeat stress enhances glutamatergic synaptic plasticity in the VTA and cocaine place conditioning. Elife 5:e15448. 10.7554/elife.15448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M. J., Beurrier C., Bonci A., Malenka R. C. (2001). Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat. Neurosci. 4, 1217–1223. 10.1038/nn757 [DOI] [PubMed] [Google Scholar]
- Trainor B. C. (2011). Stress responses and the mesolimbic dopamine system: social contexts and sex differences. Horm. Behav. 60, 457–469. 10.1016/j.yhbeh.2011.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E., Corbillé A. G., Bertran-Gonzalez J., Hervé D., Girault J. A. (2006). Inhibition of ERK pathway or protein synthesis during reexposure to drugs of abuse erases previously learned place preference. Proc. Natl. Acad. Sci. U S A 103, 2932–2937. 10.1073/pnas.0511030103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E., Corvol J. C., Pagès C., Besson M. J., Maldonado R., Caboche J. (2000). Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J. Neurosci. 20, 8701–8709. 10.1523/jneurosci.20-23-08701.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte P., Barnier J. V., Guibert B., Pagès C., Besson M. J., Hipskind R. A., et al. (1999). Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol. Cell. Biol. 19, 136–146. 10.1128/mcb.19.1.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez D. M. (1998). Stress and the developing limbic-hypothalamic-pituitary-adrenal axis. Psychoneuroendocrinology 23, 663–700. 10.1016/s0306-4530(98)00029-8 [DOI] [PubMed] [Google Scholar]
- Volkow N. D., Fowler J. S., Wang G. J., Hitzemann R., Logan J., Schlyer D. J., et al. (1993). Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse 14, 169–177. 10.1002/syn.890140210 [DOI] [PubMed] [Google Scholar]
- Volkow N. D., Fowler J. S., Wolf A. P., Schlyer D., Shiue C. Y. (1990). Effects of chronic cocaine abuse on postsynaptic dopamine receptors. Am. J. Psychiatry 147, 719–724;. 10.1176/ajp.147.6.719 [DOI] [PubMed] [Google Scholar]
- Volkow N. D., Koob G. F., McLellan A. T. (2016). Neurobiologic advances from the brain disease model of addiction. N. Engl. J. Med. 374, 363–371. 10.1056/nejmra1511480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow N., Morales M. (2015). The brain on drugs: from reward to addiction. Cell 162, 712–725. 10.1016/j.cell.2015.07.046 [DOI] [PubMed] [Google Scholar]
- Volkow N. D., Wang G. J., Fischman M. W., Foltin R. W., Fowler J. S., Abumrad N. N., et al. (1997). Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature 386, 827–830. 10.1038/386827a0 [DOI] [PubMed] [Google Scholar]
- Wang G. J., Volkow N. D., Fowler J. S., Fischman M., Foltin R., Abumrad N. N., et al. (1997). Cocaine abusers do not show loss of dopamine transporters with age. Life Sci. 61, 1059–1065. 10.1016/s0024-3205(97)00614-0 [DOI] [PubMed] [Google Scholar]
- Wheeler D. S., Ebben A. L., Kurtoglu B., Lovell M. E., Bohn A. T., Jasek I. A., et al. (2017). Corticosterone regulates both naturally occurring and cocaine-induced dopamine signaling by selectively decreasing dopamine uptake. Eur. J. Neurosci. 46, 2638–2646. 10.1111/ejn.13730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf M. E., Ferrario C. R. (2010). AMPA receptor plasticity in the nucleus accumbens after repeated exposure to cocaine. Neurosci. Biobehav. Rev. 35, 185–211. 10.1016/j.neubiorev.2010.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y., Driscoll J. R., Wilbrecht L., Margolis E. B., Fields H. L., Hjelmstad G. O. (2011). Nucleus accumbens medium spiny neurons target non-dopaminergic neurons in the ventral tegmental area. J. Neurosci. 31, 7811–7816. 10.1523/jneurosci.1504-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap J. J., Chartoff E. H., Holly E. N., Potter D. N., Carlezon W. A., Miczek K. A. (2015). Social defeat stress-induced sensitization and escalated cocaine self-administration: the role of ERK signaling in the rat ventral tegmental area. Psychopharmacology 232, 1555–1569. 10.1007/s00213-014-3796-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D., Zhang L., Lou D. W., Nakabeppu Y., Zhang J., Xu M. (2002). The dopamine D1 receptor is a critical mediator for cocaine-induced gene expression. J. Neurochem. 82, 1453–1464. 10.1046/j.1471-4159.2002.01089.x [DOI] [PubMed] [Google Scholar]