

Summary
Current models of SIRT1 enzymatic regulation primarily consider the effects of fluctuating levels of its co-substrate NAD+, which binds to the stably-folded catalytic domain. By contrast, the roles of the sizeable disordered N- and C-terminal regions of SIRT1 are largely unexplored. Here, we identify an insulin-responsive sensor in the SIRT1 N-terminal region (NTR), comprising an acidic cluster (AC) and a 3-helix bundle (3HB), controlling deacetylase activity. The allosteric assistor DBC1 removes a distal N-terminal shield from the 3HB, permitting PACS-2 to engage the AC and the transiently exposed helix 3 of the 3HB, disrupting its structure and inhibiting catalysis. The SIRT1 activator (STAC) SRT1720 binds and stabilizes the 3HB, protecting SIRT1 from inhibition by PACS-2. Identification of the SIRT1 insulin-responsive sensor and its engagement by the DBC1/PACS-2 regulatory hub provides important insight into the roles of disordered regions in enzyme regulation and the mode by which STACs promote metabolic fitness.
Graphical Abstract
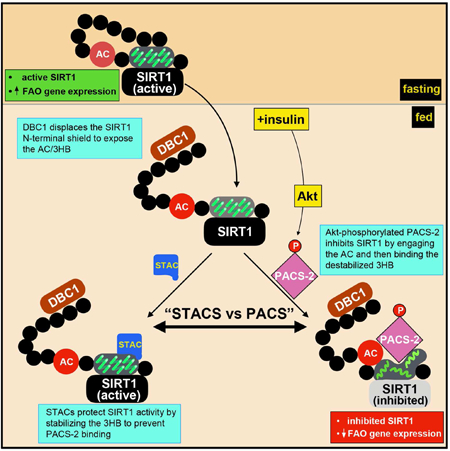
eTOC
The disordered SIRT1 N-terminal region contains an insulin-responsive sensor (IRS), comprising an acidic cluster (AC) and a 3-helix bundle (3HB), controlling enzyme activity. Insulin triggers DBC1/PACS-2 binding to the IRS. DBC1 removes the protective shield that hides the IRS, enabling PACS-2 to bind the AC and the 3HB, inhibiting SIRT1.
A remarkable property shared by nearly one-half of all vertebrate proteins is the presence of long (>30 amino acids) disordered regions (van der Lee et al., 2014). Unlike globular domains, disordered regions do not fold into autonomous rigid structures. They frequently contain flexible, 5–15 amino acid sequence motifs that can interact specifically with target sites on diverse binding partners, supporting physiologically important and distinct activities (Davey et al., 2015). Such sequence motifs are often targets for posttranslational modifications, further increasing their functionality (Tompa et al., 2014). In combination, multiple sequence motifs and their posttranslational modifications create exquisitley specific coincidence detectors that can rapidly assemble assorted cellular components to modulate function or generating hubs for controlling protein interaction networks and signaling pathways.
Enzymes are generally viewed as structured, globular proteins due to the precise spatial requirements for performing the necessary chemistry (Koshland, 1958). Nonetheless, vertebrate enzymes frequently contain appended disordered regions, which are regularly removed to render the proteins amenable for structural studies by X-ray crystallography. Thus, assessment of vertebrate enzyme regulation is often focused on the structured catalytic domains while the behavior and influences of the associated disordered regions have been largely ignored.
Sirtuins are homologs of the yeast silencing information regulator 2 protein, an NAD+-dependent histone deacetylase. Of the seven mammalian sirtuins, SIRT1 plays a critical function in DNA repair and metabolic fitness. In the liver, SIRT1 stimulates several key transcriptional programs, notably PGC-1α-dependent mitochondrial biogenesis and PGC-1α/PPARα-dependent fatty acid oxidation and ketogenesis, metabolic activities that assist in protecting organisms against diet-induced obesity (Li et al., 2014; Purushotham et al., 2009; Rodgers et al., 2005; Vega et al., 2000; Vernia et al., 2014). Small molecule sirtuin activating compounds (STACs) protect rodents from nonalcoholic fatty liver disease and diet-induced obesity, in part, by stimulating the expression of PGC-1α/PPARα target genes, in a SIRT1-dependent manner (Feige et al., 2008; Milne et al., 2007; Smith et al., 2009).
In addition to the conserved catalytic domain, SIRT1 possesses long N- and C-terminal disordered regions of largely unknown function, comprising most of the polypeptide chain. Recent studies have begun to shed some light on the disordered regions in the control of SIRT1 activity (Dai et al., 2015; Davenport et al., 2014; Hubbard et al., 2013a; Lakshminarasimhan et al., 2013). In particular, enzymatic activity requires a short segment in the C-terminal region (CTR), termed ESA (essential for sirtuin activity), which stabilizes the catalytic domain and helps to form the NAD+ binding pocket by completing the β-sheet of the Rossman fold (Dai et al., 2015; Hubbard et al., 2013a).
The lynchpin of acute SIRT1 allosteric regulation appears to be the 3-helix bundle (3HB), located in the N-terminal region (NTR). STACs bind the 3HB to allosterically increase SIRT1 activity through an unknown mechanism (Dai et al., 2015; Hubbard et al., 2013a; Lakshminarasimhan et al., 2013). Furthermore, a segment in the distal NTR has been proposed to interact with the proximal NTR to promote enzyme activity by protecting SIRT1 from unknown regulatory proteins (Ghisays et al., 2015). Therefore, to understand physiological SIRT1 regulation, it is essential to identify which cellular protein(s) are involved in controlling enzyme activity.
At present, only a handful of proteins have been discovered as potential SIRT1 regulators. Among these, DBC1 has received most attention. However, very different results for the two regions in SIRT1 that mediate DBC1 interactions and the mechanisms for enzyme repression have been put forward (Ghisays et al., 2015; Hubbard et al., 2013b; Kang et al., 2011; Kim et al., 2008; Zhao et al., 2008). PACS-2 has also been reported as a SIRT1 regulator (Atkins et al., 2014). PACS-2 is a member of the multi-functional PACS protein family, which engages a variety of motifs on client proteins, including CK2-phosphorylatable acidic clusters and helices, to modulate endomembrane traffic and ER-mitochondria communication at mitochondria-associated membranes (MAMs, (Thomas et al., 2017)). Vertebrate PACS-2 can act as a bifurcation switch, using an Akt phosphorylation site at Ser437, thereby separating its anabolic and catabolic roles (Aslan et al., 2009). Nuclear trafficking signals in vertebrate PACS-2 further expand its roles to include modulation of nuclear gene expression (Atkins et al., 2014; Thomas et al., 2017). In radiosensitive tissues, DNA damage triggers PACS-2 inhibition of SIRT1, which promotes p53 acetylation and, consequently, increases p53-dependent expression of p21 (CDKN1A) (Atkins et al., 2014). However, whether PACS-2 modulates SIRT1-dependent gene expression in metabolically active tissues is currently unknown.
Here, we use a holistic approach, combining NMR with biochemical assays and in vivo models, to uncover how PACS-2 and DBC1 create an insulin-responsive regulatory hub, which interacts with the disordered SIRT1 NTR to inhibit SIRT1-dependent activation of PGC-1α/PPARα target genes. We further show that the STAC SRT1720 protects SIRT1 from interaction with PACS-2 in vitro and in vivo, providing important insight into how small molecule inhibitors can safeguard SIRT1 activity and reduce HFD-induced steatosis and insulin resistance.
Results
PACS-2 binds the CK2 phosphorylated acidic cluster in the SIRT1 NTR
The disordered SIRT1 NTR (residues 1–236, see Fig. 1A) is necessary and sufficient for binding PACS-2 (Atkins et al., 2014). Using a Y2H assay, we determined that two segments in the distal half of the murine SIRT1 NTR engage the PACS-2 furin binding region (FBR, Fig. 1A). One interaction site resides within SIRT1132−169 and the other within SIRT1167–236. SIRT1132−169 contains a CK2-phosphorylatable acidic cluster, ES154DDDD, a motif frequently found on PACS-2 client proteins (Kang et al., 2009; Thomas et al., 2017). To verify that this motif is indeed involved in PACS-2 binding, a 5-Ala scan through residues 132–169 of SIRT1 was performed, and the resulting constructs were used as bait in a Y2H screen with the PACS-2 FBR. Both, SIRT1132-169 and SIRT1132-208 were employed to control for context-specific effects. Only substitution of the acidic cluster by alanine residues (ES154DDDD → E6A) blocked the interaction in both constructs (Figs. 1B and S1A). The mouse and human SIRT1 NTR sequences are nearly identical and a six-alanine substitution of the human SIRT1 NTR acidic cluster (ES162DEED → 6A) likewise blocked interaction with the human PACS-2 FBR (Fig. S1B).
Figure 1: PACS-2 binds the CK2-phosphorylatable acidic cluster in the SIRT1 NTR.
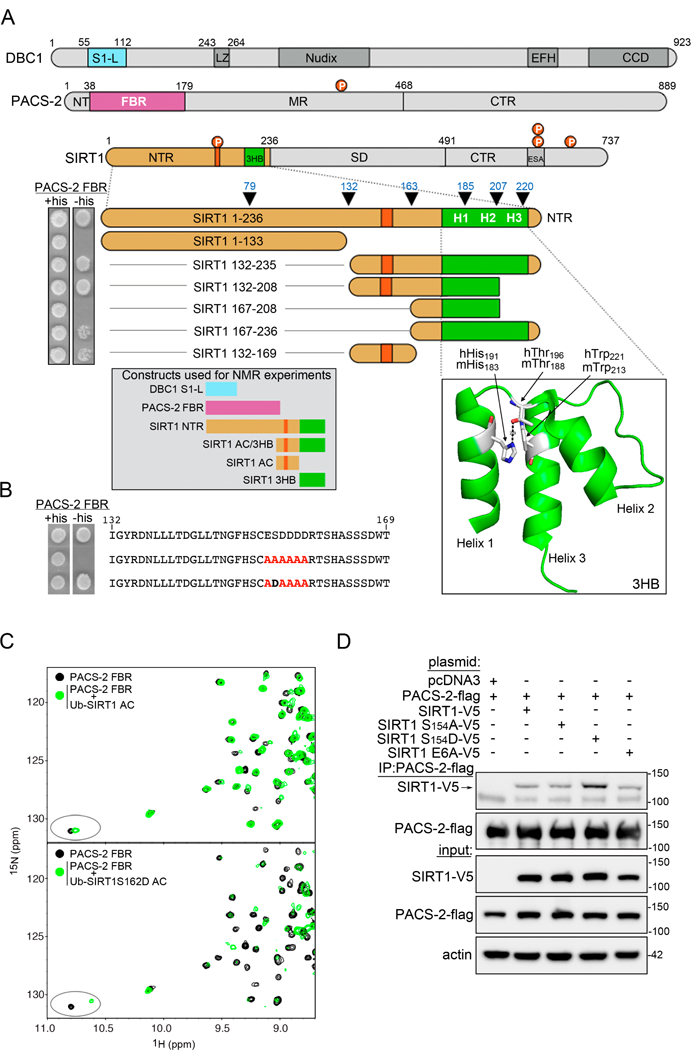
(A) Top: Schematic representation of DBC1, PACS-2 and SIRT1. Abbreviations used: S1-like domain (S1-L), leucine zipper (LZ), EF-hand (EFH), coiled-coil domain (CCD), furin binding region (FBR) N-terminal region (NTR), middle region (MR), C-terminal region (CTR), 3-helix bundle (3HB), sirtuin(catalytic) domain (SD), and essential for sirtuin activity (ESA). The SIRT1 NTR acidic cluster (aa153–158) is in orange. Red circles; PACS-2 Akt site at Ser437 and the mouse/human SIRT1 CK2 sites, Ser154/162 , Ser649/659, Ser651/661 and Ser683/693. Middle: Y2H analysis of the interaction between the PACS-2 FBR and indicated SIRT NTR deletion mutants, n=6. Line denotes removal of constructs unrelated to this study. Arrowheads denote SIRT1 NTR truncations described in Figure 2. Bottom left: Protein constructs used in the NMR experiments: DBC1 S1-L (DBC152-120), PACS-2 FBR (PACS-222-180), SIRT1 NTR (SIRT11-233), SIRT1 AC/3HB (SIRT1141-233), SIRT1 AC (SIRT1140-177), and SIRT1 3HB (SIRT1183-233). Bottom right: Structure of the human SIRT1 3HB (PDB:4ZZH) illustrating the mouse/human His183/191-Thr188/196 interaction. (B) Y2H analysis for PACS-2 FBR binding to several SIRT1132-169 AC constructs, n=3. Line denotes removal of constructs unrelated to this study. (C) Superposition of the downfield region of 1H-15N HSQC spectra of 100 µM PACS-2 FBR alone (black) or in the presence of 207 µM ubiquitin-tagged SIRT1 AC (top) or 232 µM S 162D-SIRT1 AC (bottom). The ellipse highlights a PACS-2 FBR tryptophan indole resonance which experiences a larger chemical shift change in the presence of S162D-SIRT1 AC. (D) FLAG-PACS-2 was co-expressed with V5-SIRT1 constructs in HCT116 cells. PACS-2 was immunoprecipitated (FLAG) and bound SIRT1 constructs were detected by western blot (V5), n=5.
We used NMR spectroscopy to investigate whether the PACS-2 FBR selectively binds the CK2-phosphorylatable acidic cluster in the human SIRT1 NTR. The 1H-15N HSQC spectrum of the PACS-2 FBR (PACS-222-180) displayed well-dispersed resonances of uniform intensity (Fig. S1C), indicating that this region of PACS-2 forms a stably folded structure. In contrast, the spectrum of ubiquitin-fused human SIRT1140-177, which contains the acidic cluster (SIRT1 AC, see inset in Fig. 1A), implies that this region of SIRT1 is disordered (Fig. S1D). Addition of increasing amounts of unlabeled SIRT AC into a sample of 15N-labeled PACS-2 FBR and monitoring the interaction by 1H-15N HSQC spectroscopy (Fig. 1C, top) revealed that the PACS-2 resonances are in slow exchange on the chemical shift scale, demonstrating a tight and specific interaction. The observed chemical shift changes in the PACS-2 FBR upon complexation with SIRT AC, containing the phosphomimic Ser162→Asp substitution (S162D-SIRT AC), were larger than those observed with SIRT1 AC (Fig. 1C, bottom), implying that the CK2-phosphorylated SIRT1 NTR is the preferred PACS-2 target. These results were recapitulated by Y2H, where the PACS-2 FBR interacted preferentially with SIRT1132-208, when it included the phosphomimic Ser154→Asp substitution (Figs. 1B and S1E).
To ascertain that PACS-2 indeed selectively interacts with CK2-phosphorylated SIRT1 in vivo, we co-expressed Flag-tagged PACS-2 with V5-tagged mouse SIRT1 or SIRT1 mutants, containing either a non-phosphorylatable acidic cluster (SIRT1S154A) or the phosphomimic substitution (SIRT1S154D). Consistent with the Y2H and NMR analyses, co-immunoprecipitation showed that PACS-2 bound to SIRT1S154D better than to SIRT1S154A or WT SIRT1 (Fig. 1D). However, changing the acidic cluster motif to an all alanine sequence (SIRT1E6A) did not completely abolish the interaction with PACS-2. This finding, in combination with the results from the Y2H analysis, suggests that the PACS-2 FBR interacts with a bipartite motif in the SIRT1 NTR, comprising the acidic cluster and a second site located between residues 167–236.
PACS-2 binding to the SIRT1 acidic cluster is necessary to bind the 3-helix bundle
To identify the second binding motif in the SIRT1 NTR for the PACS-2 FBR, co-immunoprecipitation assays with several murine SIRT1 N-terminal truncation mutants were carried out. Deletion of the SIRT1 N-terminal 79 or 132 amino acids (SIRT180-737 and SIRT1133-737) improved the interaction efficiency (Fig. 2A). However, an additional deletion through the AC (SIRT1164-737) reversed this effect. Interestingly, further deletion of SIRT1 residues 164–206 (SIRT1207-737), removing sequences that form the first two helices of the 3HB (see Fig. 1A), resulted in a robust interaction between PACS-2 and SIRT1. Removal of residues that form the third helix in the 3HB (SIRT1221-737) totally abrogated the PACS-2/SIRT1 interaction. The strong binding between PACS-2 and SIRT1207-737, compared to SIRT1186-737, which contains the intact 3HB, suggests that the region encompassing the third helix of the 3HB must be unfolded, but present, for efficient interaction with PACS-2.
Figure 2: PACS-2 binds the destabilized SIRT1 3-Helix bundle.
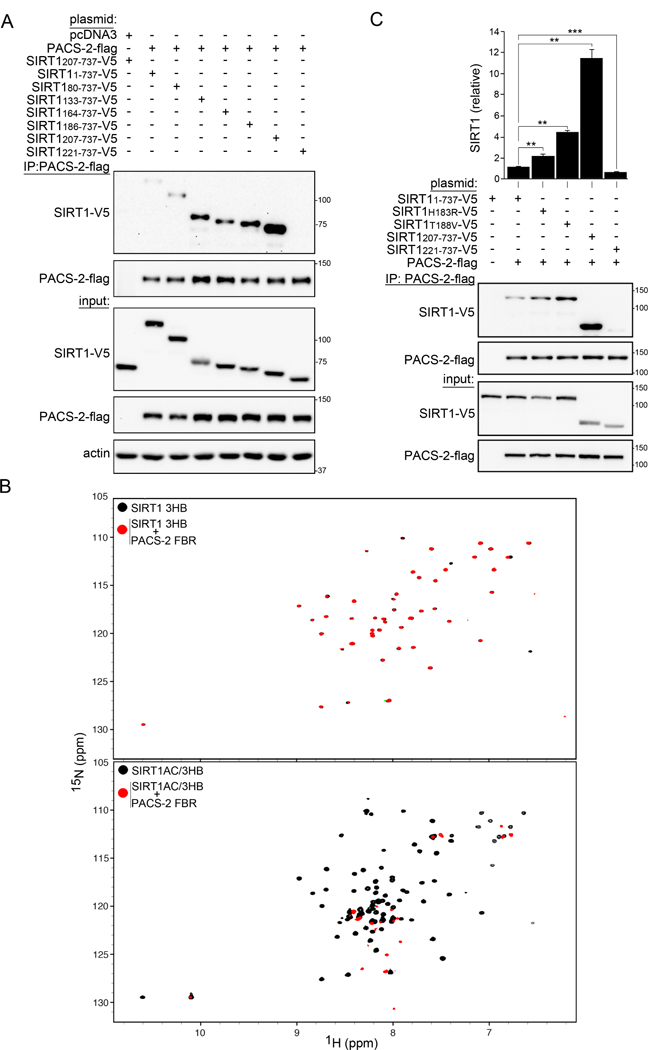
(A) FLAG-tagged PACS-2 co-expressed with V5-tagged SIRT1 constructs (Figure 1A) in HCT116 cells was immunoprecipitated (FLAG) and bound SIRT1 constructs detected by western blot (V5), n = 4. (B) Top: Superposition of 1H-15N HSQC spectra of 30 µM SIRT1 3HB alone (black) or in the presence of 200 µM PACS-2 FBR (red). Bottom: Superposition of 1H-15N HSQC spectra of 30 µM SIRT1 AC/3HB alone (black) or in the presence of 200 µM PACS-2 FBR (red). (C) Flag-PACS-2 co-expressed with the indicated V5-tagged SIRT1 constructs in HCT116 cells was immunoprecipitated (FLAG) and bound SIRT1 constructs were detected by western blot (V5). Data are mean ± SD, n = 6.
To directly test whether the structural integrity of the SIRT1 3HB affects binding to PACS-2, a set of human SIRT1 N-terminal truncations was prepared for NMR binding studies (constructs are depicted in Fig. 1A): SIRT1 3HB (SIRT1183-233, which shares 100% sequence identity with the mouse 3HB), SIRT1 AC/3HB (SIRT1141-233, the highly conserved acidic cluster (AC) plus the 3HB), and SIRT1 NTR (SIRT11-233). The 1H-15N HSQC spectrum of SIRT1 3HB exhibited well-dispersed resonances, characteristic of a folded α-helical protein (Fig. 2B top and S2A), in agreement with the crystal structure of SIRT1183-747 (Dai et al., 2015). The 1H-15N HSQC spectrum of the SIRT1 NTR, in contrast, indicated that the first ~182 amino acids of the SIRT1 N-terminus exist as a disordered, flexible polypeptide chain (Fig. S2A). Addition of the PACS-2 FBR to the SIRT1 3HB resulted in essentially an unchanged SIRT1 3HB spectrum, with broadening of very few resonances, indicative of weak and non-specific binding (Fig. 2B, top). In contrast, using SIRT1 AC/3HB with the complete bipartite binding site, resulted in instantaneous precipitation of the complex upon addition of the PACS-2 FBR (Fig. 2B, bottom). The 1H-15N HSQC spectrum of the supernatant lacked any “bound” resonances and contained only resonances exhibiting random coil characteristics, suggesting that the 3HB in SIRT1 AC/3HB no longer possesses a folded tertiary structure.
The interaction of PACS-2 with the SIRT1 3HB in vivo was probed by mutagenesis. In the crystal structure of the folded human SIRT1 3HB, a nitrogen atom of the His191 (His183 in mouse SIRT1) imidazole ring is positioned in hydrogen bonding distance to the hydroxyl of Thr196 (Thr188 in mouse SIRT1, see Fig. 1A). This interaction is likely involved in the relative positioning of helices 1 and 2 in the 3HB (Dai et al., 2015). Co-immunoprecipitation experiments using mouse SIRT1 constructs, containing His183→Arg or Thr188→Val substitutions, both of which would remove the putative H-bond, demonstrated an enhanced interaction with PACS-2 (Fig. 2C), suggesting that destabilization of the 3HB increases the binding affinity. Indeed, disruption of hydrogen bonding between His191 and Thr196 by protonation of the imidazole ring in the human 3HB was sufficient to unfold the human SIRT1 3HB structure, as evidenced by the loss of structured resonances from the 1H-15N HSQC spectrum of human SIRT1 AC/3HB at pH 5.5 and below, pH values that result in a protonated histidine side chain (Fig. S2B). Together, the above results show that PACS-2 engages the SIRT1 acidic cluster, enabling further interaction between PACS-2 and helix 3 in a partially un/folded 3HB of SIRT1.
DBC1 recruits PACS-2 to SIRT1
The enhanced interaction between PACS-2 and SIRT1 N-terminal truncations (Fig. 2) is consistent with the conclusions of a recent report that a cis interaction between the proximal and distal SIRT1 NTR protects the deacetylase from an unidentified protein modulator (Ghisays et al., 2015). We confirmed this interaction by Y2H analysis, which showed that SIRT11-54 interacted with SIRT1167-236 (Fig. S3A). We speculated that another SIRT1 regulatory protein may bind the SIRT1 NTR to displace the distal N-terminal segment, exposing the proximal AC and 3HB for PACS-2 interaction. One such candidate was DBC1, which had been suggested to bind SIRT1 and to promote repression of enzymatic activity (Kim et al., 2008; Zhao et al., 2008). Since the amount of DBC1 protein is reduced in PACS-2−/−liver, compared to WT (Fig. S3B), we reasoned that both proteins may participate in a common pathway. To test this possibility, Flag-tagged PACS-2 and V5-tagged SIRT1 were co-expressed in the absence and presence of Myc-tagged DBC1. In DBC1’s presence, the PACS-2/SIRT1 interaction was enhanced more than 8-fold for mouse or human proteins (Figs. 3A and S3C). This finding suggests that DBC1 and PACS-2 bind different sites on the SIRT1 NTR. We tested this possibility and found that PACS-2 interacted with SIRT180-737 to a greater extent than SIRT1 or SIRT152-737 (Fig. 3B, see also Fig. 2A). By contrast, DBC1 interacted to a greater extent with SIRT1 than with SIRT152-737 or SIRT180-737 (Fig. 3C). As a consequence, DBC1 failed to recruit PACS-2 to SIRT180-737 or SIRT1E6A (Fig. 3A).
Figure 3: DBC1 recruits PACS-2 to SIRT1.
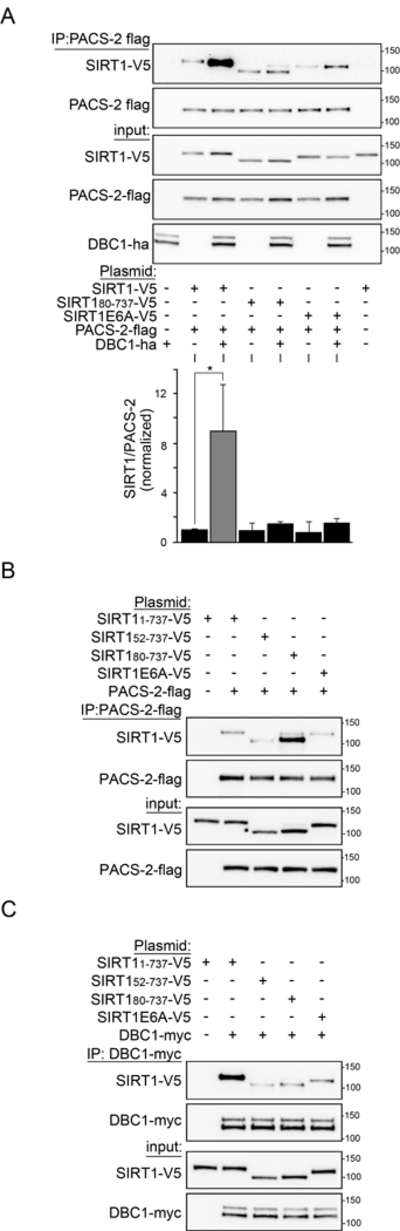
(A) FLAG-tagged PACS-2 was co-expressed with Myc-tagged DBC1 and different V5-tagged SIRT1 constructs in HCT116 cells. PACS-2 was immunoprecipitated (FLAG) and bound SIRT1 constructs were detected by western blot (V5). Data are mean ± SD, n = 4. (B) FLAG-tagged PACS-2 was co-expressed with different V5-tagged SIRT1 constructs in HCT116 cells. PACS-2 was immunoprecipitated (FLAG) and bound SIRT1 constructs were detected by western blot (V5), n = 3. (C) Myc-tagged DBC1 was co-expressed with different V5-tagged SIRT1 constructs in HCT116 cells. DBC1 was immunoprecipitated (Myc) and bound SIRT1 constructs were detected by western blot (V5), n = 3.
DBC1 and PACS-2 synergize to repress SIRT1 activity
The DBC1-dependent recruitment of PACS-2 to SIRT1 raised the possibility that DBC1 and PACS-2 may act synergistically to inhibit SIRT1 activity. This was evaluated using a specifically designed in vitro enzyme assay for SIRT1 activity (Hubbard et al., 2013a). In this assay, the minimal protein domains of both PACS-2 and DBC1 that are sufficient to bind full-length SIRT1 were used. We first confirmed a previous report that DBC11-230 was sufficient to interact with SIRT1 (Li et al., 2009) (Fig. S4A). Unfortunately, DBC11-230 was refractory for large scale recombinant protein production, necessary for structural probing by NMR. Therefore, smaller regions were expressed and purified. The S1-Like domain (S1-L, DBC152-120), which is the only segment within DBC11-230 that is predicted to be structured (Anantharaman and Aravind, 2008), exhibited a well dispersed 1H-15N HSQC spectrum, demonstrating that this domain is stably folded, possessing predominantly β-sheet character (Fig. 4A, top). DBC1 S1-L was used in an NMR binding assay with full-length human SIRT1, SIRT1 6-83, or the PACS-2 FBR. Upon addition of natural abundance full-length SIRT1, the 1H-15N HSQC spectrum of the 15N-labeled DBC1 S1-L exhibited distinct chemical shift changes, as well as peak broadening (Fig. 4A, top). Much smaller effects, on the other hand, were observed upon addition of hSIRT1 or the PACS-2 FBR (Fig. 4A, middle and bottom, respectively). These findings agree well with the results obtained from the co-IP experiments, where full-length DBC1 was found to interact strongly with full-length SIRT1 but only weakly with SIRT152-737, SIRT180-737 or PACS-2 (Figs. 3C and S4A, bottom).
Figure 4: PACS2 and DBC1 synergistically inhibit SIRT1 activity in vitro.
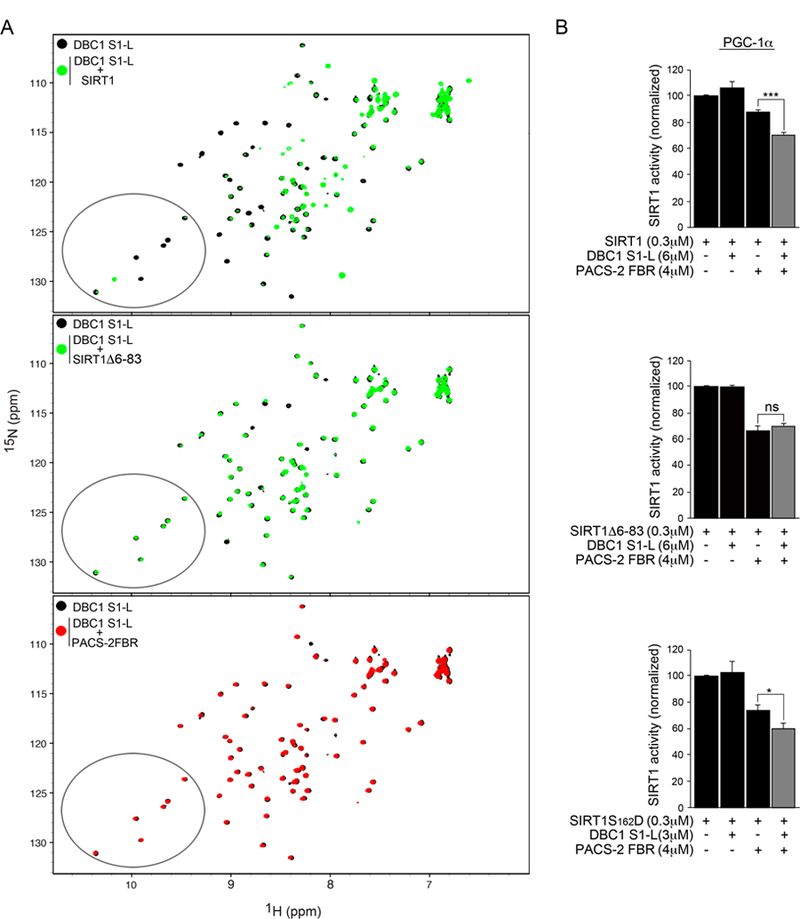
(A) Top: Superposition of 1H-15N HSQC spectra of 30 µM DBC1 S1-L alone (black) or in the presence of 70 µM full-length human SIRT1 (green). Middle: Superposition of 1H-15N HSQC spectra of 30 µM DBC1 S1-L alone (black) or in the presence of 70 µM human SIRT1 Δ6-83 (green). Bottom: Superposition of 1H-15N HSQC spectra of 30 µM DBC1 S1-L alone (black) or in the presence of 70 µM PACS-2 FBR (red). Ellipses highlight a region in the DBC1 S1-L spectrum where peak broadening and dramatic chemical shift changes in the presence of SIRT1 (top panel) but not SIRT1 Δ6-83 or PACS-2 FBR (middle and bottom panels, respectively) occur. (B) Enzyme assay using the Ac-Lys778-PGC-1α peptidyl substrate of recombinant human SIRT1, SIRT1 Δ6-83 or SIRT1S162D with the indicated combinations of recombinant DBC1 S1-L and PACS-2 FBR. Enzyme reactions were conducted as described in Methods.
The ability of purified PACS-2 FBR and DBC1 S1-L, alone or together, to inhibit SIRT1-dependent deacetylation of the PGC-1α substrate was evaluated (Fig. 4B). DBC1 S1-L alone did not interfere with SIRT1 activity, and PACS-2 FBR only minimally inhibited SIRT1 activity (~ 10% reduction). However, in combination, DBC1 S1-L and PACS-2 FBR caused a ~30% reduction in SIRT1 activity (Fig. 4B, top). By contrast, DBC1 S1-L was not required for inhibition by PACS-2 FBR when the N-terminally truncated SIRT1 6-83 construct was tested (Fig. 4B, middle). Since PACS-2 preferentially interacts with phosphorylated SIRT1 (see Fig.1), the phosphomimic SIRT1S162D mutant was also tested. This mutant was significantly more sensitive to inhibition by DBC1 S1-L and PACS-2 FBR, resulting in a ~ 42% reduction in SIRT1-dependent deacetylation of the PGC-1α substrate (Fig. 4B, bottom). Similar results were obtained using an acetylated H3K9 substrate in this enzyme assay (Fig. S4B).
The STAC SRT1720 inhibits the interaction of PACS-2 with the SIRT1 3-helix bundle
STACs have been reported to bind along helix 3 of the SIRT1 3HB (Dai et al., 2015). Since PACS-2 also interacts with helix 3, STACs, via stabilization of the 3HB, may prevent PACS-2 binding. 1H-15N HSQC spectroscopy was used to probe for binding between SIRT1 and SRT1720, the most widely investigated STAC ((Milne et al., 2007), see Figs. 5A and S5). SRT1720 binding caused dramatic chemical shift changes of the amide resonances belonging to amino acids in helix 3, including Leu220, Trp221, Val224, Ile225, and Leu228. The resonance of Ile210, which is located in the loop connecting helices 2 and 3, was also perturbed. Hydrogen bonding between the side chain of Asn226 in the 3HB and a carbonyl oxygen of STAC1 has been inferred from the crystal structure of the SIRT1/STAC1 complex (Dai et al., 2015). A similar contact may be present with SRT1720, despite the minimal structural similarity between STAC1 and SRT1720 (Dai et al., 2015; Milne et al., 2007). Indeed, addition of SRT1720 to SIRT1 3HB caused a pronounced chemical shift change in an NH2 side chain resonance pair, suggesting that SRT1720 might also engage Asn226 (Fig. 5A).
Figure 5: SRT1720 interferes with PACS-2 binding to SIRT1 in vitro and in vivo.
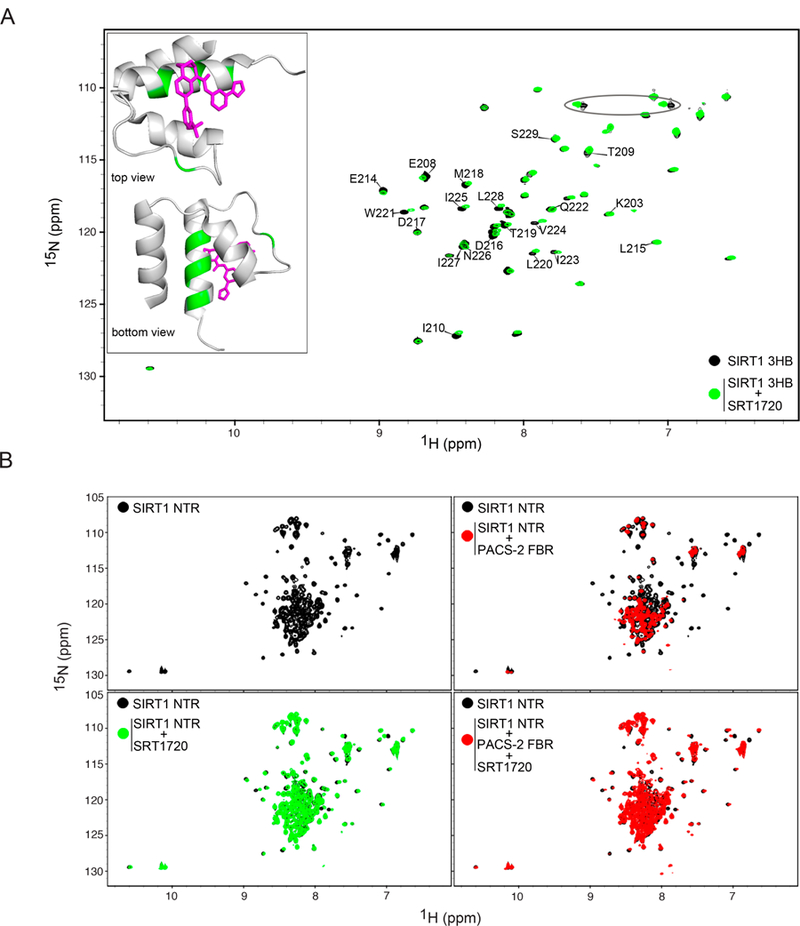
(A) Superposition of 1H-15N HSQC spectra of 100 µM SIRT1 3HB alone (black) or in the presence of 100 µM SRT1720 (green). Resonance assignments for residues in helix 3 and the linker between helices 2 and 3 are shown. The ellipse highlights a NH2 sidechain resonance pair that is affected by the interaction with SRT1720. Inset: Top and side views of SRT1720 (magenta) bound to the SIRT1 3HB (PDB:4ZZH). SIRT1 3HB residues that undergo the greatest chemical shift changes (mean + SD) are highlighted in green. (B) Superposition of 1H-15N HSQC spectra of 100 µM SIRT1 NTR alone (black) and in the presence of either 120 µM PACS-2 FBR (re d, upper right panel), 100 µM SRT1720 (green, lower left panel) or both 100 µM SRT1720 and 120 µM PACS-2 FBR (red, lower right panel).
In addition, the ability of SRT1720 to inhibit PACS-2 binding to SIRT1 was investigated. 1H-15N HSQC spectra of SIRT1 NTR in the absence or presence of SRT1720 were recorded upon addition of PACS-2 FBR (Fig. 5B). The SIRT1 NTR was used since it does not precipitate in the presence of the PACS-2 FBR like SIRT1 AC/3HB. Qualitative interpretation of the 1H-15N HSQC spectra suggests that SRT1720 binds the SIRT1 NTR in a similar manner to the SIRT1 3HB (Fig. 5A). However, given the large number of resonances in the random coil region of the SIRT11-233 spectrum, a complete assessment of its interaction with SRT1720 was not possible. Nonetheless, many helix 3 resonances, which are distinct and visible in the SIRT1 NTR spectrum, including Ile210, Leu220, Trp221, Val224, Ile225, and Leu228 are affected (Figs. 5A, 5B and S5). These resonances broadened beyond detection upon addition of the PACS-2 FBR (Fig. 5B). In contrast, pre-incubation of SIRT1 NTR with SRT1720 prevented the PACS-2-dependent resonance broadening, and resonances associated with residues in the structured 3HB were still observed in the spectrum (Fig. 5B) These data suggest that STACs enhance SIRT1 activity in vivo, in part, by preventing PACS-2 binding.
PACS-2−/−mice have elevated SIRT1 activity and are protected from diet-induced obesity
Consistent with our in vitro studies and an earlier report that PACS-2 regulates SIRT1 activity in radiosensitive tissues, we found that endogenous PACS-2 interacted with SIRT1 in liver (Fig. 6A and (Atkins et al., 2014)). Hepatic SIRT1 activity was increased in PACS-2−/−mice as evidenced by directly monitoring PGC-1α activation (deacetylation) by western blot and the SIRT1 enzyme assay (Figs. 6B and S6A). Since SIRT1 robustly induces the hepatokine FGF21 upon prolonged fasting (Badman et al., 2007; Li et al., 2014), the induction of FGF21 in liver from fasted WT and PACS-2−/−mice was measured. FGF21 expression was increased nearly 2-fold in the PACS-2−/−mice (Fig. 6C). Using primary hepatocytes isolated from WT and PACS-2−/−hepatocytes, FGF21 expression was induced by adding the PPARα agonist WY-14643 (Fig. 6D). In agreement with the in vivo studies, FGF21 expression was enhanced to a greater extent in the PACS-2−/−hepatocytes. Pre-treatment of the cells with the SIRT1 inhibitor EX-527 abrogated the increased FGF21 induction, suggesting that PACS-2 regulates the ability of SIRT1 to induce FGF21 expression in liver.
Figure 6: PACS-2−/−mice have elevated SIRT1 activity and are resistant to diet-induced obesity.
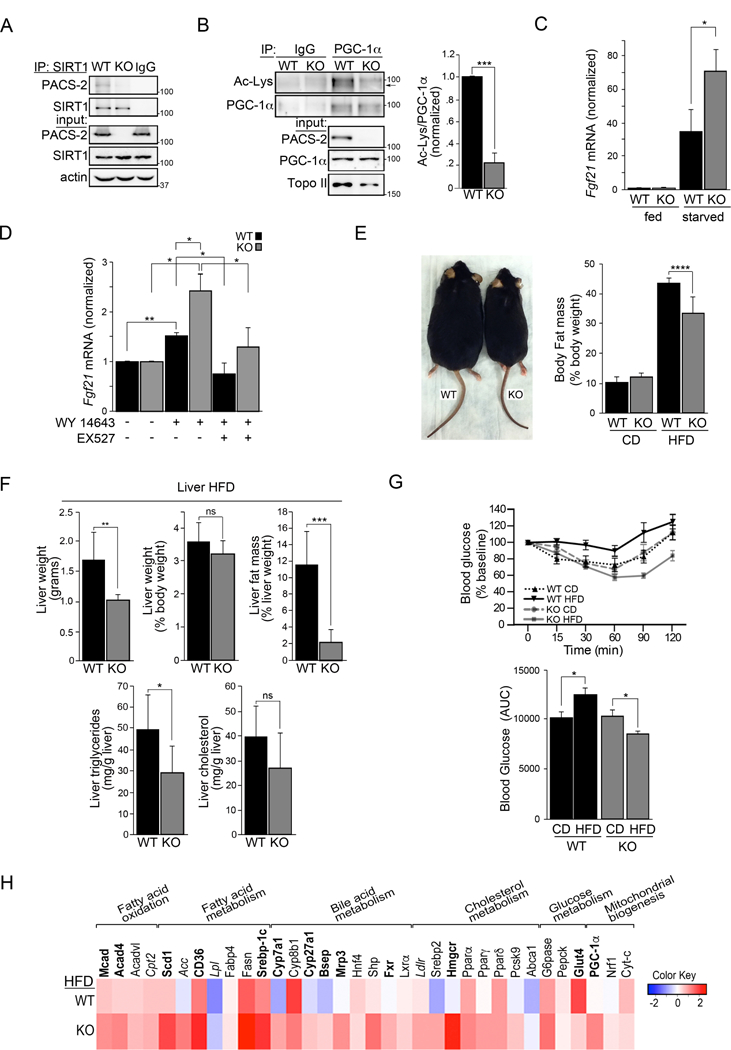
(A) Endogenous SIRT1 was immunoprecipitated from WT and PACS-2−/−liver. Co-precipitating endogenous PACS-2 was detected by western blot, n = 3. (B) Endogenous PGC-1α was immunoprecipitated from WT and PACS-2−/−liver. Acetylated-PGC-1α was detected by western blot. Data are mean ± SD, n=3. (C) RT-qPCR of FGF21 RNA from WT and PACS-2−/−mouse livers following a 14 hr fast. Data are mean ± SD, n = 4 mice per group. (D) Isolated WT and PACS-2−/−mouse primary hepatocytes were starved overnight and treated for 6 hr with 10 µM WY-14643 ± 10 µM EX-527. FGF21 was measured by RT-qPCR. Data are mean ± SD, n = 3. (E) Left: WT and PACS-2−/−mice after 8 weeks HFD. Right: Body fat mass of 8 weeks HFD WT and PACS-2−/−mice. (F) Liver weight, liver fat mass, liver triglycerides and liver cholesterol from WT (n = 6) and PACS-2−/−mice (KO, n = 13) following 8 weeks of HFD. Data are mean ± SD. (G) Top: Insulin tolerance test (ITT) response curves of WT and PACS-2−/−mice fed a CD or HFD for 8 weeks. Bottom: ITT data presented as area under the curve (AUC). Data are mean ± SD, n=6 mice per group. (H) RT-qPCR of RNA from WT and PACS-2−/−mouse livers following HFD challenge. Heatmap (Log2 scale) for HFD values normalized to CD was generated using R (n = 6 mice per group). Black bold letter, P<0.05; italics, P < 0.07, Gray letters, NS.
In line with the increased SIRT1 activity, PACS-2−/−mice should also be resistant to diet-induced obesity. Indeed, WT and PACS-2−/−mice, maintained on a control diet (CD), or challenged with an HFD for 8 weeks, demonstrated that PACS-2 loss resulted in reduced whole-body adiposity in response to the HFD (Figs. 6E and S6B-D) and protected the mice from both hepatic steatosis (Fig. 6F) and insulin resistance (Figs. 6G). RT-qPCR analyses of liver RNA from CD and HFD mice revealed that transcripts controlling fatty acid oxidation (FAO), mitochondrial biogenesis and cholesterol homeostasis were increased to a greater extent in HFD PACS-2−/−mice as compared to WT mice (Fig. 6H). To evaluate which transcription factors are likely responsible for the altered transcription profile in the PACS-2−/−mice, the overall RT-qPCR profile was interrogated using TFactS (Essaghir et al., 2010). This analysis identified several upregulated SIRT1-controlled transcription factors in HFD PACS-2−/−liver, including PPARα (Table S1).
Insulin triggers liver PACS-2 to inhibit SIRT1-dependent PGC-1α/PPARα target genes
To probe PACS-2’s ability to modulate SIRT1-dependent expression of PGC-1α/PPARα target genes, a luciferase reporter assay was used (Fig. 7A). PGC-1α stimulated PPARα-dependent expression of the PPRE (PPARα response element)-containing luciferase reporter to a greater extent in PACS-2 knockdown cells than in control cells. The SIRT1 inhibitor EX-527 abrogated the increased PGC-1α-dependent luciferase activity to a similar extent in PACS-2 knockdown or control cells, suggesting SIRT1 was responsible for the increased reporter activity in the PACS-2 knockdown cells.
Figure 7: PACS-2 regulates SIRT1-dependent expression of PGC-1α/PPARα target genes.
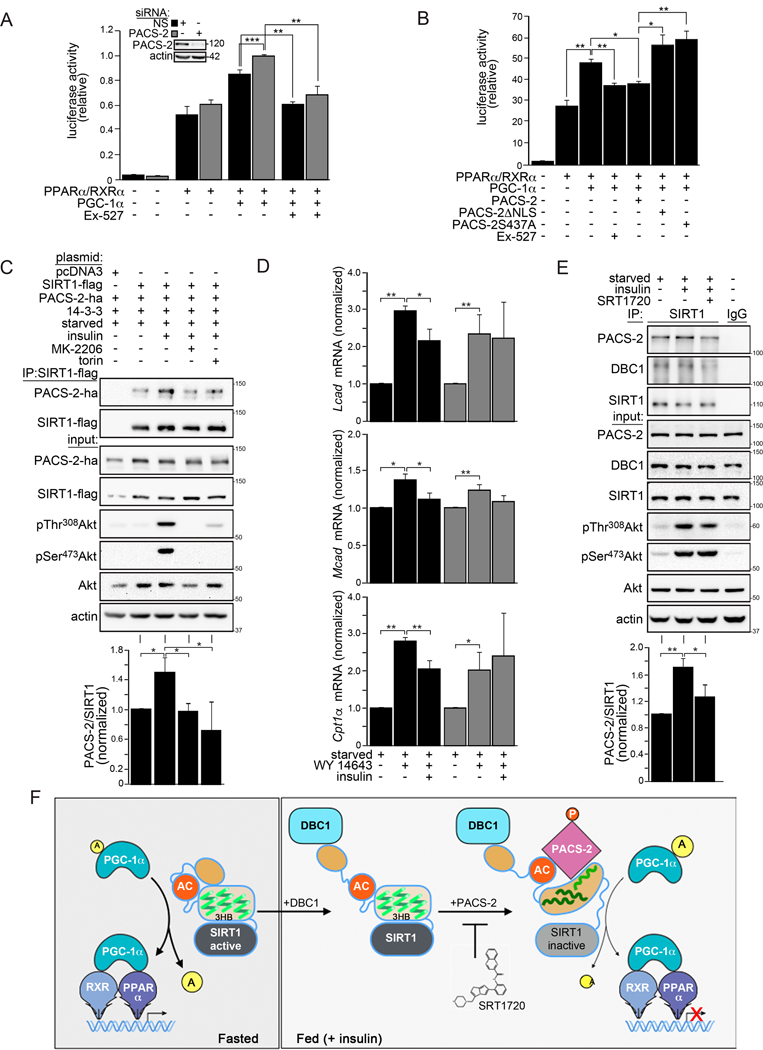
(A and B) HCT116 cells were pre-treated with non-specific (NS) or PACS-2 siRNAs or not and then transfected with plasmids expressing the PPARα response element (PPRE) luciferase reporter and the indicated constructs. Cells were then treated with 15 µM WY-14643 or 20 µM EX-527 as indicated. Luciferase activity in panels A and B was normalized with Renilla and data are the mean ± SD, n =3. Hela cells expressing the indicated proteins were starved for 14 hr treated or not with 1 µM Torin 1 or 5 µM MK-2206 and stimulated with 100 nM insulin for 30 min. SIRT1 was immunoprecipitated (FLAG) and bound PACS-2 was detected by western blot (HA); Data are mean ± SD, n = 3. (D) Isolated WT and PACS-2−/−mouse primary hepatocytes were starved overnight and treated for 6 hr with 10 µM WY-14643 followed by 100 nM insulin treatment for 4 hr. Cells were harvested and analyzed by RT-qPCR for the indicated transcripts. Data are mean ± SD, n = 3. (E) Primary hepatocytes were starved overnight, pre-treated with 10 µM SRT1720 for 1 hr and then treated with 100 nM insulin for 30 min. Endogenous SIRT1 was immunoprecipitated and bound PACS-2 and DBC1 were detected by western blot. Data are mean ± SD, n = 3. (F) Working model for the inhibition of SIRT1, orchestrated via the PACS-2 and DBC1 regulatory hub. Left: in fasting cells, active SIRT1 increases PGC-1α/PPARα-dependent expression of catabolic target genes. The SIRT1 N-terminus (tan oval) interacts in cis with an upstream segment of the NTR to shield the AC/3HB region from regulatory proteins. Right: In response to insulin, DBC1 displaces the SIRT1 N-terminal shield, exposing the AC/3HB region. Insulin/Akt signaling enables pSer437-PACS-2 to bind the SIRT1 AC (red circle) and destabilize the 3HB (green squiggles) by engaging helix 3. This inhibits enzyme activity and represses SIRT1-dependent PGC-1α/PPARα transcriptional activity. SIRT1720 stabilizes the SIRT1 3HB, thereby interfering with PACS-2-dependent inhibition of enzyme activity (light gray sirtuin domain).
Co-immunoprecipitation revealed a reduced interaction between SIRT1 and WT PACS-2 in response to serum starvation or by mutation of the Ser437 Akt site (PACS-2S437A, Fig. S7A). Consistent with these findings, PACS-2, but not PACS-2S437A, repressed SIRT1-dependent PGC-1α/PPARα reporter gene expression to the same extent as EX-527 (Fig. 7B). PACS-2 nuclear trafficking was required for this transcriptional repression, since the PACS-2 nuclear localization mutant, PACS-2 NLS, was unable to repress reporter activity (Figs. 7B and S7B). Confocal microscopy demonstrated that nucleocytoplasmic shuttling of PACS-2 was dependent upon its NLS but not the phosphorylation state of Ser437 (Fig. S7B).
We also investigated whether Akt phosphorylation of PACS-2 is important for the interaction between PACS-2 and SIRT1 and, in turn, for the insulin-dependent repression of PPARα target gene expression. Co-immunoprecipitation studies in transfected cells showed that insulin stimulated both the phosphorylation of PACS-2 Ser437 as well as the interaction between PACS-2 and SIRT1 (Figs. 7C and S7C). Pre-treatment of the cells with either the mTOR inhibitor Torin1 or the Akt inhibitor MK-2206 reduced the insulin-dependent phosphorylation of PACS-2 Ser437 and, consequently, the PACS-2/SIRT1 interaction. The importance of PACS-2 for the insulin-dependent repression of endogenous PGC-1α/PPARα target genes was tested in a model of the fasted-to-fed transition. Primary hepatocytes from WT and PACS-2−/−mice were starved and exposed to WY-14643 (fasted state), then treated or not with insulin to inhibit PPARα transcriptional activity (fed state). RT-qPCR analyses revealed that insulin treatment repressed the expression of several PCG-1α/PPARα-induced FAO genes in hepatocytes isolated from WT mice but not PACS-2−/−mice (Fig. 7D). Western blot analysis showed that insulin induced pSer473-Akt in both WT and PACS-2−/−hepatocytes, suggesting the failure of the PACS-2−/−cells to inhibit FAO gene expression resulted directly from a loss of PACS-2-dependent inhibition of PCG-1α/PPARα and not from an upstream defect in insulin signaling (Fig. S7D).
Since insulin treatment was correlated with the interaction between PACS-2 and SIRT1 we tested whether SRT1720 could prevent this interaction in primary hepatocytes. Pre-treatment with SRT1720, followed by stimulation with insulin for 30 min, showed that insulin enhanced the interaction between endogenous SIRT1 and PACS-2 and that this interaction was inhibited by SRT1720 (Fig. 7E). SRT1720 did not affect the interaction between SIRT1 and DBC1. Similar results were found using the AML12 hepatocyte cell line (Fig. S7E). Together, these results suggest that PACS-2 and DBC1 form an insulin-responsive regulatory hub, which represses SIRT1 activity in vivo (see Fig. 7F).
Discussion
Here we used a holistic approach to show that insulin acutely regulates SIRT1 activity by triggering recruitment of a PACS-2/DBC1 to the SIRT1 NTR, creating a regulatory hub. In active SIRT1, the SIRT1 NTR distal N-terminus shields the proximal AC/3HB region in the NTR from cellular interactors. DBC1 removes this intra-molecular shield, permitting access to PACS-2, which engages the AC/3HB and thus causes reduced enzyme activity (Figs. 3, 4, 7F and see Video S1). STAC binding to the 3HB stabilizes this domain structure, thereby interfering with the ability of PACS-2 to engage helix 3 of the 3HB (Figs. 5, 7E and 7F, and see Video S2).
A current model for the allosteric regulation of SIRT1 proposes that the 3HB in the NTR is a rigid structure that reversibly interacts with the catalytic domain using a ‘bend-at-the-elbow’ mechanism, such that engagement between the two domains increases enzyme activity by lowering the Km for the acetylated substrate (Dai et al., 2015; Hubbard et al., 2013b). Based on this model, it was suggested that STAC binding to the NTR stimulates SIRT1 activity by stabilizing the interaction between the 3HB and the catalytic domain (Dai et al., 2015).
In stark contrast to the above supposition, our results suggest that such a simple, binary model underestimates the intricate, dynamic and complex roles of the different motifs in the large, disordered regions of the SIRT NTR. We propose that the 3HB acts as a molecular rheostat to allosterically fine-tune SIRT1 enzymatic efficiency for different subsets of substrates by individually adjusting binding affinities, influenced by different disordered regions in the NTR. Such an image helps to explain how SIRT1 can distinguish between the > 1,500 substrates predicted for this master regulator (Zhai et al., 2017). Other factors, such as the CK2-phosphorylatable acidic cluster, which attracts PACS-2, may additionally modulate the effects mediated by the 3HB. For example, when engaged with the acidic cluster, PACS-2 is able to take advantage of the natural folding/unfolding equilibrium of the nearby 3HB to latch on to the transiently exposed helix 3, destabilizing a key module whose structure is necessary for SIRT1 activity. Thus, inhibition of SIRT1 enzymatic activity is intricately dependent on the relative stability or foldedness of protein interaction motifs in the large NTR of SIRT1. In addition, a segment near the SIRT1 N-terminal CK2-phosphorylatable acidic cluster, S164SSDWT, also influences the interaction with PACS-2 (Fig. S1A). CK2 phosphorylation of Ser164 in obese mice causes SIRT1 to redistribute to the cytoplasm by an unknown mechanism (Choi et al., 2017). Whether PACS-2 participates in this CK2-dependent relocalization of SIRT1 requires further studies beyond the scope of the present work.
Our findings suggest that DBC1 functions as an allosteric assistor, rather than a direct inhibitor of SIRT1 activity. The DBC1 S1-L domain is evolutionally related to the bacterial ribosomal protein S1 RNA binding domain (Anantharaman and Aravind, 2008). Thus, our elucidation of the critical role of the DBC1 S1-L domain in this assistor function has further uncovered a previously unrecognized protein-binding role for this domain in the interplay between PACS-2 and SIRT1. Interestingly, in contrast to PACS-2, DBC1 appears to interact with SIRT1 under both fasting and fed conditions (Figs. 7 and S7). This finding suggests that insulin/PACS-2 may change the interaction of DBC1 with SIRT1 from a passive mode to an active assistor that exposes the AC/3HB for binding by PACS-2. Importantly, the role of DBC1 and its S1-L domain may not be limited to only granting access to PACS-2, since other activators or inhibitors of SIRT1 also are capable to interact with the AC/3HB region (Brooks and Gu, 2009).
The mechanism by which resveratrol and STACs improve metabolic fitness in vivo has been highly controversial (Baur et al., 2012). While resveratrol can activate SIRT1 in vitro, its anti-aging properties appear to be mediated through interactions with unrelated proteins in vivo (Howitz et al., 2003; Park et al., 2012). SRT1720 is effective in vivo only at very high concentrations (≥100 mg/kg) and, like other STACs, was developed without knowledge of the dynamic nature of the critically important 3HB or how it interacts with cellular regulators to modulate SIRT1 activity in vivo. Our data show that SRT1720, like STAC1, binds helix 3 of the folded 3HB in the SIRT1 NTR (Fig. 5 and (Dai et al., 2015)), exhibiting the most pronounced effect on residues comprising the hydrophobic core. This suggests that SRT1720 binding stabilizes the 3HB structure (Fig. S7). This, in turn, will render it more difficult for PACS-2 to remove helix 3 from the 3HB in vitro and in vivo. Our finding that SRT1720 prevents the insulin-stimulated interaction between PACS-2 and SIRT1 in hepatocytes supports this model (Figs. 7 and S7).
Using PACS-2−/−mice in the HFD study does not permit us to determine whether loss of PACS-2 specifically in the liver was responsible for protecting mice from diet-induced obesity and insulin resistance (Figs. 6 and S6). However, our results are in good agreement with those reported using shRNA knockdown of liver PACS-2 (Arruda et al., 2014). Moreover, the increased induction of FGF21 in both liver from fasted PACS-2−/−mice as well as in isolated PACS-2−/−hepatocytes treated with WY-14643, which could be blocked by pre-incubation with EX-527, strongly supports an important role for PACS-2 in regulating SIRT1-dependent liver gene expression (Fig. 6). These findings, together with our report that PACS-2 also inhibits SIRT1 in radiosensitive tissues to protect p53 from acetylation and, in turn, p21 induction following DNA damage (Atkins et al., 2014), suggests a broad and important role for PACS-2 as an in vivo SIRT1 regulator.
The insulin-stimulated, mTOR/Akt-dependent interaction of pSer437-PACS-2 with SIRT1, together with the failure of insulin to repress expression of FAO genes in PACS-2−/−hepatocytes, suggests that PACS-2 acts downstream of insulin/mTOR/Akt to support the rapid changes in liver gene expression following the fasting-to-fed transition (Figs. 7 and S7). Both mTORC1 and mTORC2 switch hepatocyte metabolism from catabolic to anabolic pathways. While mTORC1 traffics to lysosomes where it inhibits PGC-1α /PPARα using multiple mechanisms (Jensen-Urstad et al., 2013; Kim et al., 2012; Sengupta et al., 2010), mTORC2 regulates PPARα transcriptional activity by an unknown mechanism (Hagiwara et al., 2012). Our present findings suggest that PACS-2 may hold the key: In response to insulin, MAM-localized mTORC2 activates Akt through phosphorylation of Ser473, which then phosphorylates PACS-2 at Ser437 ((Aslan et al., 2009; Betz et al., 2013; Sarbassov et al., 2005) and Figs. 7 and S7). Akt-phosphorylated PACS-2 increases ER-mitochondria contacts, reducing mitochondrial respiration and increasing steatosis in mice fed a HFD (Arruda et al., 2014; Betz et al., 2013). In addition, Akt-phosphorylated PACS-2 also traffics to the nucleus and binds SIRT1 to repress PGC-1α/PPARα activity. While Akt phosphorylation at Ser437 promotes the interaction of PACS-2 with SIRT1, it does not appear to influence PACS-2 nucleocytoplasmic shuttling as reported for Foxo proteins (Brunet et al., 1999). Thus, it is possible that spatial tuning of the cargo-binding FBR by the downstream MR regulatory region, which contains the Akt site, modulates the binding to SIRT1, as has been reported for PACS-1 (Scott et al., 2003). Nonetheless, the overall collective findings suggest that an insulin-stimulated mTORC2/Akt/PACS-2 signaling axis coordinates cytoplasmic MAM communication with SIRT1-dependent nuclear gene expression. Moreover, in this scenario, STACs will block the insulin-dependent interaction of PACS-2 with SIRT1 (Fig. 7E), implying a compelling mechanism by which STACs may increase expression of genes controlling fatty acid oxidation and ketogenesis pathways that limit diet-induced obesity (Li et al., 2014; Purushotham et al., 2009; Rodgers et al., 2005; Vega et al., 2000; Vernia et al., 2014).
In summary, our combined structural, biochemical and in vivo studies on the regulation of SIRT1 uncovered a concerted, insulin-responsive regulatory hub, through which PACS-2 and DBC1 modulate SIRT1 enzymatic activity. DBC1 binding disrupts an intra-molecular interaction between the proximal and distal segments of the disordered SIRT1 NTR, which exposes two distinct interaction motifs, the CK2-phosphorylatable acidic cluster and helix 3 of the 3HB, for PACS-2 engagement. Overall, our study uncovered the critical and distinct roles of disordered and ordered domains in the large NTR of SIRT1 regulating enzyme activity, providing structural and functional insight that will aid rational design of a new generation of STACs with greater target specificity and efficacy.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding authors; Drs. Gary Thomas (thomasg@pitt.edu) and Angela M. Gronenborn (amg100@pitt.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experimental Animals
The Oregon Health & Science University Division of Animal Care and the University of Pittsburgh Division of Laboratory Animal Resources approved all animal studies. Male C57BL/6 WT and PACS-2−/−mice (Aslan et al., 2009) were fed a chow diet (Nestle Purina, LabDiet 5001; 58% carbohydrate, 29% protein and 13% fat (3.36 kcal/g)) and then switched to a high fat diet (Research Diets, D12492; 20% carbohydrate, 20% protein and 60% fat (5.24 kcal/g)) for 8 weeks. For fasting studies, WT and PACS-2−/−mice were fasted or not overnight for 14 hr and liver RNA was prepared as described in qPCR.
Metabolic studies
Fat and lean body mass were measured using an EchoMRI 4-in-1 system (EchoMRI, Houston, TX). Data were collected when the mice were on the low fat diet and at regular intervals when the mice were on the high fat diet. Livers were removed and weighed. Hepatic fat content was measured using the Echo MRI 4-in-1 system. Livers were divided into segments and flash frozen on dry ice or stored in RNAlater (Thermo Fisher) for RNA analysis. To measure liver triglycerides and cholesterol content, 50 mg of frozen livers were homogenized in PBS, lipids were extracted with 5% TX-100, and solubilized triglycerides and cholesterol were detected using triglyceride liquicolor and cholesterol liquicolor kits as per the manufacturer’s instructions (Stanbio). Insulin tolerance tests were performed on mice first when fed the chow diet and then after 8 weeks on the high fat diet. Individually housed mice were fasted overnight (14 h), weighed and transferred to a quiet room. At Time 0, the mice were gently restrained and a drop of blood was obtained from the tail vein. Immediately afterwards, the mice received an i.p. injection of human insulin (Humalog, Eli Lilly; 0.75 U/kg body weight). Tail vein blood was sampled again 15, 30, 60, 90 and 120 min after the insulin injection. Blood glucose was measured with a One Touch Ultra Link Glucose meter (Life Scan, Inc.).
Hepatocyte isolation
Primary mouse hepatocytes were isolated from 10-to 12-week-old male C57BL/6 WT and PACS-2−/−mice by two-step perfusion with calcium and magnesium-free Hanks’ salt solution followed by a Leibovitz’s L-15 medium (Thermo Fisher, #11415064) containing Liberas™ (Roche, #5401127001). Hepatocytes were plated at 15×105 cells per 60 mm dish or 2.5×105 cells per well of a 6-well plate in William E (Thermo Fisher, #32551) containing 10% FBS, dexamethasone and insulin, and allowed to adhere for 4h on collagen-coated dish (Thermo Fisher, CB40236). After adhesion, cells were incubated in William E containing 0.2% FBS for 4h prior to the indicated treatments.
qPCR and bioinformatics
RNA was isolated from liver using TRIzol (Life Technologies, #15596–026) and from primary hepatocytes using RNeasy (Qiagen, #74104). RNA was reverse transcribed using the SuperScript III First-Strand cDNA Synthesis Kit (Invitrogen, #18080–051). qPCR reactions were performed in a StepOne Real-Time PCR System with the Power SYBR Green PCR Master Mix (Applied Biosystems, #4368706) using the primer pairs listed in Table S2. All reactions were performed in triplicate. A qPCR heatmap was generated using log 2 transformed data and gplots R package (https://www.R-project.org). Transcription factor analysis was performed using the online tool (http://www.tfacts.org/) and default settings with FDR correction using Benjamini-Hochberg at 0.05.
Tissue harvest, nuclear fractionation and immunoprecipitation
Mouse livers were homogenized in RIPA (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP-40, 1% deoxycholate, and 0.1% SDS) containing protease/phosphatase inhibitors and 1mM EDTA, 1mM dithiothreitol (DTT), and 5mM Trichostatin A (TSA) using a motorized Teflon glass homogenizer. Protein concentration was determined using the Bradford method according to manufacturer’s instructions (Bio-Rad, #500–0006). Nuclear fractions were extracted from mouse liver as described (Nagata et al., 2010). To measure SIRT1 activity, the final nuclear pellet was resuspended in NET-N buffer (20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 0.5% NP-40, 1 mM EDTA) containing protease inhibitors (0.5 mM PMSF, 0.1 µM aprotinin, E-64 and leupeptin) and phosphatase inhibitors (1 mM Na3VO4 and 20 mM NaF). For western blotting, the final nuclear pellet was resuspended in mRIPA (50 mM Tris [pH 8.0], 150 mM NaCl, 1% NP-40, 1% deoxycholate) containing protease/phosphatase inhibitors. For most immunoprecipitation experiments, cells were harvested in mRIPA containing proteinase/phosphatase inhibitors. Western blots were developed using the Pierce ECL Western Blotting Substrate (Thermo Fisher) using a FluorChem E image acquisition system (ProteinSimple) and signals were quantified using the AlphaView image analysis software package (ProteinSimple).
Cell lines, antibodies, chemicals, siRNAs and plasmids
Cell lines; HCT116 cells and U2OS cells were cultured in DMEM containing 10% fetal bovine serum (FBS) as described (Atkins et al., 2014). AML12 cells (Dr. Nejak-Bowen, Pittsburgh) were cultured in DMEM/F12 media supplemented with 10% FBS, 40 ng/ml dexamethasone and ITS (insulin, transferrin and selenium, Thermo Fisher). Antibodies; PACS-2 (#193, (Simmen et al., 2005)), phospho-Akt Substrate (RXRXXT-P, pSer437-PACS-2) (Cell Signaling Technology, #81E12B5, (Aslan et al., 2009)), actin (Millipore, MAB1501), SIRT1 (Cell Signaling Technology, #2028; Santa Cruz, sc-15404; Abcam, ab12193), DBC1 (Cell Signaling Technology, #5693), Akt (Cell Signaling Technology, #9272), pThr308-Akt (Cell Signaling Technology, #9275), pSer473-Akt (Cell Signaling Technology, #9271), PGC-1α (Santa Cruz, sc-13067), Ac-Lys (Cell Signaling Technology, #9441), Topo II beta (BD Biosciences, #611492), V5 (Invitrogen, R960–25), Flag (Sigma-Aldrich, F7425 and A2220), HA (Covance, MMS-101R), Myc (Millipore, #06–340). Chemicals; EX-527 (Tocris, #2780), SRT1720 (Selleck, S1129), MK2206 (Cayman, 11593), Torin 1 (Cayman, 10997), WY-14643 (Sigma-Aldrich, C7081), leptomycin B (Calbiochem, #431050). siRNA; Human PACS-2 siRNAs (Dharmacon Smartpool, #M-022015 or Qiagen, #SI04193112 and SI04314730) were co-nucleofected (Amaxa) into cells as described (Atkins et al., 2008). Plasmids; pPPARα, p3xPPRE-Luc and pRXR (Dr. X. Li, NIEHS), pPGC-1α (Addgene 45501, Dr. B. Spiegelman, Yale), pV5-SIRT1 (Atkins et al., 2014), pFlag-PACS-2 (Simmen et al., 2005), pFlag-pPACS-2S437A (Aslan et al., 2009), Flag-PACS-2 NLS (Atkins et al., 2014), mCherry-tagged PACS-2, PACS-2S437A and PACS-2 ΔNLS (Atkins et al., 2014), pMyc-DBC1 (Dr. J. Chung, NIH), pPnc1p (Dr. B. Hubbard, Alberta).
Luciferase assays
HCT116 cells were nucleofected (Amaxa) with the indicated siRNAs and after 48h were transfected with the indicated plasmids using Lipofectamine 2000 (Invitrogen). After 12 hr the cells were treated or not with 15 mM WY-14643 (PPARα agonist) or 20 µM EX-527 (SIRT1 inhibitor) for 24 hr. Samples were then processed using the Dual-Luciferase Reporter Assay System according to manufacturer’s instructions (Promega). Firefly and Renilla luciferase activities were measured using a Synergy 2 (BioTek) and the ratio between Firefly and Renilla signals was determined for each sample and normalized to the control.
Yeast Two-Hybrid assays
The L40 yeast strain [MATa his3, trp, ade2, LYS::(lexAop)4-HIS3 URA::(lexAop)8-lacZ GAL4] was co-transformed with prey (pVP16, Leu-selection) and bait (pBTM116, Trp-selection) plasmids and then selected for growth on Leu-/Trp-minimal plates in the presence of 1–5 mM 3-aminotriazole (3AT) as previously described (Liu et al., 1997). Quantitative β-gal assays were performed using dual transfected cells as previously described (Wan et al., 1998).
SIRT1 activity assay
SIRT1 activity was measured using the fluorometric nicotinamide assay (Hubbard et al., 2013a). Recombinant human SIRT1, hSIRT1, hSIRT1Δ6−83, SIRT1S162D, DBC1 S1-L and PACS-2 FBR were prepared as described in Cloning and Protein Purification. Peptides substrates Ac-Lys778-PGC-1α (Ac-SDPFDPASTKSK(Ac)YDSLDF-NH2) and H3K9 (Ac-TARK(Ac)STG-NH2) were from Peptide 2.0 (≥ 90% purity). Reactions were performed for one hr at 37 °C in 96-well plates using the amounts of recombinant SIRT1 constructs, DBC1 S1-L and PACS-2 FBR indicated in the figure legends together with 100 µM peptide substrate and 1 µM Pnc1p in the absence or presence of 200 µM NAD+ in PBS supplemented with 1 mM DTT. Reactions were developed with 100 µl pre-warmed OPT Developer mix [10 mM ortho-phthalaldehyde, 10 mM DTT in 30% EtOH/70% PBS (pH 7.4)] for 45 to 60 min. Fluorescent signals were detected using a Synergy 4 microplate reader (λex = 420 nm and (λem = 460 nm). For each sample, negative control reactions (no β-NAD) were performed and final measurements were calculated as follows: Fcorrected = F+NAD – F-NAD control.
Confocal microscopy
U2OS cells were transfected (FuGENE 6) with the indicated plasmids, and images were acquired on a high-resolution, wide-field Core DeltaVision System (Applied Precision). Nuclear and cytoplasmic fluorescent signal was determined using softWoRx Explorer 2.0 (Applied Precision) and the fraction of each PACS-2 construct in the nucleus was calculated as described (Atkins et al., 2014). Statistical significance was determined using a two-sided unpaired Student’s t test with unequal variance.
Cloning and Protein Purification
His6-tagged human SIRT1 and SIRT1Δ6-83 in pQE-80 were previously described Atkins, 2014 # 444}. His6-SIRT1S162D in pQE-80 was generated by site directed mutagenesis using standard methods. SIRT1183-233, SIRT1141-233, SIRT11-233, and DBC152-120 were cloned into a modified pET41a vector that includes a TEV cleavage site (Krzysiak et al., 2012). PACS222-180 was cloned into pET41 using the NdeI and XhoI sites which removed the GST tag. His6-SIRT1S162D in pQE-80 was generated by site directed mutagenesis using standard methods. The SIRT1140-177 peptide was fused to ubiquitin in pET15b. All DNA constructs were used to transform E. coli Rosetta*, which is the BL21 (DE3)* cell line (Thermo Fischer), containing the pRARE plasmid from Rosetta2 DE3 (Millipore). For isotopically labeled samples, cells were grown at 37°C in modified M9 medium, supplemented with 15N-ammonium chloride or 15N-ammonium chloride and 13C-glucose to an absorbance A600= 0.6, induced with 400 µM IPTG and grown overnight at 18°C for 16–18 hours. Cells were harvested by centrifugation (4600 x g; 10 min; 4°C). For GST tagged constructs, cells were resuspended in 20 mM HEPES pH 7.0, 500 mM NaCl, 5 mM DTT and lysed using a microfluidizer (Microfluidics). DNase (80 µg/ml) and RNase (64 µg/ml) were added to the lysate and the reaction was incubated at 4°C with stirring for 1 hr. The lysate was clarified by centrifugation (38,000xg/40 min/4°C) and applied to a GSTrap column (GE Life Sciences). Bound protein was eluted with 20 mM HEPES pH 8.0, 500 mM NaCl, 40 mM reduced glutathione. GST tagged protein was separated from any contaminating proteins by gel filtration over a Superdex 200 26/60 column (GE Life Sciences), equilibrated in 20 mM HEPES, pH 7.5, 150 mM NaCl, 0.5 mM TCEP. Digestion with TEV protease, followed by ion exchange using Q Sepharose (GE Life Sciences) was used to remove the fusion tag. Fusion-tag free protein was finally passed over a Superdex 75 16/60 column (GE Life Sciences) for buffer exchange and to remove the last traces of contaminants.
The initial preparation of PACS222-180 was performed similarly to the GST tagged constructs except that the initial cell resuspension buffer was 20mM HEPES, pH 7.5, 150 mM NaCl, 0.5 mM TCEP. PACS222-180 was purified away from crude cellular lysate by cation exchange chromatography using SP Sepharose (GE Life Sciences). The elution was then passed over a Superdex 75 16/60 column buffer exchange and to remove the last traces of contaminants
Crude cell lysate containing 6xhis-tagged constructs were prepared in the same manner as the GST-tagged constructs, except the cellular resuspension buffer was 20 mM HEPES pH 7.5, 500 mM NaCl, 20 mM imidazole, 0.5 mM TCEP. Cell lysate was applied to a HisTrap column (GE Life Sciences) and bound protein was eluted using a 30 mM-500 mM imidazole gradient of the resuspension buffer. His-tagged protein was then passed over a Superdex 200 26/60 gel filtration column equilibrated in 20 mM HEPES, pH 7.5, 150 mM NaCl, 0.5 mM TCEP. To achieve a high level of purity, the his-tagged proteins were subjected to anion exchange chromatography using Q Sepharose and buffer exchanged using a Superdex 200 16/60 column (GE Life Sciences).
For all proteins, the final gel filtration buffer was either the NMR buffer or identical to buffer used in the SIRT1 activity assay. Final protein purity was estimated at >99% by SDS-PAGE.
NMR Spectroscopy
All 2D 1H-15N HSQC spectra of SIRT1183-233, SIRT1141-233, SIRT11-233, PACS222-180, and DBC152-120 were recorded at 25°C using Bruker AVANCE 700 MHz, 800 MHz or 900 MHz NMR spectrometers equipped with z-axis gradient cryoprobes. Unless otherwise noted, spectra were collected in 20 mM HEPES pH 7.5, 100 mM NaCl, 0.5 mM TCEP. For SIRT1183-233 and SIRT1141-233/ PACS222-180 binding studies, 30 µM SIRT1 was mixed with 200 µM PACS2. For the SIRT1141-233 pH titration, spectra of 100 µM protein were recorded in 100 mM NaCl, 0.5 mM TCEP in either 20 mM HEPES pH 7.5, 20 mM HEPES pH 6.5, or 20 mM Na Citrate pH 5.5. DBC152-120 binding studies were performed by mixing 30 µM DBC1 with 70 µM SIRT1, SI RT1 6-83, or PACS222–180. Ubiquitin-SIRT1140-177 binding to PACS222-180 was monitored by mixing 100 µM PACS2 with increasing amounts of the SIRT1 peptide in 20 mM Na Acetate pH 5.0.
STAC binding experiments were performed using a 70 mM stock solution of SRT1720 (Thermo Fisher) in acidified DMSO (49.5 µl DMSO: 0.5 µl 37% HCl). For a 320 µl NMR sample, 0.46 µl of the stock solution was employed resulting in a final concentration of 100 µM. STAC binding to the 3HB was monitored by recording spectra of 100 µM SI RT1183-233 in the presence of 100 µM SRT1720. To analyze the effect of SRT1720 on the SIRT1/PACS2 interaction, spectra of 100 µM SIRT1 were recorded alone, in the presence of 100 µM SRT1 720, in the presence of 120 µM PACS2 in the presence of both 100 µM SRT1720 and 120 µM P ACS222-180, respectively.
Backbone chemical shift assignments for SIRT1 were obtained using standard 2D and 3D triple resonance experiments, including 3D HNCACB, HN(CO)CACB, HNCA, HN(CO)CA, HNCO and HN(CA)CO, recorded on a Bruker AVANCE 800MHz NMR spectrometer equipped with a z-axis gradient cryoprobe at 25°C. Non-uniform sampling wa s used during data collection using 20% of the data points and 32, 16, 16, 16, 8, and 32 scans respectively. Spectra were reconstructed using the SMILE algorithm (Ying et al., 2017), processed using NMRPipe (Delaglio et al., 1995) and analyzed using CARA (Keller, 2004) and NMRFAM-Sparky (Lee et al., 2015).
Supplementary Material
Highlights.
A critical insulin sensor is hidden in the disordered N-terminal region of SIRT1.
A shield covers the sensor, which, when removed by DBC1, permits access to PACS-2 to inhibit SIRT1.
STACs stabilize the 3-helix site in the sensor, to protect SIRT1 from PACS-2.
Insulin triggers liver PACS-2 to inhibit SIRT1-mediated catabolic gene expression.
Acknowledgements
This work was supported by NIH grants R01 DK114855 (AMG and GT), DK112844 (GT), DK070333 (DLM), R01 LM012087 and U01 HL137159 (PVB) and the University of Pittsburgh Aging Institute (TCK and SL). The authors thank T. Brosenitsch for critically reading and editing the manuscript and Drs. M. Jurczak (University of Pittsburgh), A. O’Donnell (University of Pittsburgh), K. Nejak-Bowen (University of Pittsburgh), P. Thibodeau (University of Pittsburgh), U. Shinde (OHSU), B. Hubbard (U. Alberta), X. Li (NIEHS), B. Spiegelman (Yale) and J. Chung (NIH) for reagents and helpful discussions. Mike Delk (University of Pittsburgh) is acknowledged for NMR technical support and S. Klingensmith (University of Pittsburgh) for producing the animations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declartion of Interests
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
1H, 15N, and 13C chemical shifts for the SIRT1 three-helix-bundle (3HB) will be deposited at BMRB, http://www.bmrb.wisc.edu, upon acceptance of the manuscript.
References
- Anantharaman V, and Aravind L (2008). Analysis of DBC1 and its homologs suggests a potential mechanism for regulation of sirtuin domain deacetylases by NAD metabolites. Cell Cycle 7, 1467–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda AP, Pers BM, Parlakgul G, Guney E, Inouye K, and Hotamisligil GS (2014). Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nature Med 20, 1427–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslan JE, You H, Williamson DM, Endig J, Youker RT, Thomas L, Shu H, Du Y, Milewski RL, Brush MH, et al. (2009). Akt and 14–3-3 control a PACS-2 homeostatic switch that integrates membrane traffic with TRAIL-induced apoptosis. Mol Cell 34, 497–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins KM, Thomas L, Youker RT, Harriff MJ, Pissani F, You H, and Thomas G (2008). HIV-1 Nef binds PACS-2 to assemble a multikinase cascade that triggers major histocompatibility complex class I (MHC-I) down-regulation: analysis using short interfering RNA and knock-out mice. J Biol Chem 283, 11772–11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins KM, Thomas LL, Barroso-Gonzalez J, Thomas L, Auclair S, Yin J, Kang H, Chung JH, Dikeakos JD, and Thomas G (2014). The multifunctional sorting protein PACS-2 regulates SIRT1-mediated deacetylation of p53 to modulate p21-dependent cell-cycle arrest. Cell Rep 8, 1545–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, and Maratos-Flier E (2007). Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab 5, 426–437. [DOI] [PubMed] [Google Scholar]
- Baur JA, Ungvari Z, Minor RK, Le Couteur DG, and de Cabo R (2012). Are sirtuins viable targets for improving healthspan and lifespan? Nat Rev Drug Discov 11, 443–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, and Hall MN (2013). Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci USA 110, 12526–12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CL, and Gu W (2009). How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer 9, 123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, and Greenberg ME (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868. [DOI] [PubMed] [Google Scholar]
- Choi SE, Kwon S, Seok S, Xiao Z, Lee KW, Kang Y, Li X, Shinoda K, Kajimura S, Kemper B, et al. (2017). Obesity-Linked Phosphorylation of SIRT1 by Casein Kinase 2 Inhibits Its Nuclear Localization and Promotes Fatty Liver. Mol Cell Biol 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Case AW, Riera TV, Considine T, Lee JE, Hamuro Y, Zhao H, Jiang Y, Sweitzer SM, Pietrak B, et al. (2015). Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nature Comm 6, 7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport AM, Huber FM, and Hoelz A (2014). Structural and functional analysis of human SIRT1. J Mol Biol 426, 526–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey NE, Cyert MS, and Moses AM (2015). Short linear motifs -ex nihilo evolution of protein regulation. Cell Comm Signal 13, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995). NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Essaghir A, Toffalini F, Knoops L, Kallin A, van Helden J, and Demoulin JB (2010). Transcription factor regulation can be accurately predicted from the presence of target gene signatures in microarray gene expression data. Nucleic Acids Res 38, e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, and Auwerx J (2008). Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab 8, 347–358. [DOI] [PubMed] [Google Scholar]
- Ghisays F, Brace CS, Yackly SM, Kwon HJ, Mills KF, Kashentseva E, Dmitriev IP, Curiel DT, Imai SI, and Ellenberger T (2015). The N-Terminal Domain of SIRT1 Is a Positive Regulator of Endogenous SIRT1-Dependent Deacetylation and Transcriptional Outputs. Cell Rep [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, Terracciano L, Heim MH, Ruegg MA, and Hall MN (2012). Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab 15, 725–738. [DOI] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, et al. (2003). Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425, 191–196. [DOI] [PubMed] [Google Scholar]
- Hubbard BP, Gomes AP, Dai H, Li J, Case AW, Considine T, Riera TV, Lee JE, E SY, Lamming DW, et al. (2013a). Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339, 1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard BP, Loh C, Gomes AP, Li J, Lu Q, Doyle TL, Disch JS, Armour SM, Ellis JL, Vlasuk GP, et al. (2013b). Carboxamide SIRT1 inhibitors block DBC1 binding via an acetylation-independent mechanism. Cell Cycle 12, 2233–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen-Urstad AP, Song H, Lodhi IJ, Funai K, Yin L, Coleman T, and Semenkovich CF (2013). Nutrient-dependent phosphorylation channels lipid synthesis to regulate PPARalpha. J Lipid Res 54, 1848–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Jung JW, Kim MK, and Chung JH (2009). CK2 is the regulator of SIRT1 substrate-binding affinity, deacetylase activity and cellular response to DNA-damage. PloS one 4, e6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Suh JY, Jung YS, Jung JW, Kim MK, and Chung JH (2011). Peptide switch is essential for Sirt1 deacetylase activity. Mol Cell 44, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R (2004) The Computer Aided Resonance Assignment Tutorial, CANTINA Verlag, Goldau, Switzerland. [Google Scholar]
- Kim JE, Chen J, and Lou Z (2008). DBC1 is a negative regulator of SIRT1. Nature 451, 583–586. [DOI] [PubMed] [Google Scholar]
- Kim K, Pyo S, and Um SH (2012). S6 kinase 2 deficiency enhances ketone body production and increases peroxisome proliferator-activated receptor alpha activity in the liver. Hepatology 55, 1727–1737. [DOI] [PubMed] [Google Scholar]
- Koshland DE (1958). Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc Natl Acad Sci USA 44, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzysiak TC, Jung J, Thompson J, Baker D, and Gronenborn AM (2012). APOBEC2 is a monomer in solution: implications for APOBEC3G models. Biochemistry 51, 2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshminarasimhan M, Curth U, Moniot S, Mosalaganti S, Raunser S, and Steegborn C (2013). Molecular architecture of the human protein deacetylase Sirt1 and its regulation by AROS and resveratrol. Biosci Rep 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Tonelli M, and Markley JL (2015). NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wong K, Giles A, Jiang J, Lee JW, Adams AC, Kharitonenkov A, Yang Q, Gao B, Guarente L, et al. (2014). Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology 146, 539–549 e537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Chen L, Kabra N, Wang C, Fang J, and Chen J (2009). Inhibition of SUV39H1 methyltransferase activity by DBC1. J Biol Chem 284, 10361–10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Thomas L, Warren RA, Enns CA, Cunningham CC, Hartwig JH, and Thomas G (1997). Cytoskeletal protein ABP-280 directs the intracellular trafficking of furin and modulates proprotein processing in the endocytic pathway. J Cell Biol 139, 1719–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, et al. (2007). Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata T, Redman RS, and Lakshman R (2010). Isolation of intact nuclei of high purity from mouse liver. Anal Biochem 398, 178–184. [DOI] [PubMed] [Google Scholar]
- Park SJ, Ahmad F, Philp A, Baar K, Williams T, Luo H, Ke H, Rehmann H, Taussig R, Brown AL, et al. (2012). Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 148, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, and Li X (2009). Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 9, 327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, and Puigserver P (2005). Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434, 113–118. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, and Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Scott GK, Gu F, Crump CM, Thomas L, Wan L, Xiang Y, and Thomas G (2003). The phosphorylation state of an autoregulatory domain controls PACS-1-directed protein traffic. EMBO J 22, 6234–6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Peterson TR, Laplante M, Oh S, and Sabatini DM (2010). mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 468, 1100–1104. [DOI] [PubMed] [Google Scholar]
- Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, and Thomas G (2005). PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J 24, 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Kenney RD, Gagne DJ, Frushour BP, Ladd W, Galonek HL, Israelian K, Song J, Razvadauskaite G, Lynch AV, et al. (2009). Small molecule activators of SIRT1 replicate signaling pathways triggered by calorie restriction in vivo. BMC Syst Biol 3, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G, Aslan JE, Thomas L, Shinde P, Shinde U, and Simmen T (2017). Caught in the act protein adaptation and the expanding roles of the PACS proteins in tissue homeostasis and disease. J Cell Sci 130, 1865–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P, Davey NE, Gibson TJ, and Babu MM (2014). A million peptide motifs for the molecular biologist. Mol Cell 55, 161–169. [DOI] [PubMed] [Google Scholar]
- van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, et al. (2014). Classification of intrinsically disordered regions and proteins. Chem Rev 114, 6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega RB, Huss JM, and Kelly DP (2000). The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol 20, 1868–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernia S, Cavanagh-Kyros J, Garcia-Haro L, Sabio G, Barrett T, Jung DY, Kim JK, Xu J, Shulha HP, Garber M, et al. (2014). The PPARalpha-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab 20, 512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Molloy SS, Thomas L, Liu G, Xiang Y, Rybak SL, and Thomas G (1998). PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell 94, 205–216. [DOI] [PubMed] [Google Scholar]
- Ying J, Delaglio F, Torchia DA, and Bax A (2017). Sparse multidimensional iterative lineshape-enhanced (SMILE) reconstruction of both non-uniformly sampled and conventional NMR data. J Biomol NMR 68, 101–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Z, Tang M, Yang Y, Lu M, Zhu WG, and Li T (2017). Identifying Human SIRT1 Substrates by Integrating Heterogeneous Information from Various Sources. Sci Rep 7, 4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, and Gu W (2008). Negative regulation of the deacetylase SIRT1 by DBC1. Nature 451, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.