

Abstract
Despite significant improvements in the overall survival of patients with multiple myeloma (MM) over the past 15 years, the disease remains incurable. Treatment options are limited for patients who have relapsed or are refractory to immunomodulatory drugs (IMiDs), proteasome inhibitors, and monoclonal antibodies. In these patients, immunotherapies such as checkpoint inhibitors, oncolytic vaccines, and chimeric antigen receptor (CAR) T cells provide a potentially effective alternative treatment. While checkpoint inhibitors are effective in prolonging overall survival in some patients with advanced solid cancers and Hodgkin lymphoma, they have not demonstrated significant anti-myeloma activities as a single agent in MM. In fact the combination of checkpoint inhibitors with IMiDs was recently found to increase the risk of death in myeloma patients. These challenges highlight the need for a better understanding of immune dysregulation in myeloma patients, and the mechanisms of action of- and resistance to- checkpoint inhibitors. In this review, we summarize immune dysfunction in patients with MM, and review the preclinical and clinical data regarding checkpoint inhibitors in myeloma. We conclude by proposing strategies to improve the efficacy and safety of checkpoint inhibitors in this population.
Keywords: multiple myeloma, immunotherapy, checkpoint inhibitors, CTLA4, PD-1/PD-L1
I. Multiple myeloma
Multiple Myeloma (MM) is a malignancy that is characterized by the clonal proliferation of terminally differentiated plasma cells within the bone marrow. MM represents 1% of all malignancies and 18% of hematologic malignancies in the United States accounting for an estimated 30,770 new diagnoses and 12,770 deaths in 2018 alone1. Classically, MM results in the secretion of a non-functional monoclonal immunoglobulin (Ig) that is produced by the transformed plasma cells. Production of this aberrant Ig results in several of the complications associated with MM such as renal dysfunction, neuropathy, and hyperviscosity syndrome2,3. However, in approximately 15–20% of patients the abnormal plasma cells secrete only monoclonal free light chains, and in approximately 2–3% of cases these cells secrete no monoclonal protein at all, resulting in the so-called non-secretory myeloma4,5. Myeloma cell growth in the bone marrow and the resultant cytokines produced by these transformed cells lead to the classic symptoms of active MM: osteolytic bone lesions, hypercalcemia, and anemia6.
For decades, low dose melphalan and prednisone constituted the cornerstone of MM treatment. However, complete responses with this regimen are rare, and the median time to progression is less than 15 months7,8. A significant advance in the management of MM came from the upfront use of high doses of melphalan with autologous hematopoietic stem cell transplantation (AHSCT). This treatment has allowed for improved response rates, progression free survival, and—in some trials—prolonged survival in MM patients9–12. High dose melphalan with AHSCT continues to be a fundamental therapeutic modality in younger patients after response to conventional induction therapy and/or at the time of disease progression.
The subsequent advent of new “biologic” agents in MM treatment regimens has led to marked improvement in the depth and duration of the responses obtained. The immunomodulatory drugs (i.e., thalidomide, lenalidomide, and pomalidomide), along with proteasome inhibitors (i.e., bortezomib, carfilzomib, and ixazomib) have shown significant efficacy in the management of both newly diagnosed as well as relapsed and refractory MM patients13–16. Monoclonal antibodies such as daratumumab and elotuzumab were approved by the FDA in 2015 and provide additional options for the treatment of MM17–22. Treatment of MM patients with combination regimens that contain one or more biologic agents, followed by consolidation with AHSCT has resulted in the highest response rates yet in newly diagnosed MM patients23–28.
Despite the use of the “biologic” agents and the incoporation of AHSCT, MM remains an incurable disease, with some patients relapsing within months after autologous hematopoietic stem cell transplantation. Nearly all MM patients will eventually develop resistance to currently available agents; as such there is a significant unmet medical need for the development of novel therapeutic agents against myeloma. Immunotherapy—i.e., agents which stimulate a patient’s own immune system to attack cancer cells—has recently moved to the forefront of cancer treatment, and has yielded unprecedented tumor responses and long-term survival benefits in patients with advanced solid malignancies and Hodgkin lymphoma29–32. Immunotherapy utilizes one of four major strategies: targeting tumor surface antigens with a monoclonal antibody; adoptive transfer of immune effector cells including chimeric antigen receptor T cells; boosting tumor-specific immunity using vaccines; and inhibiting tumor specific immune suppression with checkpoint inhibitors33. This review focuses on the role of checkpoint inhibitors that target the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and the programmed death 1 (PD-1)/programmed death ligand 1 (PD-L1) in the treatment of MM.
II. Overview of the human immune system
The human immune system consists of two main components: the innate immune system, and the adaptive immune system. Innate immunity serves as the initial defense mechanism against microbial invasion and is characterized by a rapid, nonspecific response to pathogens. The components of the innate immune system include natural anatomic barriers (skin and mucosa), soluble proteins, bioactive small molecules (cytokines and complement components), and several types of myeloid derived leukocytes which typically express cell-surface toll-like receptors (TLRs). All elements of the innate immune system are encoded by the host’s germline DNA. They are preformed and able to react to the first sign of pathogenic invasion.
Unlike the innate immune system, the adaptive immune response is encoded by genetic elements that undergo somatic rearrangement to form antigen-binding molecules with a unique specificity for individual antigens. The adaptive immune system is mainly derived from lymphoid progenitor cells and includes both cellular and humoral immunities. The effector cells of the adaptive immune system are B cells and T cells. The T-lymphocyte population can be further subdivided into CD4+ (T helper cells), CD8+ (cytotoxic T cells) and regulatory T cells (Tregs). Upon exposure to antigens, the T and/or B effector cells undergo clonal expansion to mount an effective response. This adaptive response produces long-lived memory T and/or B cells. These memory cells will persist in a dormant state, but can undergo rapid expansion if they ever encounter the antigen again; resulting in a more rapid, robust immune responses upon subsequent exposures to a specific antigen. B cells mediate the humoral immune response by producing antibodies against specific antigens either alone or with the help of CD4+ T cells. Cellular immunity is mainly mediated by CD8+ T cells, which eliminate pathogens and tumor cells via apoptosis or cellular lysis through the release of perforins and granzymes.
Classically, full CD4+ and CD8+ T-cell activation requires two signals. Signal 1 is provided by the interaction between the T-cell receptor (TCR) and the peptide-MHC complex on the antigen-presenting cells (APCs). Signal 2 is a costimulatory signal mediated by the interaction between the costimulatory molecule CD28 on the T cell and CD80 (B7.1) or CD86 (B7.2) on the APC34,35. Both signal 1 and signal 2 are required for full T-cell activation. In the absence of the costimulatory signal, the interaction between the TCR and the peptide-MHC complex results in T-cell anergy36–38. Cytotoxic T lymphocyte antigen 4 (CTLA4) was subsequently identified as a coinhibitory molecule that can induce the downregulation of CD28 by endocytosis and compete with CD28 for binding with CD80/CD86. CTLA4 can thus dampen the T cell response and prevent aberrant and autoreactive immune responses39–42.
Many other cell surface signaling molecules have been identified over the last 20 years (for review, see ref43) and the two-signal model has evolved into an increasingly complex regulatory system, in which both co-stimulatory and co-inhibitory molecules have been discovered. Most of these co-signaling molecules belong to either the immunoglobulin superfamily (IgSF) or the tumor necrosis factor receptor superfamily (TNFRSF). Among the IgSF receptors, the CD28 family (CD28, ICOS, CTLA4, PD1, PD1H, and BTLA) and B7 family (B71, B7H1/PDL1) are the most well-described members. Additional IgSF cosignaling receptors—type I transmembrane immunoglobulin and mucin (TIM) domaincontaining molecules, CD2/signaling lymphocytic activation molecule (SLAM) family members, Butyrophilin (BTN) and BTN-like (BTNL) family molecules, Lymphocyte activation gene 3 protein (LAG3), and CD226 family members (CD226, CRTAM, TIGIT, and CD96)—have also been reported44–47. The TNFRSF receptors contain one or more extracellular cysteine-rich domain and are divided into 4 major subfamilies: Type-V (4–1BB, OX40, CD27, GITR, CD30); Type-L (TNFR1, TNFG2, HVEM, TNFRSF3, TNFRSF25, TNFRSF6B, FAS, CD40, RANK, OPG, TRAILR1–4); Type-S (TACI, BAFFR, BCMA, TWEAKR, EDAR); and orphan.
Co-signaling molecules positively and negatively regulate T cell fate and function, and thus maintain the equilibrium between immune activation and tolerance. The level of expression of co-signaling molecules on T cells is highly variable and context dependent. In contrast to the “on/off” models of co-signaling, it was recently proposed that the repertoire of costimulatory and co-inhibitory receptors is varied continuously in response to dynamic environmental conditions: the so called “tidal model” of co-signaling43,48. This model postulates that at the onset of T-cell activation there is an abundance of co-stimulatory receptors on naïve and activated T cells, resulting in the functional responsiveness of these cells (the so-called “rising tide”). This is followed by expression of both co-stimulatory and co-inhibitory molecules; T cell functionality is ultimately directed by the relative strength of these two opposing actions (the so-called “peak tide”). Finally, the relative expression of co-inhibitory receptors increases leading to suppression of further T cell activation (the so-called “receding tide”).
CTLA4/CD28 and the programmed cell death protein 1 (PD-1)/programmed cell death ligand 1 (PD-L1) pathways are the most well characterized negative regulators of immune activation. Similar to other co-signaling molecules, CTLA4/CD28 and PD-1/PD-L1 are subject to spatiotemporal regulation and undergo bi-directional co-signaling. TCR signaling and co-signaling molecules form the immune synapse, which is composed of the central, peripheral, and distal supra-molecular activation complexes (SMAC) with functionally diverse co-signaling molecules. CD28, CD4/CD8 and the TCR form the central SMAC (cSMAC), which initiates the recruitment of signaling molecules and T cell activation49. Following activation, the co-inhibitory molecules CTLA4 and PD-1 are recruited to the immune synapse. CTLA4 and possibly PD-1 accumulate in the same region of the cSMAC as CD28 to physically exclude CD28 from the cSMAC50,51. CTLA4 induces the downregulation of CD28 by endocytosis39–42. CTLA4 and PD-1 recruit SH2 domain-containing tyrosine phosphatase 1 (SHP1), SHP2, and serine/threonine protein phosphatase 2A (PP2A), which suppress T cell activation by dephosphorylating several of the major signaling pathways that are essential for T cell activity51,52.
The interactions between co-signaling molecules and their counter-receptors (ligands) are bi-directional: the interacting molecules on each respective cell function to transduce a signal into that particular cell43. When CTLA4 on T cells binds to B7–1 and B7–2, it transmits signals into the T cell that inhibit T cell function. On the other hand, CTLA4-bound to B7–1 and B7–2 induces the expression of indoleamine 2,3-dioxygenase (IDO), which can promote the development of Treg cells53. Similarly, when PD1 on T cells binds to PD-L1 (B7-H1), it transduces inhibitory signals into T cells leading to suppressed T cell function. Addtionally, PD-L1 expressed on cancer cells has been shown to transmit an anti-apoptotic signal from bound PD154,55.
III. Immune dysfunction in multiple myeloma
As shown in Figure 1, patients with multiple myeloma show profound defects/dysfunction in both innate and adaptive immunity56. The human innate immune system includes natural killer (NK) cells, macrophages, monocytes, neutrophils, eosinophils, and basophils. NK cells recognize and kill myeloma cells by activating receptors including NKG2D, DNAX accessory molecule (DNAM-1 or CD226) and the natural cytotoxicity receptors (NCRs) Np46, Np30, Np4457,58. NK cells play an important role in tumor immune-surveillance. However, in patients with active myeloma, the expression of these activating receptors on NK cells are reduced, leading to impaired NK cell activity59,60. Additionally, PD-1 expression is upregulated on NK cells; this allows PD-L1 expressing MM cells to inhibit the cytotoxicity of NK cells through the PD-L1/PD-1 pathway61. Macrophages are increased in the bone marrow microenvironment of patients with myeloma and support the survival and proliferation of multiple myeloma cells through both contact dependent and contact independent activation of the pro-proliferative molecule STAT362,63. Neutrophils in patients with MM demonstrate reduced phagocytic activity and up-regulation of Arg-1 expression leading to an immunosuppressive effect on T lymphocytes64. Eosinophils have been shown to promote human and mouse myeloma cell growth65,66, and may in fact play a role in the biology and pathology of MM.
Figure 1:
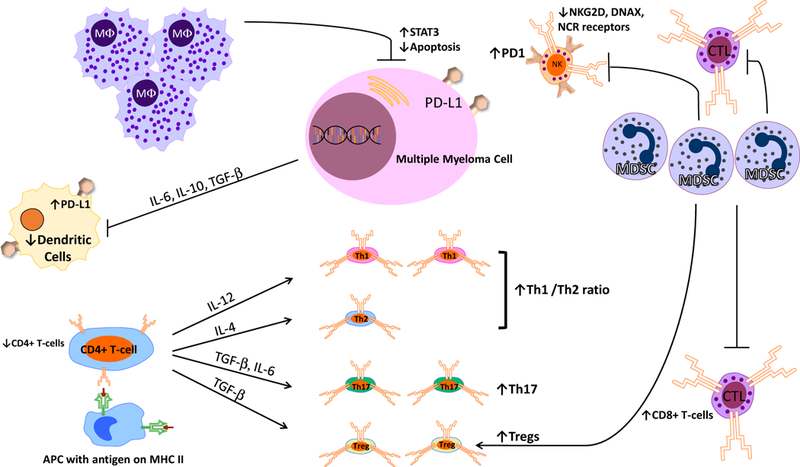
Immune Dysfunction in Multiple Myeloma (see text for details)
B and T lymphocytes mediate adaptive immunity. In MM patients, B-cell dysfunction is characterized by hypogammaglobulinemia, leading to an increased risk for infections. T-cell mediated immunity is also severely impaired in MM. MM patients demonstrate a decrease in the ratio of CD4(+)/CD8(+) T-cells in the peripheral blood: this is due to a decrease in both the absolute and relative numbers of CD4(+) T-cells, and an increase in relative numbers of CD8(+) T-cells67,68. Similarly, an increased Th1/Th2 ratio has been observed in MM patients and is associated with immune dysfunction69,70. Th17 cells—a pro-inflammatory subset of T-cells—are increased in both the peripheral blood and bone marrow of MM patients71,72. Th17 cells are particularly enriched in the bone marrow and produce significant levels of interleukin (IL)-17, which is implicated in the development of MM lytic bone lesions72. The number of immunosuppressive cells—including regulatory T and B cells and myeloid derived suppressive cells—are also significantly increased in MM patients56,73–75. T-cell mediated cytotoxicity depends upon the antigen presenting function of dendritic cells (DCs). The function of DCs is inhibited in MM patients due to the secretion of cytokines such as IL-6, IL-10 and TGF-β by myeloma cells76,77. Additionally, numbers of plasmacytoid and myeloid DCs are decreased, and levels of PD-L1 expression on plasmacytoid DCs are increased, resulting in T cell inhibition. Myeloid derived suppressor cells (MDSC) are a heterogeneous population of immature myeloid cells that are characterized by the ability to suppress anti-tumor immune responses mediated by T cells and NK cells78. There is a substantial increase in MDSCs in the peripheral blood and bone marrow of MM patients, which has been shown to play a role in disease progression and drug resistance79–81. MDSCs also promote tumor angiogenesis and growth by the secretion of cytokines and growth factors81. Notably, the percentage of PD-L1+ bone marrow MDSCs in myeloma patients in remission is unchanged from PD-L1 levels at diagnosis or at relapse; this suggests that, even with successful treatment, the bone marrow of MM patients continues to be immunosuppressed82. NKT cells express both T cell receptors and NK cell surface antigens and recognize glycolipids through CD1d. In MM patients, NKT cell’s ability to produce IFN-γ is defective leading decreased innate and adaptive immune activation83. Another T-cell subset, the γδ T cells, proliferate in the presence of IL-2 and exhibit anti-myeloma activity; their activity is also impaired in myeloma patients.
Immune dysfunction in myeloma patients is caused by several factors. The growth of the tumor within the bone marrow leads to a collapse of normal hematopoiesis. Myeloma cells produce several factors—transforming growth factor-β (TGF-β), IL-10, IL-6, and prostaglandin E2—which have a negative impact on the function of various immune cells84. For example, TGF-β induces phosphorylation of SMAD3 and suppresses IFN-γ production and antibody-dependent cellular cytotoxicity (ADCC). In addition, myeloma cells release major histocompatibility complex class I-related chain molecule A (MICA) into serum as the disease progresses. MICA functions as a ligand to the NKG2D receptor on γδ T cells, NK cells and CD8+ T cells which further dampens anti-myeloma immunity. Furthermore, lactase derived from myeloma cells upregulates VEGF through HIF-1α, which directs tumor-associated macrophages (TAMs) to differentiate to an M2-like phenotype, leading to increased tumor angiogenesis and proliferation. M2-like macrophages are the dominant form of TAMs in the bone marrow of patients with myeloma85.
The bone marrow microenvironment can also contribute to immune dysfunction in myeloma patients. Bone marrow stroma cells can produce TGF-β to suppress immune responses. Mensenchymal stem cells in myeloma patients exhibit abnormal expression of CD40/40L, VCAM1, ICAM-1, LFA-3, HO-1, HLA-DR and HLA-ABC and cause a shift in the Th17/Treg balance86. Osteoclasts produce galectin-9 (Gal-9) and a proliferation-induced ligand (APRIL), which lead to the inhibition of antitumor T cell responses through the Tim-3/Gal-9 pathway and PD-1/PD-L1 pathway87.
IV. CTLA4/CD28 and PD-1/PD-L1 pathways in multiple myeloma
CTLA-4
CTLA-4 is an inhibitory receptor expressed on T cells. The biological role of CTLA-4 is regulation of T cell responses, predominantly during initial activation in the lymph node and the prevention of autoimmunity; this has previously been illustrated by the development of massive lethal lymphoproliferation in CTLA-4 knock-out mice88,89. Recognizing the role of CTLA-4 as a negative regulator of immunity, investigators have shown that antibody blockade of CTLA-4 could result in antitumor immunity in preclinical models90. This led to the development of ipilimumab (Yervoy, Bristol-Myers Squibb) and tremelimumab which are fully humanized monoclonal antibodies targeting CTLA-4. Ipilimumab has received FDA approval for the treatment of various solid malignancies, both as a single agent and in combination with anti-PD-1 targeting therapies.
PD-1/PD-L1 Pathway
PD-1 (alias CD279), a member of the CD28 family of receptors, is expressed on the surface of antigen-activated T and B cells91. While NK cells normally do not express PD-1, NK cells from myeloma patients have been shown to express PD-161. PD-L1 (B7 homologue-1) (B7-H1) is also expressed on myeloma cells, and this level of expression is higher in the plasma cells from MM patients compared with cells from MGUS patients and healthy volunteers92. PD-L1 expression is often up-regulated at relapse or in the refractory phase93,94 and in patients who are positive for minimal residual disease95. Several groups have shown that PD-L1 expression is absent in normal plasma cells, but is expressed in myeloma cell lines and primary myeloma cells from patients with MM92,96. Compared to PD-L1 expressing myeloma cell lines, myeloma cell lines that do not express PD-L1 are more sensitive to chemotherapy and have lower levels of Bcl-2 and FasL expression93. PD-L1 expression on myeloma cells is further upregulated following in vitro treatment with IFN-γ and TLR ligands through a common pathway involving MEK/ERK and MyD8897. PD-L1 expression on MM cells is also upregulated by the IL-6 signal cascade through activation of JAK2, STAT3, and MEK1/2. IFN-γ, which is produced by CTLs and NK cells, is a strong inducer of PD-L1 expression in MM cells through the activation of the MEK/ERK pathway97,98. Finally, PD-L1 is expressed on myeloid and plasmacytoid DCs, MDSCs and nonhematopoietic cells located in the bone marrow microenvironment99.
As shown in figure 2 PD-L1 binds to PD-1 and delivers an inhibitory signal, reducing cytokine production and proliferation of T cells100. The binding of PD-1 to PD-L1 or PD-L2 decreases secretion of Th1 cytokines, inhibits T-cell proliferation, results in T-cell apoptosis, and inhibits CTL-mediated killing. Additionally, PD-1/PD-L1 binding has been shown to promote resistance to melphalan and bortezomib in myeloma cell lines through activation of the PI3K/AKT pathway101. A soluble form of PD-L1 produced through proteolytic cleavage of membrane-bound proteins from myeloma cells has recently been detected in some MM patients; higher levels of soluble PD-L1 in this population has been associated with poorer progression free and overall survival102,103.
Figure 2:
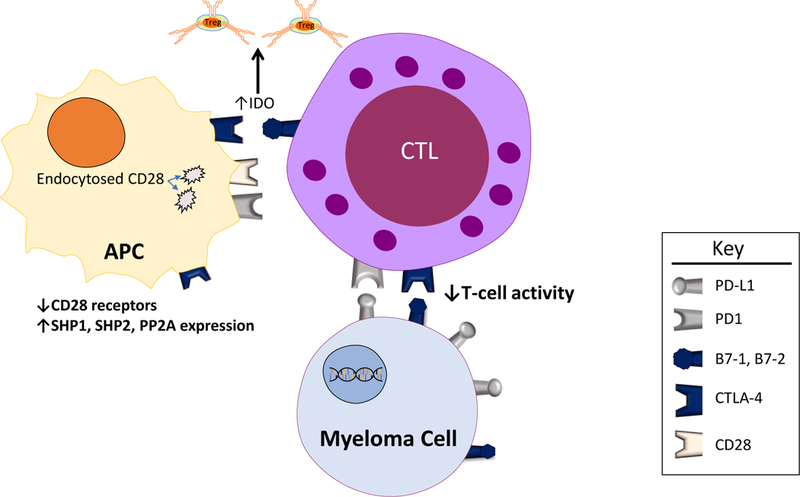
Effects of Immune Checkpoints in Myeloma. Binding of costimulatory molecules (B7–1, B7–2, or PD-L1) on myeloma cells to their respective receptors on dendritic cells results in downstream decreased immune activation. Specifically, expression of the phosphatases SHP1, SHP2, and PP2A are increased, and levels of the CD28 receptor (which is increases immune activation when bound to B7–1 or B7–2) are decreased via endocytosis of the cell surface receptors. Binding of B7–1 or B7–2 on cytotoxic T-cells with CTLA-4 on dendritic cells results in increased IDO expression which leads to increased levels of the immunosuppressive Treg cells.
V. Preclinical studies targeting CTLA4/CD28 and PD-1/PD-L1 pathways
Immunosuppresion is an important characteristic of MM pathology. Reversing this suppression could potentially restore myeloma immunosurveillance and improve disease control. Immune checkpoints are negative immunologic regulators that downregulate the magnitude of immune responses in order to protect the host from autoimmunity or damage from inflammation. This mechanism is frequently subverted by malignant cells, which escape immune surveillance by increasing inhibitory immune checkpoint ligands leading to host T cell exhaustion. Immune checkpoints have, therefore, become important therapeutic targets. Immune checkpoint inhibitors enhance the cytotoxic activity of host T cells by blocking the inhibitory signals from tumor cells. Rather than targeting the cancerous cells directly, these agents stimulate the host’s immune system to exert an antitumor effect104. The most clinically relevant checkpoints to date are CTLA-4 and PD-1/PD-L1 pathways. CTLA-4 is primarily believed to regulate immune responses early in T-cell activation while PD-1 is believed to inhibit T-cell activity in the effector phase within tissues and tumors. The use of antibodies to disrupt the receptor-ligand interactions involved in these pathways has shown remarkable results in several solid cancers (reviewed in105), and, more recently, in selected hematologic malignancies106–109.
VI. Clinical studies targeting CTLA4/CD28 and PD-1/PD-L1 pathways in multiple myeloma
Single agent activity
To date, there are no published clinical trials looking at ipilimumab or tremelimumab in MM patients. Ipilimumab was tested in a phase I trial of 28 patients with relapsed hematologic malignancies after allogenic stem cell transplant. This trial included 1 MM patient who presented with pulmonary plasmacytomas. Overall, 5 of the 22 (23%) patients who received the maximum tolerated dose of ipilimumab (10mg/kg) had a complete response while another 2 (9%) patients had a partial response including the MM patient. Notably, the response in the MM patient was durable lasting for 21 months110.
Pidilizumab (CT-011), a humanized antibody blocking PD-1, was given as a single agent in 17 patients with advanced hematologic malignancies such as AML and B-cell lymphoma in a phase I study111. Patients were treated with one of 5 dose levels - 0.2, 0.6, 1.5, 3 and 6 mg/kg. CT-011 was safe and well tolerated in this patient population with no single maximum tolerated dose defined111. The median t1/2 of CT-011 ranged from 217 to 410 hours (9–17 days) and sustained increase in the percentage of peripheral blood CD4+ cells was observed for up to 21 days after treatment. Clinical benefit with a single administration of CT-011 was found in 33% of the patients, with one complete remission. Only 1 myeloma patient was enrolled in the study; this patient received the 6mg/kg dose and achieved stable disease for >13 months.
Single agent nivolumab (Opdivo, Bristol-Myers Squibb) was administered at doses of 1 or 3 mg/kg every 2 weeks in a phase I study in patients with relapsed or refractory B-cell lymphoma (n = 31), T cell lymphoma (n = 23), or multiple myeloma (n = 27)112. Nivolumab was well tolerated with only 34% of patients experiencing immune-mediated AEs that were predominately grade 1 or 2. Only half of the patients with immune mediated AEs required immunosuppresion or interruption of nivolumab. Notably, the rate of all AEs and grade 3 or 4 AEs were lower in the MM patients (52% and 19% respectively) compared to the lymphoma patients (72% and 24% respectively)112. One patient with B-cell small lymphocytic lymphoma died of pneumonitis 19 days after receiving her initial dose of nivolumab. While nivolumab therapy resulted in an objective response rate of 36% and 40% among patients with DLBCL and FL, respectively, the best response for patients with MM was stable disease, which occurred in 17 (63%) MM patients and lasted a median of 11.4 weeks (range, 3.1 to 46.1 weeks)112.
Ribrag et al reported on the phase Ib KEYNOTE-013 study which tested the efficacy of pembrolizumab (Keytruda, Merck and Company) monotherapy for patients with relapsed/refractory multiple myeloma who had failed ≥ 2 prior lines of therapy including a proteasome inhibitor and an IMiD113. Thirty patients were treated with pembrolizumab 10mg/kg every 2 weeks or 200mg fixed dose every 3 weeks. At a median follow-up of 15 months, 28 (93%) patients discontinued the study due to disease progression. No patient experienced a response, and the best response observed was stable disease. Only 1 (3%) patient had an immune-related adverse event (grade 1 pruritis) and grade 3 treatment related AEs (myalgia) occurred in only 1 (3%) patient. There were no grade 4 TRAEs or deaths due to TRAEs113. These data are summarized in table 1.
Table 1:
Summary of Single Agent Checkpoint Inhibitor Trials. (ORR: overall response rate, DC: disease control rate, PR: partial response, SD: stable disease).
| Agent | Target | Trial Population | N (MM Patients) | Overall Endpoint | Response Rate (%) | # of Myeloma Responders(%) | Response Rate MM(%) | Best Response in MM patients | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Ipilimumab | CTLA4 | Relapsed Hematologic Malignancies | 22 (1) | ORR | 32 | 1 | 100 | PR | 108 |
| Pidilizumab | PD-1 | Advanced Hematologic Malignancies | 17 (1) | DC | 33 | 1 | 100 | PR | 109 |
| Nivolumab | PD-1 | Relapsed Refractory Hematologic Malignancies | 81 (27) | DC | 16 | 1 | 4 | SD | 110 |
| Pembrolizumab | PD-1 | Relapsed Refractory MM | 30 (30) | ORR | 0 | 0 | 0 | SD | 111 |
Combination therapy.
In preclinical studies, IMiDs were found to enhance the effects of PD-1/PD-L1 inhibitors on T cell- and NK cell- mediated cytotoxicity114. The combination of IMiDs and PD-1/PD-L1 inhibitors led to increased cell death of MM cells61,114,115. Notably, this effect was not associated with an increase in PD-1/PD-L1 expression on the effector cells114,116.These encouraging preclinical results led to clinical trials investigating the combination therapy of PD-1/PD-L1 inhibitors with IMiDs in patients with multiple myeloma which are summarized in table 2.
Table 2:
Summary of Combination Trials with Checkpoint Inhibitors. (ORR: overall response rate, sCR: stringent complete response, CR: complete response, irAEs: Immune related adverse events, NR: not reported).
| Combination | N | ORR (%) | Best Response i | irAEs(%) | Grade 3–4 irAEs | Ref(s) |
|---|---|---|---|---|---|---|
| Pembrolizumab, Pomalidome, Dexamethasone | 48 | 60 | CR | 33 | 10 | 116 |
| Pembrolizumab, Lenalidomide, Dexamethasone | 62 | 44 | sCR | 13 | 0 | 117 |
| Pembrolizumab, Pomalidome, Dexamethasone | 125 | 34 | CR | 58 | 18 | 118,119 |
| Pembrolizumab, Lenalidomide, Dexamethasone | 151 | 64 | NR | 68 | 44 | 119,120 |
Because nivolumab continues to bind to the T cell PD-1 receptor for over 2 months after administration, Pianko et al recently reported on the efficacy and toxicity of therapy immediately after treatment with nivolumab in 19 relapsed myeloma patients117. Twelve of the patients were treated with FDA-approved standard therapies after nivolumab and 7 patients were given experimental therapy or enrolled on another clinical trial. Interestingly, of the 12 patients receiving standard therapies, 7 (58%) achieved a PR or better, 2 (17%) achieved a VGPR, and 1 (8%) patient who received radiation therapy for a plasmacytoma reached a CR that lasted for >30 months112,117.
Badros et al reported on a single-center, phase II study of the combination of pembrolizumab, pomalidomide and dexamethasone in relapsed/refractory MM. A total of 48 patients were enrolled in the study. Patients had previously received both an IMiD and proteasome inhibitor and had a median of 3 lines of prior therapy. Furthermore, 70% of enrolled patients had received an autologous hematopoietic stem cell transplant and 62% had high-risk cytogenetics. Pembrolizumab was given at 200mg IV every 2 weeks, pomalidomide 4mg daily 21 days on and 1 week off, and dexamethasone 40mg weekly in 28-day cycles. Overall, 60% of treated patients showed objective responses including CR in 8% of patients, VGPR in 19%, and PR in 33% of patients. Median duration of response was 14.7 months. At median follow up of 15.6 months, progression-free survival was 17.4 months. Grade 3–4 adverse events occurred in 42% of patients including neutropenia in 42% of patients, anemia in 21% of patients, hyperglycemia in 21% of patients and pneumonia in 15% of patients. Pneumonitis occurred in 13% and hypothyroidism in 10%, but grade 3 or 4 levels were only seen in 2% and 4% respectively. Analyses of pretreatment bone marrow samples revealed a non-statistically significant trend (p = o.o6) for increased expression of PD-L1 in responders and a moderately statistically significant positive correlation between PFS and increased T-lymphocyte infiltrates, irrespective of PD-1 expression (p =0.05)118.
Keynote-023 (NCT02036502) was a phase I trial that tested the combination of pembrolizumab, lenalidomide, and low-dose dexamethasone in patients who had previously progressed on ≥ 2 prior therapies including a proteasome inhibitor and an IMiD. As of May 2017, a total of 62 patients with RRMM were enrolled in the study and 50 patients were evaluable for response. The MTD/MAD was pembrolizumab 200 mg fixed dose in combination with lenalidomide 25 mg and dexamethasone 40 mg. Of the evaluable patients 22 (44%) responded to treatment with 2 (4%) patients achieving a sCR, 6 (12%) patients achieving a VGPR, and 14 (28%) patients achieving a PR. Additionally, another 25 (50%) patients met criteria for SD for an overall disease control rate of 94% in the relapsed refectory setting119. Responses were durable, with a median duration of 18.7 months in the responders. Interestingly, while the overall PFS of the patients who were lenalidomide refractory was shorter (6.3 vs 7.2 months), duration of response was significantly longer at 24.9 months in the 13 patients who were lenalidomide refractory and achieved a PR or better. Only 8 patients (13%) experienced immune mediated adverse events (most commonly thyroid dysfunction), but there were 2 deaths in the trial: one from hepatic venoocclusive disease, and one from ischemic stroke119. This data suggests that PD-1 blockade with pembrolizumab in combination with lenalidomide and dexamethasone is associated with a tolerable safety profile and promising antimyeloma activity in heavily pretreated patients with RRMM.
The Keynote-183 phase III trial randomized patients with relapsed/refractory multiple myeloma who had received at least 2 lines of prior treatment to receive pomalidomide and low-dose dexamethasone with or without pembrolizumab (200 mg every 3 weeks). At the data cutoff of June 2, 2017, a total of 249 patients were enrolled: 125 randomized to the pembrolizumab arm and 124 patients randomized to pomalidomide and dexamethasone alone. The objective response rate (ORR) was 34% versus 40% in the pembrolizumab and control arms, respectively. The median time-to-progression (TTP) was 8.1 months versus 8.7 months, respectively (HR, 1.14; 95% CI, 0.75–1.74)120. At the median follow-up of 8.1 months, there were 29 deaths in the pembrolizumab arm versus 21 deaths in the control arm, with the pembrolizumab group showing a greater than 50% increase in the relative risk of death compared with the control arm. The rate of severe, grade 3–5 toxicity was 83% in the pembrolizumab arm versus 65% in the control group. The rates of serious adverse events (AEs) were 63% versus 46%, respectively. Causes of death unrelated to disease progression in the pembrolizumab cohort included myocarditis, Stevens-Johnson syndrome, myocardial infarction, pericardial hemorrhage, cardiac failure, respiratory tract infection, neutropenic sepsis, sepsis, multiple organ dysfunction, and respiratory failure120,121.
Similarly, the Keynote-185 phase III trial randomized newly diagnosed, treatment naïve MM patients who were ineligible for autologous stem cell transplant to receive lenalidomide and low-dose dexamethasone with or without pembrolizumab (200 mg every 3 weeks). A total of 301 patients were included in the safety and efficacy analysis at the data cutoff date of June 2, 2017. The ORR was 64% in the pembrolizumab arm versus 62% in the control arm. The median TTP had not yet been reached in either arm (HR, 0.55; 95% CI, 0.20–1.50) at the time of data cutoff. At the median follow-up of 6.6 months, there were 19 deaths in the pembrolizumab arm compared with 9 deaths in the control arm (HR for OS, 2.06; 95% CI, 0.93–4.55). The relative risk of death in the pembrolizumab arm was more than double that of the control group. The rate of severe, grade 3–5 toxicity was 72% in the investigational arm versus 50% in the control arm. The rates of serious AEs were 54% versus 39%, respectively. Causes of death unrelated to disease progression identified in the pembrolizumab cohort included intestinal ischemia, cardiorespiratory arrest, suicide, pulmonary embolism, cardiac arrest, pneumonia, sudden death, myocarditis, large intestine perforation, and cardiac failure121,122. Because of the increased risk for death observed in pembrolizumab arm in both the Keynote-183, and 185 studies, the FDA halted these trials on July 3, 2017 (Merck, the manufacturer of Pembrolizumab, had ceased enrolling new patients as of June 12, 2017).
Development of immune-related adverse events (irAEs) is associated with response to checkpoint inhibitors in some disease settings123–126. However, data from Keynote-183 and Keynote-185 yielded inconsistent results regarding the relationship between irAEs and response to checkpoint inhibition. As reported by Krauss et al. at the 2018 ASCO annual meeting, the Keynote-183 trial had 70 patients (56%) in the pembrolizumab, pomalidomide and dexamethasone arm develop an irAE. These patients had an ORR of 37%, which was not significantly different from the 31% ORR in the 55 patients (44%) who did not have irAEs. Of the 21 patients that experienced Grade ≥3 irAEs in the pembrolizumab containing arm, only 6 (29%) achieved a PR or better. These data suggest no association between irAEs and ORR. However, in Keynote-185, 102 patients (68%) in the pembrolizumab containing arm had one or more irAEs, and this was associated with an ORR of 73%. Response rate in the 54 patients (36%) who had grade ≥3 irAEs was similar at 70%. Both of these values are significantly higher than the response rate in the 49 patients who did not have any irAEs (45%), suggesting an association between response and irAEs in this trial121. The reason for these disparate findings remains unclear. One possibility is that patients with newly diagnosed myeloma, such as those in the Keynote-185 study, retain greater capacity to mount an immune response by virtue of having not been exposed to the dampening effects of prior therapy. Interestingly, both of these studies showed significant irAEs for their respective control arms (45% in Keynote183, and 44% in Keynote-185) which seems to correlate with increased response121. These data confirm that IMiDs are indeed immunomodulators and that their ability to induce an immune response is preserved in both newly diagnosed and relapsed refractory MM.
Combinations therapy with nivolumab in MM patients is currently being investigated in several clinical trials. Checkmate-039 is a phase I study that aims to establish the tolerability of nivolumab and the combination of nivolumab and daratumumab, with or without an IMiD (pomalidomide and dexamethasone) in subjects with relapsed or refractory MM127. CA204142 is a phase II, multiple cohort study of elotuzumab in combination with pomalidomide, low-dose dexamethasone, and nivolumab, in patients with relapsed or refractory multiple myeloma who have previously progressed on a lenalidomide containing regimen. Checkmate-602 is a phase III, open-label, randomized trial evaluating the combination of nivolumab, elotuzumab, pomalidomide, and dexamethasone for patients with relapsed/refractory multiple myeloma128. Checkmate-602 is supported by preclinical models which have shown that the combination of elotuzumab and anti-PD-1 resulted in increased tumor-infiltrating NK cells and activation of CD8+ T-cells129. All 3 trials were placed on partial clinical holds in October 2017 by the FDA as part of the review of the data on pembrolizumab. On December 5, 2017, the holds placed on the phase I CheckMate-039 and phase II CA204142 trials were lifted by the FDA; while the partial hold on the phase III CheckMate-602 trial study remained in place until June 2018.
The MEDI4736-MM-003 trial (NCT02807454) was designed to study the efficacy of the PD-L1 inhibitor Duvalumab (Iminzi, Astra-Zeneca) in combination with daratumumab with and without pomalidomide and dexamethasone in RRMM130. However, the FDA placed this trial on partial hold in September 2017; the study was terminated shortly thereafter. Only 37 patients were enrolled in the study at the time of discontinuation. A phase I study (NCT02716805) to assess the safety and tolerability of tremelimumab and durvalumab administered with high dose chemotherapy and autologous stem cell transplant in high risk MM was also terminated after only 6 patients had been enrolled.
The investigational PD-1 inhibitor JNJ-63723283 was being tested in combination with daratumumab in the MMY-2036 study (NCT03357952) in patients with relapsed refractory MM who had received ≥3 lines of prior therapy, including an IMiD and proteasome inhibitor. At the same time, the combination of daratumumab and the PD-L1 inhibitor atezolizumab (Tecentriq, Roche/Genentech) was being tested in NSCLC (NCT0302342). Interim analysis of this trial by the data monitoring committee suggested no improvement in efficacy with the combination compared to atezolizumab alone, and increased rate of mortality in the combination arm. Based on these results GenMab announced in May of 2018 that Janssen would stop studies of daratumumab in combination with anti-PD-1/PD-L1.
V. Future directions in treatment with immune checkpoint inhibitors
As detailed above, checkpoint inhibitors have proven challenging in the treatment of multiple myeloma. Targeting the CTLA4 and/or PD-1/PD-L1 pathways can re-activate and boost antitumor immunity, resulting in disease regression and prolonged survival in solid tumors. However, checkpoint inhibitors have not been effective as a single agent in multiple myeloma patients. Furthermore, current combinations of checkpoint inhibitors with IMiDs have yielded disappointing results and were associated with an increased risk of death in randomized phase III studies. While checkpoint inhibitors remain an attractive therapy for MM, further research is needed to improve efficacy and limit toxicitywith these agents. We proposed three main areas for future research endeavors.
1. A comprehensive understanding of the unique immune dysfunctions in multiple myeloma.
Unlike solid tumors, multiple myeloma is a malignancy of plasma cells which produce monoclonal immunoglobulin proteins. These plasma cells and abnormal immunoglobulin proteins cause profound immune dysfunction that is unique to MM. For instance, it was reported that T cells in multiple myeloma demonstrated senescent phenotypes rather than the exhausted phenotypes typically seen in solid tumors131. Additionally, 75% of myeloma patients showed clonal expansion of T cells that express low levels of PD-1 or CTLA-4, but also express markers consistent with telomere-independent senescence132. The presence of senescence demonstrates the need to evoke the expansion of activated, myeloma-reactive T cells and provides a rationale for combination therapy. Similarly, it is very likely that the functions and phenotypes of other immune cells such as Tregs, MDSCs, and NK cells could be different in multiple myeloma patients when compared to patients with other malignancies. In conclusion, a more thorough understanding of the unique immune dysfunctions of myeloma patients is warranted.
Secondly, the repertoire of co-signaling molecules on T cells, antigen presenting cells, and tumor cells is highly versatile and context dependent. The potential effects of administration of CTLA4 or PD-1/PD-L1 antibodies could have on the expression and levels of other co-signaling molecules are largely unknown. It is possible that administration of CTLA4 antibody and PD1/PD-L1 antibodies could lead to compensatory up-regulation of other immune checkpoints (as has been seen in other cancers)133,134, or increases in MDSCs and/or Tregs cells.
Finally, many other factors can impact the efficacy of CTLA4 or PD-1/PD-L1 targeted therapy. For instance, the responses to PD-1 targeted therapy are quite different among relapsed/refractory MM, newly diagnosed MM, and smoldering MM patients. Factors such as patient population (age, co-morbidities, disease), prior therapy, and history of stem cell transplant can affect CTLA4 or PD-1/PD-L1 targeted therapy.
Ratner et al. recently reported rapid progression of adult T cell leukemia/lymphoma after antiPD-1 therapy in the first three patients of their phase II trial135. Interestingly, Ishitsuka et al. performed a similar phase II trial in Japan with 8 patients who were treated with at least one dose of nivolumab and none of these patients showed rapid disease progression136. It was postulated that these conflicting results might be caused by differences in the patients’ disease status and therefore a difference in the immune environment. The 3 patients in Ratner’s report had ATLL with smoldering or very slow progression. In contrast, the patients in the Japan cohort had aggressive disease or chronic type with unfavorable prognostic factors. These findings suggest that disease status and the immune environment may play a role in PD-1 treatment.
Recently, it was shown that gut microbiome and antibiotic use affect the efficacy of PD-1 based immunotherapy against epithelial cancers (melanoma, sarcoma, non-small cell lung cancer, and renal cell carcinoma)137–139. Patients with advanced NSCLC, RCC or urothelial carcinoma who were prescribed antibiotics within 2 months before or 1 month after the first administration of PD-1/PD-L1 antibody demonstrated a shorter PFS and OS137. Metagenomics of patient stool samples revealed a correlation between treatment responses to checkpoint inhibitors and the relative abundance of Akkermansia muciniphila. The presence of A. muciniphila was associated with increased recruitment of tumor infiltrating CCR9+CXCR3+CD4+ T lymphocytes and enhanced efficacy of PD-1 blockade in an IL-12 dependent manner. When feces from NSCLC patients who responded to anti-PD-1 therapy were transplanted into germ-free or antibiotic-treated mice, the animals had improved response to PD-1 blockade. Conversely, feces from patients who did not respond to anti-PD-1 did not improve response to PD-1 blockade in germ-free mice. However, when these mice received oral A. muciniphila alone or combined with Enterococcus hirae (which stimulates goblet cells), tumor growth slowed137. Similarly, analysis of stool in melanoma patients prior to treatment with anti-PD-1 immunotherapy showed higher levels of Bifidobacterium longum, Collinsella aerofaciens, and Enterococcus faecium in the responders. Once again, transfer of fecal material from the responders into germ free mice slowed the growth of implanted B16.SIY melanoma cells compared to mice who received fecal material from the non-responders138. It is very likely that chemotherapy and antibiotic use in myeloma patients could affect composition of the gut microbiome and diminish the efficacy of checkpoint inhibitors. These data need to be weighed against the recent data that showed a survival benefit with prophylactic antibiotic use in myeloma patients undergoing induction chemotherapy140.
2. Identification and development of effective combination therapies with checkpoint inhibitors.
Most of the combination chemoimmunotherapy trials to date examine combinations of checkpoint inhibitors with IMiDs. As previously noted, IMiDs reduce PD-1 expression on T cells and NK cells as well as PD-L1 expression on myeloma cells and MDSCs. Therefore, cotreatment with an IMiD and anti-PD1/PD-L1 could lead to reduced target antigens on the effector cells and thus less antibody binding. Furthermore, this downregulation of PD-1/PD-L1 could lead to a compensatory up-regulation of other immune checkpoints—a mechanism that has been previously described in other cancers133,134—which could contribute to the lack of activity seen with these agents in combination. Further investigation in this area is clearly warranted to explore other potential combinations of therapy that could capitalize on checkpoint inhibition.
Combination of radiotherapy and anti-PD-1/PD-L1 antibody.
It is noteworthy that only one patient achieved a CR with nivolumab single agent therapy112. This patient developed a rib plasmacytoma while on nivolumab. Nivolumab was stopped and the patient underwent local radiation. After completion of radiation therapy, the patient was restarted on nivolumab for 2 months and achieved a CR that lasted for >14 months112. This single case suggests that the combination of radiotherapy and anti-PD-1/PD-L1 monoclonal antibody may be useful for patients with MM; an effect that was previously reported in several solid cancers 141–143. In these instances, treatment with radiation is hypothesized to convert tumors to a more immunogenic phenotype by upregulating antigenic expression and co-stimulatory molecules, by increasing the secretion and production of immunostimulatory cytokines, and by increasing recruitment of APCs and effectors into the tumor microenvironment144.
Combination of tumor vaccines with checkpoint inhibitors.
The addition of tumor vaccines to treatment with checkpoint inhibitors could prime antigen presenting cells and T cells for antitumor immunity. Rosenblatt et al administered a multiple myeloma vaccine made from patient-derived tumor cells fused with autologous dendritic cells to patients following AHSCT and demonstrated immunologic and clinical responses145,146. In vitro studies suggest that PD-1 blockade by pidilizumab (CT-011) acts synergistically when co-administered with a DC/Myeloma fusion vaccine147. This combination is currently be evaluated in a phase II clinical trial following AHSCT148.
Combination of chemotherapy with checkpoint inhibitors and using checkpoint inhibitors in the post autologous transplant setting.
One of the major immune dysfunctions in myeloma patients results from the presence of immune suppressor cell populations such as Tregs and MDSCs which can persist even after patients achieve successful remission82. Therefore, one strategy to enhance the efficacy of checkpoint inhibitors is to use cytotoxic therapy to deplete immune suppressor cells thus enhancing the reconstitution of myeloma immunity149. Similarly, autologous hematopoietic stem cell transplant was reported to be able to deplete regulatory T cells and cause concurrent expansion of myeloma-specific clones145,150. Therefore, the combination of a checkpoint inhibitor with a DC/tumor vaccine following AHSCT has been hypothesized to enhance the efficacy of checkpoint inhibitors in myeloma148.
Combination of CAR-T cells with checkpoint inhibitors.
Chimeric antigen receptor (CAR)-T cell therapy has become a very promising treatment for relapsed/refractory MM. This therapy involves the infusion of large amounts of myeloma cell-specific effector T cells151. Early phase clinical trials have shown impressive efficacy in heavily pretreated, highly relapsed refractory patients152–154, but it remains to be seen whether these responses will be durable. Combination of CAR-T cell therapy with checkpoint inhibitors could lead to enhanced antimyeloma effects and increased duration of response. Indeed, preclinical models have demonstrated augmented anti-tumor activity with the combination of CAR-T cell therapy and a PD-1 antibody155. However, several potential concerns of combining these treatment modalities exist. These include: increased T-cell anergy or apoptosis of CAR-T cells when bound to CTLA4 or PD-1/PD-L1, and increased risk for adverse events such as cytokine release syndrome or CNS toxicity with overexcitation of immune effectors149,156.
Combination of various checkpoint inhibitors.
The immune response is regulated by highly dynamic, overlapping, and numerous coinhibitory molecules. It has previously been demonstrated that at 3 and 12 months after autologous transplant, CTLA-4, LAG3, and TIM-3 are expressed on T cells isolated from patients with MM150. A preclinical study indicated that blocking PD-L1 together with other immune checkpoints (CTLA-4, LAG-3 or TIM-3) promotes the survival MM-bearing mice following low dose total body irradiation157. Similarly, the inhibitory immunoreceptor T cell immunoglobulin and ITIM domains (TIGIT) have been shown to have greater expression in MM patients then PD-1. Recent data has shown that inhibition of TIGIT leads to significantly improved OS in animal models of MM compared with anti-PD-1158,159. Moving forward, it will be critical to understand the clinical effects of blocking these pathways alone and in combination with PD-1 blockade. Currently, the combination of CTLA4 and PD-1 inhibitors is being evaluated in a phase Ib/IIa trial of patients with diffuse large B-cell lymphoma, T-cell lymphoma, or multiple myeloma who are at high risk for relapse following autologous stem cell transplant (NCT02681302).
Combination of COX2 inhibitors with anti-PD-1/PD-L1 antibody.
The enzyme cyclo- oxygenase-2 (COX-2) catalyzes the synthesis of prostaglandin E2. Both COX-2 and PGE2 can directly mediate pro-tumor activities and recruit and induce MDSCs in the tumor microenvironment160. Several preclinical models have demonstrated that inhibition of COX-2 or PGE2 combined with PD-1 pathway blockade acts synergistically161–163. This suggests that COX inhibitors could be useful adjuncts for immune-based therapies, including PD-1 blockade in cancer patients. Similarly, entinostat, a selective class I and class IV histone deacetylase inhibitor, has been shown to significantly reduce arginase-1, iNOS and COX-2 levels and to neutralize MDSCs. The combination of entinostat and PD-1 inhibition yielded enhanced antitumor effects in murine models of lung and renal cell carcinoma compared to PD-1 inhibition alone164.
Combination of interferon or interferon activator with checkpoint inhibitors.
Interferon was the first drug used to stimulate the immune system. Its efficacy was only modest however, and it currently does not have a role in myeloma treatment. Recent publications highlight a potential role for interferon in combination with other immune modulators—such as checkpoint inhibitors. Specifically, it has been shown that ablation of histone demethylase LSD1 activates type I interferon, which stimulates anti-tumor T cell immunity. This elicits significant responses to checkpoint blockade in mouse melanomas which were previously refractory to anti-PD-1 therapy165. Similarly, the combination of interferon and anti-CTLA4 antibody had superior efficacy compared to anti-CTLA4 antibody alone in a phase Ib trial of melanoma patients166.
Supplementation of gut microbiome with PD-1/PD-L1 treatment.
It was recently demonstrated that the gut microbiome influences the efficacy of PD-1 antibody treatment in solid tumors137–139. How concurrent antibiotic use and the gut microbiome affect the efficacy of checkpoint inhibitors in multiple myeloma is largely unknown. It is possible that, similar to solid tumors, oral supplementation of A. muciniphila could restore or enhance the efficacy of checkpoint inhibitors in multiple myeloma; prior exposures to antibiotics could therefore be potentially deleterious to myeloma patients being considered for immunotherapy. However, this must be weighed against the potential benefit of prophylactic antibiotics which was recently demonstrated in a large phase III trial of newly diagnosed MM patients140
3. Development of clinical/biomarkers for immune monitoring and predicting immune-related adverse events.
Looking forward, it will be critical to develop clinical/biomarkers that can be used to monitor immune responses during checkpoint inhibitor treatment, predict treatment responses to checkpoint inhibitors, and detect irAEs early before they become severe. In solid cancers, neoantigens, MHC antigens, PD-1 expression level, microsatellite instability, and mutational load have been proposed to correlate with treatment response167. Tumor necrosis factor, interferon-gamma, and various cytokines were found to be predictive markers for PD-1 treatment in advanced non-small cell lung cancer168. Level of peripheral blood eosinophils, IL-17 level, gene expression profiling, and digestive infiltrate by neutrophils have been found to be associated with checkpoint inhibitor toxicities in solid malignancies169.
VII. Conclusions.
Multiple myeloma remains an incurable disease. Immunotherapy using checkpoint inhibitors holds great promise in reversing the suppressive anti-myeloma immune response in MM patients. Thus far, the efficacy and safety profile of checkpoint inhibitors have been disappointing in MM. However, a full understanding of the immune defects present in the MM patient is required to appreciate the unique state of immune impairment that this disease entails. With this understanding, new combinations of agents and novel targets of immune therapy can be nominated to provide innovative therapies for this disease. Adoptive T cell transfer therapy has demonstrated very promising efficacy in myeloma treatment.
Highlights.
Multiple myeloma is associated with profound defects/dysfunction in both innate and adaptive immunity. PD-L1 expression is up-regulated in myeloma cells and at relapse or in the refractory phase, which results in the suppression of cytotoxic T cell activity. While checkpoint inhibitors are effective in prolonging overall survival in some patients with advanced solid cancers and Hodgkin lymphoma, they have not demonstrated significant anti-myeloma activities as a single agent in MM. The combination of checkpoint inhibitors with immunomodulatory agents was recently found to increase the risk of death in myeloma patients. Additional studies are needed for a better understanding of immune dysregulation in myeloma patients, and the mechanisms of action of- and resistance to- checkpoint inhibitors
Acknowledgement
Funding
This work is supported by Duke Cancer Institute Fund, NIH R44CA199767, and NIH R01CA197792.
Footnotes
Competing interests
The authors declare no competing conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
VIII. References
- 1.Surveillance Research Program, N. C. I. Fast Stats: An interactive tool for access to SEER cancer statistics, <https://seer.cancer.gov/faststats> (2018).
- 2.Latov N Pathogenesis and therapy of neuropathies associated with monoclonal gammopathies. Ann Neurol 37 Suppl 1, S32–42 (1995). [DOI] [PubMed] [Google Scholar]
- 3.Batuman V The Pathogenesis of Acute Kidney Impairment in Patients With Multiple Myeloma. Advances in Chronic Kidney Disease 19, 282–286, doi: 10.1053/j.ackd.2012.04.009 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Zhang J et al. Light chain multiple myeloma, clinic features, responses to therapy and survival in a long-term study. World Journal of Surgical Oncology 12, 234, doi: 10.1186/1477-7819-12-234 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chawla SS et al. Clinical Course and Prognosis of Non-Secretory Multiple Myeloma. Eur J Haematol, doi: 10.1111/ejh.12534 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Fonseca R International Myeloma Working Group molecular classification of multiple myeloma: spotlight review 23, 2210–2221, doi: 10.1038/leu.2009.174 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palumbo A et al. Oral melphalan, prednisone, and thalidomide in elderly patients with multiple myeloma: updated results of a randomized controlled trial. Blood 112, 3107–3114, doi:blood-2008-04-149427 [pii] 10.1182/blood-2008-04-149427 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Wijermans P et al. Phase III Study of the Value of Thalidomide Added to Melphalan Plus Prednisone in Elderly Patients With Newly Diagnosed Multiple Myeloma: The HOVON 49 Study. J Clin Oncol, doi:JCO.2009.26.1610 [pii] 10.1200/JCO.2009.26.1610. [DOI] [PubMed] [Google Scholar]
- 9.Child JA et al. High-dose chemotherapy with hematopoietic stem-cell rescue for multiple myeloma. N Engl J Med 348, 1875–1883, doi: 10.1056/NEJMoa022340 348/19/1875 [pii] (2003). [DOI] [PubMed] [Google Scholar]
- 10.Blade J et al. High-dose therapy intensification compared with continued standard chemotherapy in multiple myeloma patients responding to the initial chemotherapy: long-term results from a prospective randomized trial from the Spanish cooperative group PETHEMA. Blood 106, 3755–3759, doi:2005-03-1301 [pii] 10.1182/blood-2005-03-1301 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Attal M et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Francais du Myelome. N Engl J Med 335, 91–97 (1996). [DOI] [PubMed] [Google Scholar]
- 12.Attal M et al. Single versus double autologous stem-cell transplantation for multiple myeloma. N Engl J Med 349, 2495–2502, doi: 10.1056/NEJMoa032290 349/26/2495 [pii] (2003). [DOI] [PubMed] [Google Scholar]
- 13.Richardson PG et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 348, 2609–2617, doi: 10.1056/NEJMoa030288 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Richardson PG et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 352, 2487–2498, doi: 10.1056/NEJMoa043445 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Kuhn DJ et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 110, 3281–3290, doi:blood-2007-01-065888 [pii] 10.1182/blood-2007-01-065888 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ludwig H et al. Survival and years of life lost in different age cohorts of patients with multiple myeloma. J Clin Oncol 28, 1599–1605, doi: 10.1200/jco.2009.25.2114 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Usmani SZ et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood 128, 37–44, doi: 10.1182/blood-2016-03-705210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palumbo A et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N Engl J Med 375, 754–766, doi: 10.1056/NEJMoa1606038 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Dimopoulos MA et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N Engl J Med 375, 1319–1331, doi: 10.1056/NEJMoa1607751 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Lokhorst HM et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N Engl J Med 373, 1207–1219, doi: 10.1056/NEJMoa1506348 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Richardson PG et al. Elotuzumab in combination with lenalidomide and dexamethasone in patients with relapsed multiple myeloma: final phase 2 results from the randomised, open-label, phase 1b-2 dose-escalation study. The Lancet. Haematology 2, e516–527, doi: 10.1016/s2352-3026(15)00197-0 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lonial S et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N Engl J Med 373, 621–631, doi: 10.1056/NEJMoa1505654 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Barlogie B et al. Incorporating bortezomib into upfront treatment for multiple myeloma: early results of total therapy 3. Br J Haematol 138, 176–185, doi:BJH6639 [pii] 10.1111/j.1365-2141.2007.06639.x (2007). [DOI] [PubMed] [Google Scholar]
- 24.Reeder CB et al. Cyclophosphamide, bortezomib and dexamethasone induction for newly diagnosed multiple myeloma: high response rates in a phase II clinical trial. Leukemia 23, 1337–1341, doi:leu200926 [pii] 10.1038/leu.2009.26 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knop S et al. Bortezomib, IV cyclophosphamide, and dexamethasone (VelCD) as induction therapy in newly diagnosed multiple myeloma: results of an interim analysis of the German DSMM Xia trial. J Clin Oncol 27, abstr 8516 (2009). [Google Scholar]
- 26.Palumbo A et al. Bortezomib as induction before autologous transplantation, followed by lenalidomide as consolidation-maintenance in untreated multiple myeloma patients. J Clin Oncol 28, 800–807, doi:JCO.2009.22.7561 [pii] 10.1200/JCO.2009.22.7561 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Rajkumar SV et al. Lenalidomide plus high-dose dexamethasone versus lenalidomide plus low-dose dexamethasone as initial therapy for newly diagnosed multiple myeloma: an open-label randomised controlled trial. Lancet Oncol 11, 29–37, doi:S1470-0 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar SK et al. Bortezomib, dexamethasone, cyclophosphamide and lenalidomide combination for newly diagnosed multiple myeloma: phase 1 results from the multicenter EVOLUTION study. Leukemia, doi:leu2010116 [pii] 10.1038/leu.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drake CG, Lipson EJ & Brahmer JR Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nature reviews Clinical oncology 11, 24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menon S, Shin S & Dy G Advances in cancer immunotherapy in solid tumors. Cancers 8, 106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y Cancer immunotherapy: harnessing the immune system to battle cancer. The Journal of clinical investigation 125, 3335–3337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen R et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 35, 2125–2132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez-Otero P, Paiva B, Engelhardt M, Prosper F & San Miguel JF Is immunotherapy here to stay in multiple myeloma? Haematologica 102, 423–432, doi: 10.3324/haematol.2016.152504 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.June CH, Ledbetter JA, Gillespie MM, Lindsten T & Thompson CB T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol Cell Biol 7, 4472–4481 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bretscher P & Cohn M A theory of self-nonself discrimination. Science 169, 1042–1049 (1970). [DOI] [PubMed] [Google Scholar]
- 36.Mueller DL, Jenkins MK & Schwartz RH Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu Rev Immunol 7, 445–480, doi: 10.1146/annurev.iy.07.040189.002305 (1989). [DOI] [PubMed] [Google Scholar]
- 37.Lafferty KJ & Cunningham AJ A new analysis of allogeneic interactions. The Australian journal of experimental biology and medical science 53, 27–42 (1975). [DOI] [PubMed] [Google Scholar]
- 38.Linsley PS, Clark EA & Ledbetter JA T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc Natl Acad Sci U S A 87, 50315035 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rudd CE, Taylor A & Schneider H CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev 229, 12–26, doi: 10.1111/j.1600-065X.2009.00770.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linsley PS et al. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med 174, 561–569 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Azuma M et al. B70 antigen is a second ligand for CTLA-4 and CD28. Nature 366, 76–79, doi: 10.1038/366076a0 (1993). [DOI] [PubMed] [Google Scholar]
- 42.Hathcock KS et al. Identification of an alternative CTLA-4 ligand costimulatory for T cell activation. Science 262, 905–907 (1993). [DOI] [PubMed] [Google Scholar]
- 43.Chen L & Flies DB Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 13, 227–242, doi: 10.1038/nri3405 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarter K et al. Btn2a2, a T cell immunomodulatory molecule coregulated with MHC class II genes. Journal of Experimental Medicine, jem. 20150435 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kane LP T cell Ig and mucin domain proteins and immunity. The Journal of Immunology 184, 2743–2749 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lozano E, Joller N, Cao Y, Kuchroo V & Hafler DA The CD226/CD155 interaction regulates the proinflammatory (Th1/Th17)/anti-inflammatory (Th2) balance in humans. The Journal of Immunology, 1300945 (2013). [DOI] [PMC free article] [PubMed]
- 47.Vigano S, Perreau M, Pantaleo G & Harari A Positive and negative regulation of cellular immune responses in physiologic conditions and diseases. Clinical and Developmental Immunology 2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu Y, Yao S & Chen L Cell surface signaling molecules in the control of immune responses: a tide model. Immunity 34, 466–478, doi: 10.1016/j.immuni.2011.04.008 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saito T, Yokosuka T & Hashimoto-Tane A Dynamic regulation of T cell activation and co-stimulation through TCR-microclusters. FEBS Lett 584, 4865–4871, doi: 10.1016/j.febslet.2010.11.036 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Yokosuka T et al. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity 33, 326–339, doi: 10.1016/j.immuni.2010.09.006 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Yokosuka T et al. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med 209, 1201–1217, doi: 10.1084/jem.20112741 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saito T & Yamasaki S Negative feedback of T cell activation through inhibitory adapters and costimulatory receptors. Immunol Rev 192, 143–160 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Munn DH, Sharma MD & Mellor AL Ligation of B7–1/B7–2 by human CD4+ T cells triggers indoleamine 2,3-dioxygenase activity in dendritic cells. J Immunol 172, 4100–4110 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Keir ME, Butte MJ, Freeman GJ & Sharpe AH PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 26, 677–704, doi: 10.1146/annurev.immunol.26.021607.090331 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Azuma T et al. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 111, 3635–3643, doi: 10.1182/blood-2007-11-123141 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tamura H Immunopathogenesis and immunotherapy of multiple myeloma. Int J Hematol 107, 278–285, doi: 10.1007/s12185-018-2405-7 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Carbone E et al. HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood 105, 251–258, doi: 10.1182/blood-2004-04-1422 (2005). [DOI] [PubMed] [Google Scholar]
- 58.El-Sherbiny YM et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res 67, 8444–8449, doi: 10.1158/0008-5472.can-06-4230 (2007). [DOI] [PubMed] [Google Scholar]
- 59.von Lilienfeld-Toal M et al. Reduced immune effector cell NKG2D expression and increased levels of soluble NKG2D ligands in multiple myeloma may not be causally linked. Cancer immunology, immunotherapy : CII 59, 829–839, doi: 10.1007/s00262-0090807-3 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fauriat C, Mallet F, Olive D & Costello RT Impaired activating receptor expression pattern in natural killer cells from patients with multiple myeloma. Leukemia 20, 732733, doi: 10.1038/sj.leu.2404096 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Benson DM Jr. et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 116, 2286–2294, doi: 10.1182/blood-2010-02-271874 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng Y et al. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood 114, 3625–3628, doi: 10.1182/blood-2009-05-220285 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim J et al. Macrophages and mesenchymal stromal cells support survival and proliferation of multiple myeloma cells. Br J Haematol 158, 336–346, doi: 10.1111/j.1365-2141.2012.09154.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parrinello N et al. Neutrophils Of Multiple Myeloma Are Dysfunctional and Immunosuppressive. Blood 122, 3138–3138 (2013).24030378 [Google Scholar]
- 65.Wong TW et al. Induction of malignant plasma cell proliferation by eosinophils. PLoS One 8, e70554, doi: 10.1371/journal.pone.0070554 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wong D et al. Eosinophils and megakaryocytes support the early growth of murine MOPC315 myeloma cells in their bone marrow niches. PLoS One 9, e109018, doi: 10.1371/journal.pone.0109018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pessoa de Magalhaes RJ et al. Analysis of the immune system of multiple myeloma patients achieving long-term disease control by multidimensional flow cytometry. Haematologica 98, 79–86, doi: 10.3324/haematol.2012.067272 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mills KH & Cawley JC Abnormal monoclonal antibody-defined helper/suppressor T-cell subpopulations in multiple myeloma: relationship to treatment and clinical stage. Br J Haematol 53, 271–275 (1983). [DOI] [PubMed] [Google Scholar]
- 69.Murakami H, Ogawara H & Hiroshi H Th1/Th2 cells in patients with multiple myeloma. Hematology 9, 41–45, doi: 10.1080/10245330310001652437 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Beyer M et al. In vivo peripheral expansion of naive CD4+CD25high FoxP3+ regulatory T cells in patients with multiple myeloma. Blood 107, 3940–3949, doi: 10.1182/blood-2005-09-3671 (2006). [DOI] [PubMed] [Google Scholar]
- 71.Noonan K et al. A novel role of IL-17-producing lymphocytes in mediating lytic bone disease in multiple myeloma. Blood 116, 3554–3563, doi: 10.1182/blood-2010-05-283895 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Prabhala RH et al. Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood 115, 5385–5392, doi: 10.1182/blood-2009-10-246660 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Korde N, Kristinsson SY & Landgren O Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): novel biological insights and development of early treatment strategies. Blood 117, 5573–5581, doi: 10.1182/blood-2011-01-270140 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Giannopoulos K, Kaminska W, Hus I & Dmoszynska A The frequency of T regulatory cells modulates the survival of multiple myeloma patients: detailed characterisation of immune status in multiple myeloma. Br J Cancer 106, 546–552, doi: 10.1038/bjc.2011.575 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Muthu Raja KR et al. Increased T regulatory cells are associated with adverse clinical features and predict progression in multiple myeloma. PLoS One 7, e47077, doi: 10.1371/journal.pone.0047077 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ratta M et al. Dendritic cells are functionally defective in multiple myeloma: the role of interleukin-6. Blood 100, 230–237 (2002). [DOI] [PubMed] [Google Scholar]
- 77.Brown RD et al. Dendritic cells from patients with myeloma are numerically normal but functionally defective as they fail to up-regulate CD80 (B7–1) expression after huCD40LT stimulation because of inhibition by transforming growth factor-beta1 and interleukin-10. Blood 98, 2992–2998 (2001). [DOI] [PubMed] [Google Scholar]
- 78.Talmadge JE & Gabrilovich DI History of myeloid-derived suppressor cells. Nat Rev Cancer 13, 739–752, doi: 10.1038/nrc3581 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ramachandran IR et al. Myeloid-derived suppressor cells regulate growth of multiple myeloma by inhibiting T cells in bone marrow. J Immunol 190, 3815–3823, doi: 10.4049/jimmunol.1203373 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Favaloro J et al. Myeloid derived suppressor cells are numerically, functionally and phenotypically different in patients with multiple myeloma. Leuk Lymphoma 55, 28932900, doi: 10.3109/10428194.2014.904511 (2014). [DOI] [PubMed] [Google Scholar]
- 81.Gorgun GT et al. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 121, 2975–2987, doi: 10.1182/blood-2012-08-448548 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Castella B et al. Immune Checkpoint Blockade Combinations As Promising Strategy for Cancer Immunotherapy in Multiple Myeloma Patients. Blood 128, 2059–2059 (2016). [Google Scholar]
- 83.Song W et al. Generation of antitumor invariant natural killer T cell lines in multiple myeloma and promotion of their functions via lenalidomide: a strategy for immunotherapy. Clinical Cancer Research 14, 6955–6962 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pratt G, Goodyear O & Moss P Immunodeficiency and immunotherapy in multiple myeloma. Br J Haematol 138, 563–579, doi: 10.1111/j.1365-2141.2007.06705.x (2007). [DOI] [PubMed] [Google Scholar]
- 85.Chen H et al. Increased M2 Macrophages in Multiple Myeloma Patients with Progressive Disease and Down-Regulated Polarization with the JAK2 Inhibitor Ruxolitinib. Blood 124, 4106–4106 (2014). [Google Scholar]
- 86.André T et al. Immune impairments in multiple myeloma bone marrow mesenchymal stromal cells. Cancer Immunology, Immunotherapy 64, 213–224, doi: 10.1007/s00262-014-1623-y (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.An G et al. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: therapeutic implication. Blood 128, 1590–1603, doi: 10.1182/blood-2016-03-707547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tivol EA et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547 (1995). [DOI] [PubMed] [Google Scholar]
- 89.Waterhouse P et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270, 985–988 (1995). [DOI] [PubMed] [Google Scholar]
- 90.Leach DR, Krummel MF & Allison JP Enhancement of antitumor immunity by CTLA-4 blockade. Science 271, 1734–1736 (1996). [DOI] [PubMed] [Google Scholar]
- 91.Agata Y et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol 8, 765–772 (1996). [DOI] [PubMed] [Google Scholar]
- 92.Bae J et al. Function and expression of checkpoint inhibitors and immune agonists on immune cells in monoclonal gammopathy of undetermined significance (MGUS), smoldering multiple myeloma (SMM) and MM and tumor-specific T lymphocytes. Journal of Clinical Oncology 35, 11577–11577, doi: 10.1200/JCO.2017.35.15_suppl.11577 (2017). [DOI] [Google Scholar]
- 93.Tamura H et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia 27, 464–472, doi: 10.1038/leu.2012.213 (2013). [DOI] [PubMed] [Google Scholar]
- 94.Dhodapkar MV et al. Clinical, genomic, and imaging predictors of myeloma progression from asymptomatic monoclonal gammopathies (SWOG S0120). Blood 123, 78–85, doi: 10.1182/blood-2013-07-515239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Paiva B et al. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia 29, 2110–2113, doi: 10.1038/leu.2015.79 (2015). [DOI] [PubMed] [Google Scholar]
- 96.Hallett WH, Jing W, Drobyski WR & Johnson BD Immunosuppressive effects of multiple myeloma are overcome by PD-L1 blockade. Biol Blood Marrow Transplant 17, 1133–1145, doi: 10.1016/j.bbmt.2011.03.011 (2011). [DOI] [PubMed] [Google Scholar]
- 97.Liu J et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD 88-, TRAF6-, and MEK-dependent pathway. Blood 110, 296–304, doi: 10.1182/blood-2006-10-051482 (2007). [DOI] [PubMed] [Google Scholar]
- 98.Lee SJ et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett 580, 755–762, doi: 10.1016/j.febslet.2005.12.093 (2006). [DOI] [PubMed] [Google Scholar]
- 99.Sponaas AM et al. PDL1 Expression on Plasma and Dendritic Cells in Myeloma Bone Marrow Suggests Benefit of Targeted anti PD1-PDL1 Therapy. PLoS One 10, e0139867, doi: 10.1371/journal.pone.0139867 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Carter L et al. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol 32, 634–643, doi: (2002). [DOI] [PubMed] [Google Scholar]
- 101.Ishibashi M et al. Myeloma Drug Resistance Induced by Binding of Myeloma B7-H1 (PD-L1) to PD-1. Cancer immunology research 4, 779–788, doi: 10.1158/2326-6066.cir-15-0296 (2016). [DOI] [PubMed] [Google Scholar]
- 102.Huang S-Y et al. Soluble PD-L1: a biomarker to predict progression of autologous transplantation in patients with multiple myeloma. Oncotarget 7, 62490 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang L et al. Serum levels of soluble programmed death ligand 1 predict treatment response and progression free survival in multiple myeloma. Oncotarget 6, 41228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Quezada SA & Peggs KS Exploiting CTLA-4, PD-1 and PD-L1 to reactivate the host immune response against cancer. Br J Cancer 108, 1560–1565, doi: 10.1038/bjc.2013.117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nicodemus CF Antibody-based immunotherapy of solid cancers: progress and possibilities. Immunotherapy 7, 923–939 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zinzani PL et al. Safety & tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood, blood-2016–2012-758383 (2017). [DOI] [PMC free article] [PubMed]
- 107.Ansell SM et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. New England Journal of Medicine 372, 311–319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Younes A et al. Nivolumab for classical Hodgkin’s lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: a multicentre, multicohort, single-arm phase 2 trial. The Lancet Oncology 17, 1283–1294 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Armand P et al. Programmed death-1 blockade with pembrolizumab in patients with classical Hodgkin lymphoma after brentuximab vedotin failure. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 34, 3733–3739 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Davids MS et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. N Engl J Med 375, 143–153, doi: 10.1056/NEJMoa1601202 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Berger R et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res 14, 3044–3051, doi: 10.1158/1078-0432.ccr-07-4079 (2008). [DOI] [PubMed] [Google Scholar]
- 112.Lesokhin AM et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J Clin Oncol 34, 2698–2704, doi: 10.1200/jco.2015.65.9789 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ribrag V et al. PEMBROLIZUMAB MONOTHERAPY FOR RELAPSED/REFRACTORY MULTIPLE MYELOMA (RRMM): PHASE 1B KEYNOTE-013 STUDY. EHA EHA Learning Center; (2017). [Google Scholar]
- 114.Gorgun G et al. Lenalidomide Enhances Immune Checkpoint Blockade-Induced Immune Response in Multiple Myeloma. Clin Cancer Res 21, 4607–4618, doi: 10.1158/1078-0432.ccr-15-0200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Luptakova K et al. Lenalidomide enhances anti-myeloma cellular immunity. Cancer immunology, immunotherapy : CII 62, 39–49, doi: 10.1007/s00262-012-1308-3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Luptakova K et al. Lenalidomide Decreases PD-1 Expression, Depletes Regulatory T-Cells and Improves Cellular Response to a Multiple Myeloma/Dendritic Cell Fusion Vaccine In Vitro. Blood 116, 492–492 (2010). [Google Scholar]
- 117.Pianko MJ et al. Efficacy and toxicity of therapy immediately after treatment with nivolumab in relapsed multiple myeloma. Leuk Lymphoma 59, 221–224, doi: 10.1080/10428194.2017.1320713 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Badros A et al. Pembrolizumab, pomalidomide, and low-dose dexamethasone for relapsed/refractory multiple myeloma. Blood 130, 1189–1197, doi: 10.1182/blood-2017-03-775122 (2017) [DOI] [PubMed] [Google Scholar]
- 119.Ocio EM et al. Pembrolizumab (Pembro) plus lenalidomide (Len) and low-dose dexamethasone (Dex) for relapsed/refractory multiple myeloma (RRMM): Efficacy and biomarker analyses. Journal of Clinical Oncology 35, 8015–8015, doi: 10.1200/JCO.2017.35.15_suppl.8015 (2017). [DOI] [Google Scholar]
- 120.Mateos M-V et al. A phase 3 randomized study of pembrolizumab (Pembro) plus pomalidomide (Pom) and dexamethasone (Dex) for relapsed/refractory multiple myeloma (RRMM): KEYNOTE-183. Journal of Clinical Oncology 36, 8021–8021, doi: 10.1200/JCO.2018.36.15_suppl.8021 (2018). [DOI] [Google Scholar]
- 121.Krauss AC et al. FDA analysis of pembrolizumab trials in multiple myeloma: Immune related adverse events (irAEs) and response. Journal of Clinical Oncology 36, 8008–8008, doi: 10.1200/JCO.2018.36.15_suppl.8008 (2018). [DOI] [Google Scholar]
- 122.Usmani SZ et al. A phase 3 randomized study of pembrolizumab (pembro) plus lenalidomide (len) and low-dose dexamethasone (Rd) versus Rd for newly diagnosed and treatment-naive multiple myeloma (MM): KEYNOTE-185. Journal of Clinical Oncology 36, 8010–8010, doi: 10.1200/JCO.2018.36.15_suppl.8010 (2018). [DOI] [Google Scholar]
- 123.Attia P et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol 23, 6043–6053, doi: 10.1200/jco.2005.06.205 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Weber JS et al. Safety Profile of Nivolumab Monotherapy: A Pooled Analysis of Patients With Advanced Melanoma. J Clin Oncol 35, 785–792, doi: 10.1200/jco.2015.66.1389 (2017). [DOI] [PubMed] [Google Scholar]
- 125.Downey SG et al. Prognostic factors related to clinical response in patients with metastatic melanoma treated by CTL-associated antigen-4 blockade. Clin Cancer Res 13, 6681–6688, doi: 10.1158/1078-0432.ccr-07-0187 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Postow MA, Sidlow R & Hellmann MD Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N Engl J Med 378, 158–168, doi: 10.1056/NEJMra1703481 (2018). [DOI] [PubMed] [Google Scholar]
- 127.Lesokhin AM et al. Nivolumab in combination with daratumumab, with or without pomalidomide and dexamethasone, for relapsed/refractory multiple myeloma: 2 cohorts of the phase 1 CheckMate 039 safety study. Journal of Clinical Oncology 35, TPS3102-TPS3102, doi: 10.1200/JCO.2017.35.15_suppl.TPS3102 (2017). [DOI] [Google Scholar]
- 128.Lonial S et al. CheckMate 602: An open-label, randomized, phase 3 trial of combinations of nivolumab, elotuzumab, pomalidomide and dexamethasone in relapsed/refractory multiple myeloma. Journal of Clinical Oncology 35, TPS8052-TPS8052, doi: 10.1200/JCO.2017.35.15_suppl.TPS8052 (2017). [DOI] [Google Scholar]
- 129.Bezman NA et al. PD-1 blockade enhances elotuzumab efficacy in mouse tumor models. Blood advances 1, 753–765 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Richardson PG et al. Durvalumab (DURVA) plus daratumumab (DARA) in patients (pts) with relapsed and refractory multiple myeloma (RRMM). J Clin Oncol 35 (2017). [Google Scholar]
- 131.Zelle-Rieser C et al. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. Journal of hematology & oncology 9, 116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Suen H et al. Multiple myeloma causes clonal T-cell immunosenescence: identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia 30, 1716 (2016). [DOI] [PubMed] [Google Scholar]
- 133.Koyama S et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nature communications 7, 10501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shayan G et al. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology 6, e1261779 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ratner L, Waldmann TA, Janakiram M & Brammer JE Rapid Progression of Adult T-Cell Leukemia-Lymphoma after PD-1 Inhibitor Therapy. N Engl J Med 378, 1947–1948, doi: 10.1056/NEJMc1803181 (2018). [DOI] [PubMed] [Google Scholar]
- 136.Ishitsuka K, Utsunomiya A & Ishida T PD-1 Inhibitor Therapy in Adult T-Cell Leukemia–Lymphoma. The New England journal of medicine 379, 695 (2018). [DOI] [PubMed] [Google Scholar]
- 137.Routy B et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97, doi: 10.1126/science.aan3706 (2018). [DOI] [PubMed] [Google Scholar]
- 138.Matson V et al. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gopalakrishnan V et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Drayson MT et al. Tackling Early Morbidity and Mortality in Myeloma (TEAMM): Assessing the Benefit of Antibiotic Prophylaxis and Its Effect on Healthcare Associated Infections in 977 Patients. Blood 130, 903–903 (2017).28637661 [Google Scholar]
- 141.Levy A, Massard C, Soria JC & Deutsch E Concurrent irradiation with the anti-programmed cell death ligand-1 immune checkpoint blocker durvalumab: Single centre subset analysis from a phase 1/2 trial. Eur J Cancer 68, 156–162, doi: 10.1016/j.ejca.2016.09.013 (2016). [DOI] [PubMed] [Google Scholar]
- 142.Twyman-Saint Victor C et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520, 373 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hiniker SM et al. A prospective clinical trial combining radiation therapy with systemic immunotherapy in metastatic melanoma. International Journal of Radiation Oncology* Biology* Physics 96, 578–588 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ko EC & Formenti SC Radiotherapy and checkpoint inhibitors: a winning new combination? Therapeutic advances in medical oncology 10, 1758835918768240 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rosenblatt J et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin Cancer Res 19, 3640–3648, doi: 10.1158/1078-0432.ccr-13-0282 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Garcia-Marquez MA, Shimabukuro-Vornhagen A, Theurich S & von Bergwelt-Baildon M Vaccination with dendritic cell-tumor fusion cells in multiple myeloma patients: a promising strategy? Immunotherapy 5, 1039–1042, doi: 10.2217/imt.13.96 (2013). [DOI] [PubMed] [Google Scholar]
- 147.Rosenblatt J et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. Journal of immunotherapy (Hagerstown, Md. : 1997) 34, 409–418, doi: 10.1097/CJI.0b013e31821ca6ce (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Karp Leaf R, Cho HJ & Avigan D Immunotherapy for Multiple Myeloma, Past, Present, and Future: Monoclonal Antibodies, Vaccines, and Cellular Therapies. Curr Hematol Malig Rep 10, 395–404, doi: 10.1007/s11899-015-0283-0 (2015). [DOI] [PubMed] [Google Scholar]
- 149.Rosenblatt J & Avigan D Targeting the PD-1/PD-L1 axis in multiple myeloma: a dream or a reality? Blood 129, 275–279, doi: 10.1182/blood-2016-08-731885 (2017). [DOI] [PubMed] [Google Scholar]
- 150.Chung DJ et al. T-cell Exhaustion in Multiple Myeloma Relapse after Autotransplant: Optimal Timing of Immunotherapy. Cancer immunology research 4, 61–71, doi: 10.1158/2326-6066.cir-15-0055 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rotolo A, Caputo V & Karadimitris A The prospects and promise of chimeric antigen receptor immunotherapy in multiple myeloma. Br J Haematol 173, 350–364, doi: 10.1111/bjh.13976 (2016). [DOI] [PubMed] [Google Scholar]
- 152.Raje NS et al. bb2121 anti-BCMA CAR T-cell therapy in patients with relapsed/refractory multiple myeloma: Updated results from a multicenter phase I study. Journal of Clinical Oncology 36, 8007–8007, doi: 10.1200/JCO.2018.36.15_suppl.8007 (2018). [DOI] [Google Scholar]
- 153.Fan F et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. Journal of Clinical Oncology 35, LBA3001-LBA3001, doi: 10.1200/JCO.2017.35.18_suppl.LBA3001 (2017). [DOI] [Google Scholar]
- 154.Cohen AD et al. (Am Soc Hematology, 2017).
- 155.Moon EK et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res 20, 4262–4273, doi: 10.1158/1078-0432.ccr-13-2627 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cherkassky L et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest 126, 3130–3144, doi: 10.1172/jci83092 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jing W et al. Combined immune checkpoint protein blockade and low dose whole body irradiation as immunotherapy for myeloma. Journal for immunotherapy of cancer 3, 2, doi: 10.1186/s40425-014-0043-z (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Guillerey C et al. TIGIT immune checkpoint blockade restores CD8+ T cell immunity against multiple myeloma. Blood, blood-2018–2001-825265 (2018). [DOI] [PubMed]
- 159.Yadav M et al. Tigit, CD226 and PD-L1/PD-1 Are Highly Expressed By Marrow-Infiltrating T Cells in Patients with Multiple Myeloma. Blood 128, 2102–2102 (2016). [Google Scholar]
- 160.Yan G et al. A RIPK3-PGE2 circuit mediates myeloid-derived suppressor cell-potentiated colorectal carcinogenesis. Cancer Res, doi: 10.1158/0008-5472.can-17-3962 (2018). [DOI] [PubMed] [Google Scholar]
- 161.Hou W, Sampath P, Rojas JJ & Thorne SH Oncolytic Virus-Mediated Targeting of PGE2 in the Tumor Alters the Immune Status and Sensitizes Established and Resistant Tumors to Immunotherapy. Cancer Cell 30, 108–119, doi: 10.1016/j.ccell.2016.05.012 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mao Y et al. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res 20, 4096–4106, doi: 10.1158/1078-0432.ccr-14-0635 (2014). [DOI] [PubMed] [Google Scholar]
- 163.Veltman JD et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer 10, 464, doi: 10.1186/1471-2407-10-464 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Orillion A et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin Cancer Res 23, 5187–5201, doi: 10.1158/1078-0432.ccr-17-0741 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sheng W et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 174, 549–563.e519, doi: 10.1016/j.cell.2018.05.052 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Brohl AS et al. A phase IB study of ipilimumab with peginterferon alfa-2b in patients with unresectable melanoma. Journal for immunotherapy of cancer 4, 85 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yuasa T, Masuda H, Yamamoto S, Numao N & Yonese J Biomarkers to predict prognosis and response to checkpoint inhibitors. International journal of clinical oncology 22, 629–634, doi: 10.1007/s10147-017-1122-1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Boutsikou E et al. Tumour necrosis factor, interferon-gamma and interleukins as predictive markers of antiprogrammed cell-death protein-1 treatment in advanced non-small cell lung cancer: a pragmatic approach in clinical practice. Therapeutic advances in medical oncology 10, 1758835918768238, doi: 10.1177/1758835918768238 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Manson G, Norwood J, Marabelle A, Kohrt H & Houot R Biomarkers associated with checkpoint inhibitors. Annals of oncology : official journal of the European Society for Medical Oncology 27, 1199–1206, doi: 10.1093/annonc/mdw181 (2016). [DOI] [PubMed] [Google Scholar]